

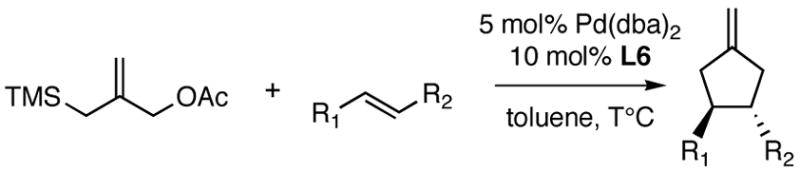
Natural products containing five-membered rings are found throughout the chemical literature. Transition metal-catalyzed [3+2] trimethylenemethane (TMM) cycloadditions provide direct routes to substituted cyclopentanes.1 In 1979, we first reported that Pd-TMM complexes generated from 3-acetoxy-2-trimethylsilylmethyl-1-propene (1) and catalytic palladium react with electron-deficient olefins to produce exo-methylene cyclopentanes (eqn 1).2 We have shown that this palladium-catalyzed [3+2] TMM cycloaddition reaction is a highly chemo-, regio- and diastereoselective process.3,4 This methodology has also been extended to [4+3] and [6+3] cycloadditions,5,6 and has been employed in a number of total syntheses.3,7–9 Unfortunately, the formation of stereogenic centers in the cycloadducts produced in this chemistry has mainly been limited to the use of chiral auxiliaries.10 The sole example of a palladium-catalyzed asymmetric [3+2] sulfone substituted TMM cycloaddition was reported from the Hayashi lab with ee’s ranging from 4–78%.11
(1).
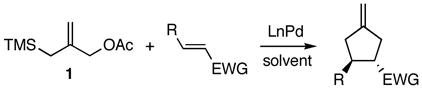
The difficulty in desigining a catalyst for the asymmetric Pd-TMM cycloaddition reaction is illustrated in our proposed stepwise mechanism (Scheme 1).12,13 The formation of the zwitterionic intermediate is generated by insertion of LnPd into the C-O bond followed by attack of the displaced acetate anion onto the trimethylsilyl group, which deposits its electrons onto the TMM fragment.14 The enantiodetermining step is most likely the initial nucleophilic attack. This step occurs distal to the set of coordinated chiral ligands. Our early work on the beneficial effect of phosphite and phosphorustriamine ligands on the efficiency of the cycloaddition induced us to examine palladium complexes of chiral phosphoramidites15a as asymmetric catalysts.
Scheme 1.
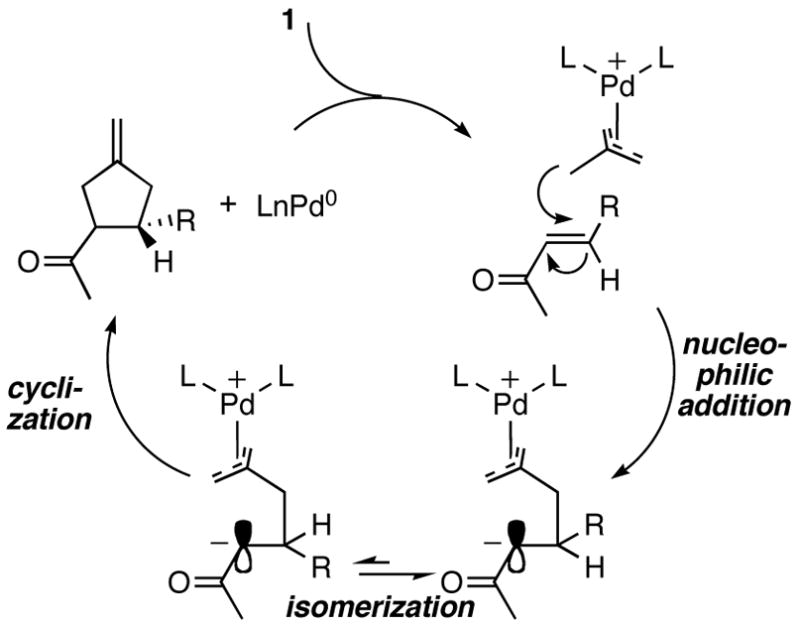
Initally, we examined the reaction of 1 with methyl cinnamate in the presence of 5 mol% Pd(dba)2 and 10 mol% ligand (L1–L6) in toluene at 23 °C (Scheme 2). Interestingly, catalysts formed from L1, L4, or L5 gave 0% yield; however, catalysts formed from L2 gave 78% yield and 29% ee. We thought an increase in the rigidity of L2 may lead to an increase in % ee. Indeed, the synthesis of L615b and its application in the asymmetric TMM [3+2] cycloaddition gave 80% yield and 58% ee of the desired cyclopentane product.
Scheme 2.
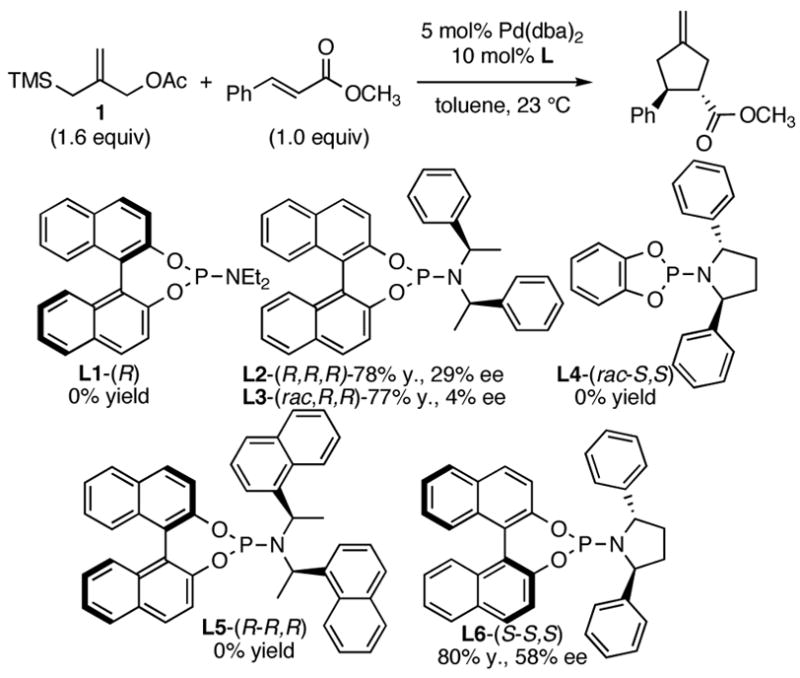
Although toluene has typically been used in previous TMM [3+2] reactions, we decided to screen a number of solvents to examine the effect on the yield and selectivity of this process (Table 1). Reactions conducted in THF, ether, and DME gave good yields (68–80%) with modest (48–55% ee, entries 2–4) selectivities, while reactions conducted in DCM, CH3CN, DMF, or dioxane gave low yields of the desired product (entries 5–8). Thus, toluene appears optimal.
Table 1.
Solvent Effect on Asymmetric TMM [3+2] Cycloadditions
 | |||
|---|---|---|---|
| Entry | Solvent | % Yielda | %ee |
| 1 | toluene | 80 | 58 |
| 2 | THF | 68 | 48 |
| 3 | DME | 80 | 53 |
| 4 | ether | 69 | 55 |
| 5 | DCM | 10 | 32 |
| 6 | CH3CN | 0 | 0 |
| 7 | DMF | 0 | 0 |
| 8 | dioxane | 35 | 39 |
Determined by GC analysis with an internal standard.
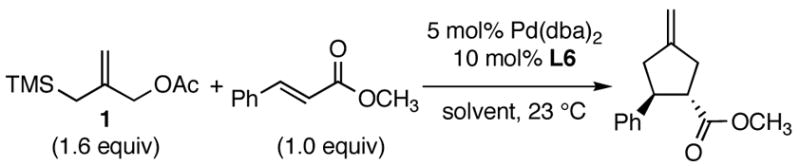
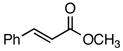
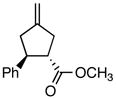
Using the optimized catalytic conditions, we examined the initial scope of the TMM accepting olefin (Table 2). All cyclopentane products were formed in >19:1 trans:cis selectivity. The reaction of methyl cinnamate with 1 at 0 °C increased the enantioselectivity from 58% (at 23 °C) to 62%. Cooling the reaction to −25 °C failed to produce >5% product as determined by GC analysis. Employing the more reactive (E)-benzalacetone allowed the reaction to be performed at −25 °C, which increased selectivity to 82% ee (entry 2). Lowering the reaction temperature to −50 °C failed to produce any noticeable product by GC analysis. The selectivity of this reaction does not rely on the presence of the phenyl group as trans-nonenone reacts with yields and ee’s that are comparable to benzalacetone (entry 3). Switching the methyl group to an ethyl (entry 4) or phenyl (entry 5) gives yields and ee’s that are comparable to benzalacetone. Use of electron-rich or electron-poor aromatic enones slightly lessened the enantioselectivity with respect to the parent chalcone. Nitriles were compatible functionalities and gave enantioselectivities comparable to esters (entry 7).
Table 2.
Initial Scope of Palladium-Catalyzed [3+2] TMM Cycloaddition Reactions.
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | T(°C) | Yield (%)a | ee(%) |
| 1 |
|
|
23
0 |
80
81 |
58
62 |
| 2 |
|
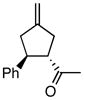
|
23
−25 |
76
63 |
72
82 |
| 3 |
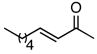
|
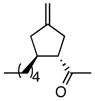
|
23
−25 |
72
79 |
73
84 |
| 4 |
|
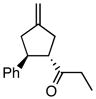
|
23
−25 |
84
72 |
73
83 |
| 5 |
|
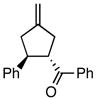
|
−25 | 83 | 80 |
| 6 |
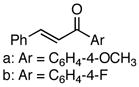
a: Ar = C6H4-4-OCH3 b: Ar = C6H4-4-F |
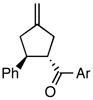
|
a: −25
b: −25 |
73
80 |
72
74 |
| 7 |
|
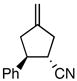
|
0 | 78 | 58 |
Isolated yields
The reaction of 1 with various tetralone derivatives in the presence of Pd(dba)2 and L6 gave high yields and ee’s of the corresponding exo-methylene cyclopentanes. The reaction of benzylidene tetralone at 23 °C gave 99% yield and 85% ee (entry 1). Reducing the temperature to −25 °C gave 94% yield and 92% ee. Exchanging the phenyl group for six- and five-member aromatics did not have a large effect on the reaction (entries 2–3). Cycloalkyl substituted tetralones were also good substrates (entries 4–5); however, cyclohexylidene tetralone failed to react under our conditions at room temperature.
In summary, we have developed conditions to effect an asymmetric palladium-catalyzed [3+2] cycloaddition reaction between in situ generated Pd-TMM intermediates and various olefins, in spite of the large distance between the chiral ligands and the asymmetric bond-forming event. Future work will focus on extending this methodology to include [4+3] and [6+3] cycloadditions, as well as developing catalysts that promote asymmetric [3+2n] cycloaddition reactions with substituted TMM precursors.
Supplementary Material
Experimental details for procedures and spectral data for all unknown compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Table 3.
Palladium-Catalyzed [3+2] TMM Cycloaddition Reactions of Aryl- and Alkylidene Tetralones.
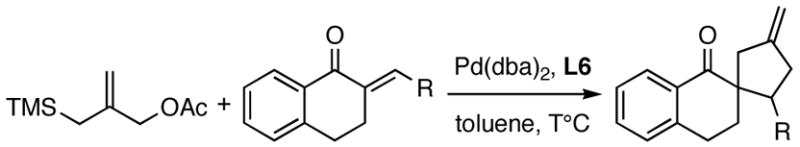 | ||||
|---|---|---|---|---|
| Entry | R | T(°C) | Yield (%)a | ee(%) |
| 1 |
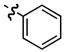
|
23
−25 |
99
94 |
85
92 |
| 2 |
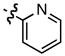
|
23
−25 |
78
87 |
83
86 |
| 3 |
|
23
−25 |
71
60 |
73
82 |
| 4 |
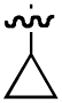
|
23
−25 |
95
70 |
79
87 |
| 5 |
|
0 | 53 | 78 |
Isolated yields.
Acknowledgments
We thank the NIH (GM13598) and the NSF for their generous support of our programs. Palladium salts were a generous gift from Johnson-Matthey. Mass spectra were provided by the Mass Spectrometry Regional Center at the University of California-San Francisco, supported by the NIH Division of Research Resources. JPS thanks the NIH (GM069269-02) for a postdoctoral fellowship.
References
- 1.Lautens M, Klute W, Tam W. Chem Rev. 1996;96:49. doi: 10.1021/cr950016l. [DOI] [PubMed] [Google Scholar]
- 2.Trost BM, Chan DMT. J Am Chem Soc. 1979;101:6429. [Google Scholar]
- 3.Trost BM. Pure Appl Chem. 1988;60:1615. [Google Scholar]
- 4.Trost BM, Nanninga TN, Satoh T. J Am Chem Soc. 1985;107:721. [Google Scholar]
- 5.Trost BM, MacPherson DT. J Am Chem Soc. 1987;109:3483. [Google Scholar]
- 6.Trost BM, Soane PR. J Am Chem Soc. 1987;109:615. [Google Scholar]
- 7.Trost BM, Crawley ML. Chem Eur J. 2004;10:2237. doi: 10.1002/chem.200305634. [DOI] [PubMed] [Google Scholar]
- 8.Trost BM, Higuchi RI. J Am Chem Soc. 1996;118:10094. [Google Scholar]
- 9.Trost BM, Parquette JR. J Org Chem. 1994;59:7568. [Google Scholar]
- 10.Trost BM, Yang B, Miller ML. J Am Chem Soc. 1989;111:6482. [Google Scholar]
- 11.Yamamoto A, Ito Y, Hayashi T. Tetrahedron Lett. 1989;30:375. [Google Scholar]
- 12.Gordon DJ, Fenske RF, Nanninga TN, Trost BM. J Am Chem Soc. 1981;103:5974.Singleton has shown that the mechanism may be a concerted process for certain substrates: Singleton DA, Schulmeier BE. J Am Chem Soc. 1999;121:9313–9317.
- 13.Trost BM, Chan DMT. J Am Chem Soc. 1983;105:2326. [Google Scholar]
- 14.Su CC, Chen JT, Lee GH, Wang Y. J Am Chem Soc. 1994;116:4999. [Google Scholar]
- 15.Feringa BL. Acc Chem Res. 2000;33:346. doi: 10.1021/ar990084k.For the lone report of L6 in asymmetric catalysis see: Choi YH, Choi JY, Yang HY, Kim YH. Tetrahedron: Asymmetry. 2002;13:801.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details for procedures and spectral data for all unknown compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.