

Abstract
Stereoselective synthesis of octahydro-5,6,6a-triazaacenaphthalenes 29 and 34 having the anti relationship of the angular hydrogens flanking the pyrrolidine nitrogen confirmed suspicions that the relative configuration of the left hand tricyclic guanidine fragment of batzelladine F should be revised to have the syn relationship of these hydrogens. Several strategies were examined for coupling tricyclic guanidine fragments to prepare potential structures for batzelladine F. Eventually a convergent synthesis strategy was devised, whose central step was a fragment-coupling tethered-Biginelli reaction (Scheme 17). Using this approach we synthesized four potential structures of batzelladine F, 35–38. None of these compounds, nor their enantiomers, were identical to natural batzelladine F. Reinvestigation of mass spectra of natural batzelladine F, and fragments 88 and 89 obtained upon saponification of batzelladine F, demonstrated that the originally proposed connectivity of this alkaloid was also incorrect. The revised connectivity, 90, of natural batzelladine F depicted in Scheme 21 is proposed.
Introduction
In 1997, Patil and co-workers reported the isolation of batzelladines F–I from a red Jamaican sponge incorrectly identified at the time as Batzella sp;2,3 the structures 1–4 assigned originally to these alkaloids are shown in Figure 1. Each of these natural products contain two tricyclic guanidines and were reported to induce dissociation of the tyrosine kinase p56lck from CD4. It was postulated that disruption of this interaction could be used to treat autoimmune diseases. As part of our larger program in the synthesis of guanidinium alkaloids,4 we were attracted to batzelladine F for two reasons. First, we were interested in preparing compounds that inhibit specific protein-protein interactions involving large surface areas, and, second, we sought to define totally the relative and absolute configuration of batzelladine F, much of which was unclear at the outset of this work.5
Figure 1.

Proposed Structures of Batzelladines F–I.
The original structural proposal 1 for batzelladine F was based on the following analysis. The relative configuration of the right-hand tricyclic portion (C18 through C28) of batzelladine F was assigned by comparison of its 13C NMR spectra to that of the tricyclic guanidine alkaloid batzelladine D, the structure of which had been defined by total synthesis.6,7 The configuration of the left-hand tricyclic portion (C1 through C9) was assigned on the basis of NOE data.8 The lengths of the nonyl chain at C25, and the length of the chain connecting the tricyclic guanidines, were assigned by MS fragmentation patterns, but this analysis was not discussed, nor were the data presented. There was no information that related the relative configurations of the two tricyclic guanidine fragments, specified the configuration at C16, or defined the absolute configuration.
In 1999, Snider and Murphy independently published syntheses of tricyclic guanidines related to the left-hand portion of batzelladine F that have a syn relationship of the methine hydrogens flanking the pyrrolidine nitrogen.9 The 13C NMR data of these analogs supported a revision of the configuration of the natural product at C4 and C7 from anti to syn. However, rigorous verification of this proposal would require the synthesis of a related anti-fused tricyclic guanidine and the demonstration that its spectra were distinct from those of the natural product.
In this contribution, we first describe our synthesis of 2a,8a-anti tricyclic guanidines corresponding to the originally proposed relative configuration of these units of batzelladine F and proof that the original configurational assignment of the right-hand guanidine fragment of this alkaloid was incorrect (Section I). We then describe our preparation of analogous 2a,8a-syn tricyclic guanidines (Section II) and the evolution of our strategy for the total synthesis of linked tricyclic guanidines corresponding to the proposed constitution and revised relative configuration of batzelladine F (Section III–Sections IV). We then demonstrate that the originally reported connectivity of the natural product was also incorrect, leading to a new proposal for the structure of batzelladine F (Section V). In the following contribution, we detail our synthesis of this new structure, and studies that define all aspects of the structure of batzelladine F.10
Results and Discussion
I. Synthesis of 2a,8a-Anti-decahydrotriazaacenaphthalenes Corresponding to the Originally Proposed Structure of Batzelladine F
Believing that both tricyclic guanidine fragments of batzelladine F possessed the same anti relationship between the angular hydrogens at carbons 4 and 7, and carbons 20 and 23 (batzelladine numbering), we conceived of a divergent strategy that would allow us to construct each half of the molecule from a common intermediate. We chose the ester bond as a logical first disconnection, thus, generating alcohol 5 and acid 6 as synthetic objectives. Each of these intermediates was conceived as arising from an intermediate such as 7a or 7b, which differ in the side chain at C9. Employing a disconnection developed during our total synthesis of batzelladine D,6 intermediates 7a and 7b were simplified to a common precursor 8 bearing a masked aldehyde that would allow for attachment of different side chains by olefination reactions. Anti amino alcohol 8 was seen as arising from β-hydroxy ester 9.
The preparation of common intermediate 8 began with Weinreb amide 10,11 to which Grignard reagent 1112 was added to afford β-hydroxy ketone 12 in 76% yield (Scheme 2). This intermediate was subjected to Evans’ variant of the Tishchenko reduction to set the anti relationship and differentiate the hydroxyl groups of the resulting 1,3-diol.13 In this way, mono-protected diol 13 was obtained in 92% yield from ketone precursor 12 as a single stereoisomer. Nitrogen was installed in the form of an azide, by Mitsunobu inversion of alcohol 13 with hydrazoic acid to provide azide 14 in 95% yield.14,15 The 1,3-anti configuration was then re-established by methanolysis of propionate 14, followed by Mitsunobu inversion of the product alcohol with p-nitrobenzoic acid16 to deliver aryl ester 15 in 78% yield for the two steps. Ozonolysis of unsaturated azido ester 15 in a mixture of MeOH, CH2Cl2 and NaHCO3 provided aldehyde 16, setting the stage for attachment of the different tricyclic guanidine side chains.17
Scheme 2.

After aldehyde 16 failed to react cleanly with the ylide derived from a simple hexyl phosphonium salt, we turned to the Kocienski modification of the Julia olefination.18 This procedure required the synthesis of an appropriate sulfone partner (Scheme 3). Thus, diol 17 was mono-protected by reductive opening of the corresponding benzylidine acetal to provide alcohol 18 in 94% yield and 96% ee. 19,20 Activation of the alcohol as a tosylate allowed displacement with mercapto tetrazole 19. Oxidation of the resulting sulfide with m-chloroperbenzoic acid (m-CPBA) in a mixture of CH2Cl2 and pH 7 buffer afforded sulfone 20 in 72% yield for the three steps.
Scheme 3.

Aldehyde 16 was then elaborated to amino alcohol 22 as summarized in Scheme 4. Union of sulfone 20 with aldehyde 16 was accomplished by addition of a THF solution of the aldehyde to a small excess of the lithium reagent derived from sulfone 20 at −50 °C. Alkene 21 was obtained in ~75% yield as a 3:2 mixture of stereoisomers. This product was contaminated with residual sulfone 20, which could be removed more readily after hydrolysis of the ester. The azide and double bond were then reduced concurrently with hydrogen over palladium poisoned with ethylenediamine.21 As expected, the benzyl ether was stable to this reduction. In this fashion, amino alcohol 22 was obtained in 73% overall yield from aldehyde 16.
Scheme 4.

Employing chemistry developed during our earlier synthesis of the guanidine core of batzelladine D,7 the conversion of amino alcohol 22 to the corresponding tricyclic guanidine 26 took place smoothly (Scheme 5).22 Guanylation of amine 22 with reagent 23,23 followed by hydrolysis of the acetal provided guanidine hemi aminal 24. This labile intermediate was immediately condensed with allyl acetoacetate to provide Biginelli product 25 in 45% yield as an 8:1 mixture of stereoisomers.24 After separation of the minor syn stereoisomer by chromatography, mesylation of the anti product and ring closure provided tricyclic guanidine 26 in 78% yield.
Scheme 5.

With allyl ester 26 in hand, we sought to investigate its decarboxylation. Such transformations of simple vinylogous carbamates were precedented, typically involving heating the free acid to >100 °C in the presence of an acid or catalyst such as cyanide.25 We were delighted to find that reaction of ester 26 with catalytic (PPh3)4Pd and pyrrolidine in a mixture of THF and MeOH resulted within an hour in cleavage of the allyl ester and concomitant decarboxylation.26 The intermediate enamine absorbed one equivalent of MeOH to yield hemi aminal 27. Without isolation, this intermediate was reduced with NaBH3CN in AcOH to afford saturated tricyclic guanidine 28 in 67% yield. Stereoselection in this reduction results from axial delivery of hydride to the N-amidinyl iminium ion derived from 27.27 Finally, the benzyl ether of 28 was removed by hydrogenolysis to provide alcohol 29.
The facility with which guanidine ester 26 decarboxylates after deallylation can be explained by postulating an intermediate such as 31, which would arise from tautomerization of 30 (Scheme 7). The breaking bond in guanidnium carboxylate 31 would be aligned well with the iminium π-system.
Scheme 7.

We were now in a position to compare the NMR spectra of our tricyclic guanidine products, batzelladine F, and Murphy’s syn-fused model compound 33 (Table 1).28 The 13C NMR spectra of the two anti-fused tricyclic guanidines 29 and 3429 match each other almost perfectly yet differ somewhat from those of the natural product. In contrast, the 13C NMR data for 33 match those of the natural product closely. It was clear from this comparison that the proposed anti relationship of the angular hydrogens of the left-hand guanidine fragment of batzelladine F was incorrect: the actual configurational relationship is indeed syn.
Table 1.
Comparison of 13C Chemical Shifts of Batzelladine F and Tricyclic Model Compounds.a
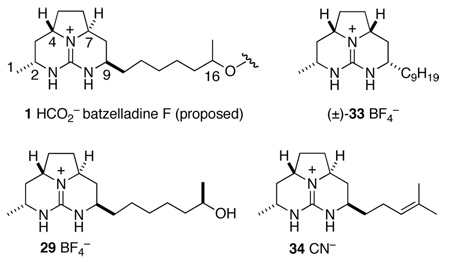 | ||||
|---|---|---|---|---|
| cabon # | 29 | 34 | 1 | 33 |
| 1 | 21.7 | 21.7 | 20.7 | 20.7 |
| 2 | 48.9 | 48.6 | 47.2 | 47.3 |
| 4 | 56.5 | 56.5 | 57.5 | 57.5 |
| 7 | 56.4 | 56.3 | 57.4 | 57.5 |
| 9 | 53.1 | 52.8 | 51.6 | 51.6 |
13C NMR in CD3OD (500 MHz).
II. Synthesis of 2a,8a-Syn-decahydrotriazaacenaphthalenes
Although the relative configuration of the left-hand tricyclic portion of batzelladine F was now firmly established as syn, no information was available regarding the relative configuration of the two guanidine fragments, the relative configuration of the methyl substituent of the linker, nor the absolute configuration. Thus, there were eight compounds 35–38 (four pairs of enantiomers) that fit all of the available data for batzelladine F (Figure 2).
Figure 2.


Plausible Structures for Batzelladine F as of 1999.
Proposed revised constitution of batzelladine F (90) and the configurational uncertainties yet to be resolved.
We turned next to develop a convenient synthesis of the left-hand tricyclic guanidine portions of 35–38. The approach we chose was founded on the chemistry we had developed earlier to synthesize the tricyclic moiety of batzelladine B,22 in this case a tethered Biginelli reaction would be employed to combine fragments 41 and 42 (Scheme 8). Both of these intermediates we saw being available from the same starting material, methyl (R)-3-hydroxybutarate (44).
Scheme 8.

The synthesis of β-keto ester 52 began with (R)-iodide 45, which was prepared in three routine steps from ester 44 (Scheme 9).30 Alkylation of this iodide with the lithium enolate of tert-butyl acetate proceeded well on a small scale.31 On larger scale, the desired product 46 was contaminated with varying amounts of the dialkylation product 47, which was difficult to remove. Reduction of this mixture with LiAlH4 provided a mixture of primary alcohols, from which the desired product 48 could be isolated cleanly.32 Conversion of alcohol 48 to iodide 49 set the stage for elaboration to a β-keto ester. Thus, addition of the dianion of methyl acetoacetate (50) to iodide 49 proceeded cleanly, delivering 51 in 68% yield.33 Transesterification of this product with allyl alcohol gave allyl ester 52 in nearly quantitative yield.34
Scheme 9.

Guanidine hemi aminal 41 was assembled by the sequence summarized in Scheme 10. Hydroxybutyrate 44 was converted first to Weinreb amide 53 in 80% yield using the Merck procedure (Scheme 10).35 Addition of Grignard reagent 11 to this amide provided hydroxy ketone 54, which was reduced with Et2BOMe and NaBH4 to provide syn-diol 55 as a single stereoisomer.36 Double Mitsunobu reaction of this intermediate with hydrazoic acid, followed by hydrogenation of the diazide product over Pd·C provided diamine 43 in 80% yield. Conversion of this intermediate to Troc-protected guanidine 57 was accomplished with reagent 56 in 82% yield.37
Scheme 10.

To set the stage for the Biginelli condensation, the Troc group and the dimethyl acetal were removed from guanidine 57 simultaneously by reaction with Zn dust in aqueous AcOH. However, removal of residual zinc salts from the resulting crude guanidine hemi aminal 41 proved to be problematic. For example, saturation of the reaction with H2S formed a thick precipitate, presumably ZnS, which was difficult to filter. Moreover, the polar nature of guanidine salt 41 caused it to adhere to these salts. At most, 40% of the desired product could be recovered from this reaction. Therefore, we prepared the benzyl imino thiocarbamate 58 (see Supporting Information for details), and condensed it with diamine 43 to form the Cbz-protected guanidine 59 in 82% yield. Hydrogenolysis of this intermediate removed the Cbz group. Hydrolysis of the resulting guanidinium acetal proceeded cleanly, albeit slowly, at room temperature in aqueous acetic acid to provide bicyclical hemi aminal 41 in nearly quantitative yield over the two steps.
With the two tethered Biginelli reaction partners in hand, the construction of 2a,8a-syndecahydrotriazaacenaphthalene 39 proceeded uneventfully (Scheme 11). Guanidine 41 condensed smoothly with an excess of β-keto ester 52, under previously optimized conditions,22 to provide tricyclic guanidine 60 in 81% yield as a 5:1 mixture of syn and anti stereoisomers. As in the anti series, the unnecessary ester appendage was removed by brief treatment of Biginelli product 60 with (Ph3P)4Pd and pyrrolidine, followed by reduction of the intermediate hemi aminal with NaBH4 in AcOH. Work-up of this reaction with 1 N HCl then removed the silyl group. After chromatography on SiO2, the product was treated with aqueous NaBF4 to provide guanidine alcohol 39 having a BF4− counter ion in 66% overall yield from ester 60. As expected, the diagnostic methine resonances in the 13C NMR spectrum of syn-decahydrotriazaacenaphthalene 39 matched almost exactly those signals of batzelladine F and Murphy’s model compounds (see Supporting Information).
Scheme 11.

III. Synthesis of the Revised Proposed Structure of Batzelladine F: -Bromo Acid Strategy
The preparation of the right-hand 2a,8a-anti-decahydrotriazaacenaphthalenecarboxylate is summarized in Scheme 12. This synthesis began with hemi aminal 61,7 which was condensed with tert-butyl acetoacetate under conditions optimized for anti stereoselection. This reaction provided anti bicyclic guanidine 62, which was isolated as the acetate salt after chromatography on silica gel. Conversion of this intermediate to the corresponding mesylate was complicated by N-sulfonylation of the guanidine. Eventually, we found that exchanging the acetate counter ion of guanidine 62 for tetrafluoroborate allowed for clean mesylation of the alcohol upon reaction with MsCl and Et3N in CH2Cl2. This mesylate cyclized in hot CHCl3 in the presence of excess Et3N to provide tricyclic guanidine ester 63 in 62% overall yield. As expected,7 hydrogenation of 63 over Rh·Al2O3 proceeded with little stereoselection. The resulting mixture of esters was immediately deprotected with HCO2H to provide the corresponding acids. These isomers were separated by reverse-phase HPLC to provide stereoisomer 64, having the same relative configuration as batzelladine D and the right-hand guanidine fragment of batzelladine F, in 30% yield along with 48% of stereoisomer 65.
Scheme 12.

With the two tricyclic guanidine fragments available, their union was investigated. Using the more abundant isomer 65 as a model, a variety of coupling reagents were evaluated, including diimides, DEAD–Ph3P, and activated esters.38 The only conditions that showed promise involved the use of 2-chloro-1-methylpyridinium iodide (Mukaiyama's salt) and DMAP in MeCN.39 Coupling of acid 65 and alcohol 39 in this way provided ester 66 in 48% yield after purification by HPLC (Scheme 13).
Scheme 13.

With a successful coupling procedure in hand, we turned to prepare the hexacyclic diguanidine 35 depicted in Figure 2 (Scheme 14). The desired coupling of guanidine alcohol 39 and guanidinium carboxylate 64 did not take place at 50 °C or 75 °C.38 However, when this coupling reaction was conducted at 100 °C, ester 68 was formed in 57% yield. Unfortunately, upon characterization, it became apparent that 68 had undergone epimerization at C19. Particularly diagnostic was the 1H NMR signal of C19, which appeared as a doublet of doublets at δ 2.38 (dd, J = 10.3, 10.3 Hz, CD3OD); whereas authentic batzelladine F shows the corresponding resonance at δ 3.06 (dd, J = 4.6, 3.3 Hz, CD3OD). It was subsequently discovered that acid 64 rapidly epimerizes under these reaction conditions, and the resulting epimeric acid, 67, couples to alcohol 39 rapidly at 50 °C. These results lead us to believe that epimerization preceeded coupling. The failure of 64 to couple under these conditions and its epimerization under more forcing conditions is readily attributed to the hindered axial nature of the carboxylic acid substituent.
Scheme 14.

We turned to explore whether the ester could be epimerized after coupling. In our synthesis of batzelladine D, an attempt was made to epimerize a related ester by kinetic protonation of an enolate intermediate.7 Although yields and selectivities were low in this earlier study, we felt that this strategy warranted re-investigation. The desired configuration of the diguanidine ester would arise if the enolate of ester 68 protonated from the less-hindered β face.
Before embarking on such an ambitious undertaking, we sought to explore the epimerization step in a model system. We anticipated that success would require that at least some of the NH hydrogens of the guanidine groups be masked. Thus, the tricyclic guanidine ester 69 depicted in equation 1 was assembled by reduction of α,β-unsaturated ester 63 with NaBH3CN in AcOH, followed by N-benzylation by reaction with KHMDS and benzyl bromide. We were disappointed to find that reaction of this ester with a variety of bases (LDA, LiNEt2, KHMDS, BuLi), followed by quenching with a deuterium source resulted in no deuterium incorporation (equation 1). In contrast, reaction of guanidine ester 69 with excess tert-butyllithium at −78 °C in THF, followed by quenching the reaction with CD3OD resulted in 100% deuteration. However, 1H-NMR showed to our surprise that deuterium had been introduced at the benzylic methylene group.40

Unable to generate an enolate by deprotonation, we sought to access this intermediate in a slightly more circuitous manner. We anticipated that a bromine substituent α to the ester would allow the enolate required for epimerization to be generated by reduction. The synthesis of the bromo acid required to investigate this strategy began with unsaturated ester 63, which was treated with N-bromosuccinimide (NBS) in MeOH to generate bromo ether 71 in quantitative yield (Scheme 15). This intermediate was reduced immediately with NaBH3CN in AcOH to provide α-bromo ester 72. This reduction was plagued by competing elimination of “BrOMe” to return unsaturated ester 63, which under the reaction conditions was reduced to ester 73.41 Cleavage of the tert-butyl group of ester 72 with 98% HCO2H, and purification of the product by HPLC furnished the desired acid 74 in 47% overall yield. Although not established rigorously, the configuration indicated for 74 would arise from diaxial addition of MeOBr to 63 and axial reduction of the N-amidinyl iminium ion generated from 71.
Scheme 15.

With guanidine α-bromo acid 74 available, its coupling to guanidine alcohol 39 was investigated (Scheme 16). Using Mukaiyama's salt, coupling proceeded to a small extent (~20%, HPLC-MS analysis) after 24 h at 50 °C. Attempts to force the reaction by increasing the temperature to 85 °C provided a mixture of diguanidines 75 and 76 in which both elimination of HBr and coupling had taken place. These double bond isomers could be separated by HPLC, and were readily distinguished by 1H NMR spectroscopy.42
Scheme 16.

The formation of 76 finally suggested a potentially straightforward route for accessing batzelladine F: couple the guanidine fragments prior to saturating the right-hand tricyclic guanidine fragment. We proceeded to test the last stage of this strategy with diguanidine 76, which was now available (Scheme 17). Hydrogenation of 76 over Rh·Al2O3 provided stereoisomeric diguanidines 35 and 77, which could be separated by HPLC. Although compound 35 showed 1H and 13C NMR spectra consistent with those of natural batzelladine F, it was substantially different from the natural product by HPLC analysis.
Scheme 17.

Given that we had only a 25% chance of making the correct relative stereoisomer at the outset, it was not surprising that 35 was not batzelladine F. At this point, we were faced with two issues. First was the problem of efficiently linking the guanidine fragments and installing the desired configuration at C19. Second was the identity of the natural product. Although it was not immediately clear how the former could be solved, a late stage catalytic hydrogenation strategy should permit us to prepare the remaining three stereoisomers, allowing for elucidation of the relative and absolute configuration of the natural product.
IV. Fragment Coupling Biginelli Strategy for the Synthesis of the Revised Proposed Structure of Batelladine F and Stereoisomers
Having decided to pursue a synthesis strategy with saturation of the C19–C27 double bond as the final step, we realized that there was a shorter route to such late-stage intermediates than the α-bromo acid coupling strategy. This approach is outlined retrosynthetically in Scheme 17. As in our synthesis of batzelladine D, we envisaged the right-hand tricyclic guanidine fragment of 1 evolving from pentacyclic diguanidine 78 by ring closure followed by hydrogenation. This intermediate would be the product of a highly convergent tethered Biginelli condensation between β-keto ester 79 and guanidine hemi-aminal 61. Ester 79 would arise from alcohol 80.
To pursue the strategy outlined in Scheme 17, we began with guanidine alcohol 81 whose tricyclic guanidine moiety is enantiomeric to the guanidine fragment of alcohol 39. Alcohol 81, which was prepared analogously to 39,43 was acylated with methyl acetoacetate in the presence of DMAP to provide β-keto ester 82 in quantitative yield (Scheme 18).34 This material was combined with an excess of guanidine aminal 61 and morpholinium acetate in 2,2,2-trifluoroethanol and heated at 60 °C for 48 h. Pentacyclic diguanidine 83 was obtained in 64% yield after separation from residual 61 and minor amounts (≤10%) of isomer 84.
Scheme 18.

To complete the synthesis of the hexacyclic diguandine, we needed to close the final ring, and saturate the C19–C27 double bond (Scheme 19). To this end, the triflouroacetate counter ions of intermediate 83 were exchanged for tetrafluoroborate by washing a CHCl3 solution of 83 with aqueous NaBF4. This diguanidine salt was converted to its mesylate derivative by reaction with MsCl and Et3N in CH2Cl2, and the mesylate intermediate was cyclized in hot CHCl3 in the presence of excess Et3N to deliver 85 in 55% overall yield. Finally, hydrogenation of hexacyclic diguanidine 85 over Rh·Al2O3 in acidic MeOH provided batzelladine F isomers 37 and 86, neither of which was identical to the natural product by HPLC comparisons.
Scheme 19.

The stereoisomers of the proposed structure of batzelladine F that remained to be synthesized were 36 and 38, which were epimeric at C16 to the isomers prepared thusfar (Figure 2). The synthesis of these compounds, which paralleled that of 37, is detailed in the Supporting Information. To our chagrin, these isomers were also distinct from an authentic sample of batzelladine F by HPLC comparisons.
V. What is Batzelladine F?
We had made one enantiomer of what we believed to be the four “possible” structures of batzelladine F that differed in relative configuration, however none were identical by HPLC comparisons to the natural product. Attention next turned to the remote possibility that the guanidinium counter ions were complicating these comparisons. Batzelladine F was isolated as its diformate salt, and retains these counter ions strongly. Thus, we converted synthetic stereoisomer 37, a ditrifluoroacetate salt, to its diformate salt by washing a CHCl3 solution of 37 with aqueous formic acid-sodium formate solution (0.5 M each). Complete conversion to the formate salt was confirmed by 19F NMR analysis by observing disappearance of the signal at −76.8 ppm. As expected this diformate salt had the same HPLC retention time as the corresponding ditrifluoroacetate salt.
Having confirmed that the counter ion would be unlikely to interfere with HPLC comparisons, our focus turned to a careful examination of the small sample of authentic batzelladine F that we had obtained from the SKB sample collection. Although some impurities were apparent, it was clear from its 1H NMR spectrum that this sample had not decomposed appreciably. In particular, the major component retained the original axial orientation of the ester as evidenced by the chemical shift and coupling constants of the methine hydrogen adjacent to the ester: δ 3.06 (dd, J = 4.6, 3.3 Hz, CD3OD). As expected, the major peak in the HPLC chromatogram of the authentic sample possessed the expected exact mass by electrospray ionization (m/z 625.5161; 625.5169 calcd for C37H65N6O2). However, the mass spectrometry (MS) fragmentation pattern of authentic batzelladine F differed from those of the isomers we had prepared. Our samples showed peaks at m/z = 304 and 322 resulting from the expected McLafferty fragmentation of the ester bond, whereas the synthetic sample lacked these fragments and showed peaks at m/z = 276 and 350.44
Now suspicious of the constitution of natural batezelladine F, we subjected both synthetic isomer 37 and authentic batzelladine F to basic methanolysis followed by MS analysis. Synthetic 37 yielded the expected peaks at m/z = 294 and 364, corresponding to alcohol 81 and ester 87 (Scheme 20). Yet the authentic sample yielded peaks at m/z = 322 and 336 (Scheme 21). As one product was 28 amu heavier than expected, whereas the other was 28 amu lighter, it became apparent that the lengths of the guanidine side chains in the originally proposed structure were incorrect. It seemed likely that the methanolysis products of batzelladine F are 88 and 89. Union of these fragments yields structure 90 (Figure 2), which fits all of the NMR and mass spectrometric data for batzelladine F.
Scheme 20.

Scheme 21.

Conclusion
Stereoselective synthesis of octahydro-5,6,6a-triazaacenaphthalenes having the anti relationship of the angular hydrogens flanking the pyrrolidine nitrogen confirmed suspicions9 that the relative configuration of the right hand tricyclic guanidine fragment of batzelladine F had been assigned incorrectly;2 this unit should be revised to have the anti relationship of these hydrogens. A convergent synthesis strategy was devised, whose central step was a fragment-coupling tethered-Biginelli reaction, that allowed one enantiomer of the eight hexacyclic diguanidines depicted in Figure 2 to be prepared. These synthetic products differed from batzelladine F, therefore the mass spectrum of natural batzelladine F was reinvestigated. This analysis established that the originally proposed connectivity of this alkaloid also was incorrect, leading us to propose the revised costitution 90 of natural batzelladine F.
As the absolute configuration of the two tricyclic guanidine fragments of batzelladine F was still unknown and the relative configuration at C18 (Figure 2), eight possibilities (four enantiomer pairs) remained for the structure of batzelladine F. The total synthesis of one enantiomer of these four diastereomers and the comparisons of these products with the natural marine isolate that firmly established 90 as the correct constitution and revealed the full three-dimensional structure of batzelladine F are described in the accompanying article.10b
Supplementary Material
Reaction schemes for the preparation of compounds reported only in Supporting Information, experimental details and tabulated characterization data for new compounds, HPLC chromatograms of repuified natural batzelladine F and synthetic batzelladine analogs 36, 37, 38, 86, S14 and S16, ESI mass spectrometric analysis of synthetic batzelladine analog 37 and authentic batzelladine F, copies of 1H and 13C NMR spectra of new compounds, and complete reference 17 (125 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 1.

Scheme 6.

Acknowledgment
We thank NIH (HL-25854) for financial support, Pharmacia and the ACS Organic Division for fellowship support (F. C.), Dr. John Greaves for mass spectrometric analyses, and Dr. J. A. Chan of SmithKline Beecham for providing an authentic sample of batzelladine F, without which the structural revision would not have been possible. NMR and mass spectra were determined at UC Irvine with instruments purchased with the assistance of NSF and NIH.
References
- 1.Department of Medicinal Chemistry, Genentech, Inc., 1 DNA Way. South San Francisco, CA 94080.
- 2.Patil AD, Freyer AJ, Taylor PB, Carté B, Zuber G, Johnson RK, Faulkner DJ. J. Org. Chem. 1997;62:1814–1819. [Google Scholar]
- 3.The identity of the sponge has been revised: Braekman JC, Daloze D, Tavares R, Hajdu E, Van Soest RWM. J Nat. Prod. 2000;63:193–196. doi: 10.1021/np990403g.
- 4.For a recent review, see: Aron ZD, Overman LE. Chem. Commun. 2004:253–265. doi: 10.1039/b309910e.
- 5.(a) For reviews summarizing the isolation, structure and synthesis of batzelladine alkaloids, see: Berlinck RGS, Kossuga MH. J. Nat. Prod. 2005;22:516–550. doi: 10.1039/b209227c. and earlier reviews in this series. (b) For the recent isolation of a new batzelladine alkaloid, see: Gallimore WA, Kelly M, Scheuer PJ. J. Nat. Prod. 2005;68:1420–1423. doi: 10.1021/np050149u. (c) For a recent synthetic study, see: Arnold MA, Duron SG, Gin DY. J. Am. Chem. Soc. 2005;127:6924–6925. doi: 10.1021/ja0519029.
- 6.Snider BB, Chen J, Patil AD, Freyer AJ. Tetrahedron Lett. 1996;37:6977–6980. [Google Scholar]
- 7.Cohen F, Overman LE, Sakata SKL. Org. Lett. 1999;1:2169–2172. doi: 10.1021/ol991269u. [DOI] [PubMed] [Google Scholar]
- 8.This data was not presented in the paper or the Supporting Information.
- 9.(a) Snider BB, Busuyek MV. J. Nat. Prod. 1999;62:1707–1711. doi: 10.1021/np990312j. [DOI] [PubMed] [Google Scholar]; (b) Black GP, Murphy PJ, Thornhill AJ, Walshe NDA, Zanetti C. Tetrahedron. 1999;55:6547–6554. [Google Scholar]
- 10.(a) Our synthesis of the natural product has been outlined: Cohen F, Overman LE. J. Am. Chem. Soc. 2001;123:10782–10783. doi: 10.1021/ja017067m. Cohen F, Overman LE. following article in this issue.
- 11.Coffey DS, McDonald AI, Overman LE, Stappenbeck F. J. Am. Chem. Soc. 1999;121:6944–6945. [Google Scholar]
- 12.Lee TV, Porter JR. Org. Synth. 1995;72:189–195. [Google Scholar]
- 13.Evans DA, Hoveyda AH. J. Am. Chem. Soc. 1990;112:6447–6449. [Google Scholar]
- 14.Mitsunobu O. Synthesis. 1981:1–32. [Google Scholar]
- 15.Hassner A, Fibiger R, Andisik DJ. J. Org. Chem. 1984;49:4237–4244. [Google Scholar]
- 16.Martin SF, Dodge JA. Tetrahedron Lett. 1991;32:3017–3020. [Google Scholar]
- 17.Without this basic additive, the bis-dimethyl acetal was isolated in quantitative yield. See: Woodward RB, et al. J. Am. Chem. Soc. 1981;103:3210–3213.
- 18.Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998:26–28. [Google Scholar]
- 19.Seebach D, Zuger M. Helv. Chem. Acta. 1982;65:495–503. [Google Scholar]
- 20.Volkov AA, Zlotski SS, Kravets EK, Spirknin LV. Chem. Heterocycl. Compd. (English Translation) 1985;21:861–863. [Google Scholar]
- 21.Sjiki H, Hattori K, Hirota K. J. Org. Chem. 1998;63:7990–7992. [Google Scholar]
- 22.Franklin AS, Ly SK, Mackin GH, Overman LE, Shaka AJ. J. Org. Chem. 1999;64:1512–1519. doi: 10.1021/jo981971o. [DOI] [PubMed] [Google Scholar]
- 23.Bernatowicz MZ, Wu Y, Matsueda GR. J. Org. Chem. 1992;57:2497–2502. [Google Scholar]
- 24.Stereoselection in the tethered Biginelli condensation has been thoroughly investigated: McDonald AI, Overman LE. J. Org. Chem. 1999;64:1520–1528. doi: 10.1021/jo981972g.
- 25.Reuman M, Eissenstat MA, Weaver JD., III Tetrahedron Lett. 1994;35:8303–8306. and references therein. [Google Scholar]
- 26.A model compound similar to 26 bearing a methyl ester was completely resistant to both basic and acidic hydrolysis.
- 27.Snider BB, Chen J, Patil AD, Freyer AJ. Tetrahedron Lett. 1996;37:6977–6980. [Google Scholar]
- 28.Black GP, Murphy PJ, Walshe NDA. Tetrahedron. 1998;54:9481–9488. [Google Scholar]
- 29.Synthesized from unsaturated azido benzoate 15 in a fashion analogous to 29; see Supporting Information.
- 30.Overman LE, Rabinowitz MH. J. Org. Chem. 1993;58:2335–3237. [Google Scholar]
- 31.Heathcock CH, Pietter S, Ruggeri RB, Ragan JA, Kath JC. J. Org. Chem. 1992;57:2554–2566. [Google Scholar]
- 32.Several lengthier synthesis of this compound are reported: Nokami J, Taniguchi T, Ogawa Y. Chem. Lett. 1995:43–44. Solladié G, Lohse O. J. Org. Chem. 1993;58:4555–4563. Ernst B, Wagner B. Helv. Chim. Acta. 1989;72:165–171.
- 33.Huckin SN, Weiler L. J. Am. Chem. Soc. 1974;96:1082–1087. [Google Scholar]
- 34.Taber DF, Amedio JC, Patel YK. J. Org. Chem. 1985;50:3618–1619. [Google Scholar]
- 35.Williams JM, Jobson RB, Yasuda N, Marchesini G, Dolling U–H, Grabowski EJJ. Tetrahedron Lett. 1995;36:5461–5464. [Google Scholar]
- 36.Chen KM, Hardmann GE, Prasad K, Repic O, Shapiro MJ. Tetrahedron Lett. 1987;28:155–158. [Google Scholar]
- 37.Atkins PR, Glue SEJ, Kay IT. J. Chem. Soc., Perkin Trans. 1. 1973:2644–2646. [Google Scholar]
- 38.These reactions were conducted on a small scale (<1 mg) and analyzed by HPLC MS.
- 39.Mukayama T, Usui M, Shimada E, Saigo K. Chem. Lett. 1975:1045–1048. [Google Scholar]
- 40.Molecular mechanics modeling shows that the ester group of intermediates of this type exists preferntially in a conformation in which the methine hydrogen and the carbonyl π-system are nearly orthogonal. This feature is likely responsible for the failure to generate the ester enolate of 69.
- 41.Other reductive conditions (NaBH(OAc)3, AcOH; NaBH4, EtOH, 98% HCO2H, Et3SiH, TFA) either failed to reduce 71 or led to decomposition.
- 42.The signals in the 1H NMR spectrum of 76 corresponding to the left-hand tricyclic portion were nearly identical to that of 68. The two most diagnostic peaks were the vinyl methyl singlet at δ 2.26 and the allylic methine at δ 4.25 (dd, J = 11.7, 5.2 Hz). Both of these resonances were absent from the spectrum of 75, and were replaced with an allylic methine at δ 4.49 (q, J = 6.3 Hz) and a multiplet at δ 2.45–2.30 corresponding to the allylic methylene.
- 43.Details are provided in the Supporting Information.
- 44.For a discussion of the McLafferty rearrangement in other batzelladine alkaloids see Patil AD, Kumar NV, Kokke WC, Bean MF, Freyer AJ, De Brosse C, Mai S, Truneh A, Faulkner DJ, Carte B, Breen AL, Hertzberg RP, Johnson RK, Westley JW, Potts BCM. J. Org. Chem. 1995;60:1182–1188. Braekman JC, Daloze D, Tavares R, Hajdu E, Van Soest RWM. J. Nat. Prod. 2000;63:193–196. doi: 10.1021/np990403g.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reaction schemes for the preparation of compounds reported only in Supporting Information, experimental details and tabulated characterization data for new compounds, HPLC chromatograms of repuified natural batzelladine F and synthetic batzelladine analogs 36, 37, 38, 86, S14 and S16, ESI mass spectrometric analysis of synthetic batzelladine analog 37 and authentic batzelladine F, copies of 1H and 13C NMR spectra of new compounds, and complete reference 17 (125 pages). This material is available free of charge via the Internet at http://pubs.acs.org.