

This communication details the asymmetric metallophosphite-catalyzed 1,3-silylacylation of nitrones (eq 1). This reaction provides access to enantiomerically enriched N-aryl α-amino ketones and, to the best of our knowledge, constitutes the first example of direct C-acylation of nitrones.
(1).
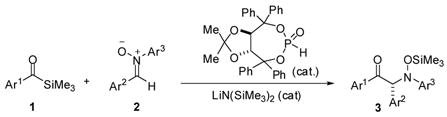
α-Amino ketones are useful building blocks in organic chemistry.1 The addition of stoichiometric acyl anion equivalents to imines represents a useful synthetic method that introduces this versatile functional group.2–5 In a seminal advance, Murry and Frantz described thiazolium carbene-catalyzed aza-benzoin additions between aldehydes and acyl imines generated in situ from tosylamides.6 In 2005, Miller and co-workers developed an asymmetric variant using a thiazolylalanine-derived catalyst and electron-deficient aryl aldehydes as the acyl donors.7 Good enantioselectivities were obtained (~75–85%), which increased to 98% for many products upon recrystallization, albeit at the expense of yield. The enantiomeric excess of the products was found to be dependent upon reaction time as racemization occurred under the basic reaction conditions (excess R3N).
Our laboratory has recently developed metallophosphites8,9 as a new family of umpolung catalysts for the enantioselective C–acylation of aldehydes and alkenes.10–12 We were interested in testing the notion that these catalysts could be employed for the asymmetric C–acylation of C=N π bonds as well. We hypothesized that strong nucleophilicity and low basicity of metallophosphites could allow us to develop an asymmetric acylation that would include electron-neutral and electron-rich substrates.
A potential complication with the use of imines in the projected application was the appreciable endothermicity of the requisite turnover-enabling [1,4]-O→N silyl transfer (Scheme 1, 4 → 5).13 We hypothesized that nitrones14 could be superior azomethine electrophiles due to the reestablishment of thermoneutrality via a [1,5]-O→O silyl transfer (6 → 7). Indeed, in the examination of a range of imine and imine-derived electrophiles as coupling partners with acyl silanes, productive coupling was only observed with nitrone electrophiles (Figure 1). An N-aryl moiety was optimal and the N-o-methoxyphenyl (N-OMP) derivative 19 was initially selected as a substrate for optimization.
Scheme 1.
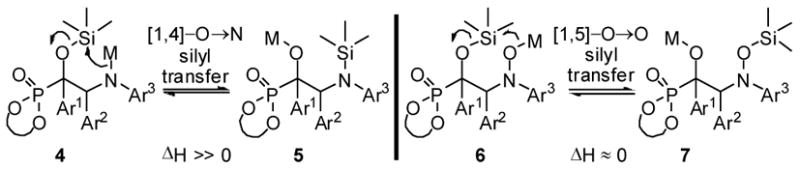
Energetics of Si Transfer: Imines vs. Nitrones
Figure 1.
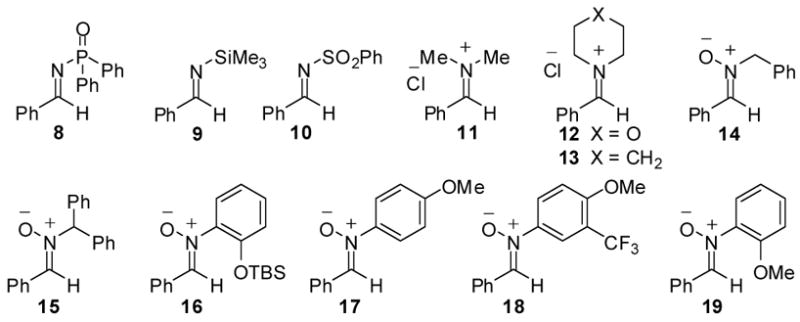
Imines and Nitrones Examined for C-Acylation
Using nitrone 19 and acyl silane 1, typical reaction variables were examined (Table 1). An evaluation of bases indicated that a lithium counterion was required for reaction, with LiN(SiMe3)2 and n-BuLi giving the best results (Table 1). In a solvent screen, 2-MeTHF and diethyl ether gave similar yields, with the former providing optimal levels of enantiocontrol. Reactions performed in toluene, CH2Cl2, THF, or tBuOMe failed to give any desired product.
Table 1.
Screen of Reaction Conditionsa
| entry | base | solvent | yieldb | e.r.c |
|---|---|---|---|---|
| 1 | NaN(SiMe3)2 | 2-MeTHF | 0% | n.d.d |
| 2 | KH | 2-MeTHF | 0% | n.d. |
| 3 | EtMgBr | 2-MeTHF | 0% | n.d. |
| 4 | DBU | 2-MeTHF | 0% | n.d. |
| 5 | KOtBu | 2-MeTHF | 0% | n.d. |
| 6 | sec-BuLi | 2-MeTHF | 8% | n.d. |
| 7 | LiN(SiMe3)2 | 2-MeTHF | 52% | 97:3 |
| 8 | n-BuLi | 2-MeTHF | 49% | 97:3 |
| 9 | n-BuLi | Et2O | 46% | 91:9 |
Acyl silane 1 (1.0 equiv), nitrone 19 (1.5 equiv), (R,R)-TADDOL-phosphite (0.25 equiv), base (0.20 equiv) in 3 mL of solvent at room temperature. Ar1 = p-MeOPh for entries 1–7; Ar = Ph for entries 8–9.
Yield determined by 1H NMR spectroscopy versus an internal standard.
Determined by CSP-SFC.
n.d. = not determined.
After optimization of the base, solvent, and temperature, the yield of 3a reached a plateau of ~50%. An irreversible redox reaction between the phosphite and nitrone that formed phosphate and imine was implicated in the moderate yields. Neither increasing the catalyst loading nor slow addition of the nitrone ameliorated the problem; however, a modest modification of reaction stoichiometry, using a slight excess of the acyl silane relative to the nitrone, minimized the addition of phosphite to the nitrone and allowed 3a to be prepared in >90% yield (1H NMR).
An evaluation of coupling partners revealed that reactions with both electron neutral and electon-rich aryl acyl silanes proceeded equally well. Electron-rich, –poor, and –neutral aryl nitrones with several N-aryl groups were also nicely tolerated (Table 2).
Table 2.
Scope of Asymmetric Nitrone Acylationa
| Entry | Product | Yield (%)b | e.r.c |
|---|---|---|---|
| 1 |
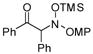
3a |
68 | 97:3 |
| 2 |
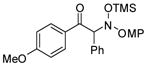
3b |
77 | 98.5:1.5 |
| 3 |
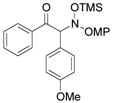
3c |
84 | 97:3 |
| 4 |
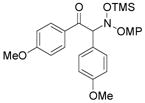
3d |
94 | 98.5:1.5 |
| 5 |
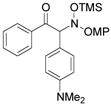
3e |
76 | 96.5:3.5 |
| 6 |
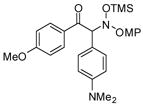
3f |
36 | 97:3 |
| 7 |
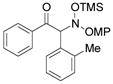
3g |
77 | 97:3 |
| 8 |
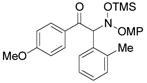
3h |
86 | 98:2 |
| 9d |
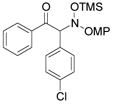
3i |
82 | 98:2 |
| 10d |
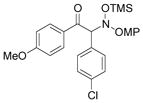
3j |
93 | 98.5:1.5 |
| 11e |
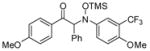
3k |
76 | 98:2 |
| 12e |
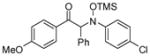
3l |
65 | 95:5 |
Ar1C(O)SiMe3 (1.5 equiv), Ar2CHN(O)Ar3 (1.0 equiv), (R,R)-TADDOL-phosphite (0.25 equiv.), LiN(SiMe3)2 (0.23 equiv.) in 6 mL of 2-MeTHF at room temperature unless otherwise stated. OMP = o-MeOPh.
Yield of isolated, analytically pure 3 as judged by 1H NMR spectroscopy and combustion analysis.
e.r. determined by CSP-SFC.
Phosphite (0.20 equiv), LHMDS (0.17 equiv).
Conducted at 0 ºC.
In most cases, the reaction proceeded with good isolated yields and excellent enantiocontrol. Products were purified to analytical purity by flash chromatography on SiO2 gel that had been deactivated with 5% Et3N/hexanes. Failure to deactivate the silica led to formation of the achiral α-ketimine via HOSiMe3 elimination. The somewhat lower yields for 3a, 3f, and 3l arise from this artifact of purification. At this point, these additions are applicable only to aryl acyl silanes and nitrones derived from aromatic aldehydes: our efforts with a variety of aliphatic substrates have failed to yield coupling products.
α-N-silyloxyamino ketone 3j was prepared on a 16-gram scale employing a somewhat lower catalyst loading (eq 2). We saw virtually the same yield and enantioselection as on a 0.1 g scale. Excess acyl silane could also be recovered by chromatography. Crystallization of 3j permitted assignment of the absolute stereochemistry as (R) by X-ray diffraction and increased the product e.r. to 99.5:0.5.
(2).
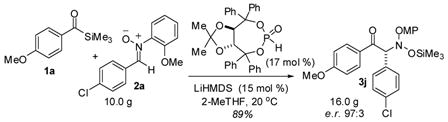
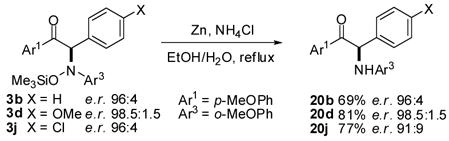
The product N–O bond can be reductively cleaved without loss of configuration using Zn metal in EtOH/aq NH4Cl to reveal the α-N-arylamino ketone (eq 3).15
(3).
In conclusion, we have described the first enantioselective addition of acyl silanes to nitrone electrophiles. The particular requirements for successful catalysis in this system are uniquely met by providing an energetically accessible pathway for silyl transfer. These additions typically proceed in good yield with high enantioselectivity to give protected α-N-arylamino ketones and are amenable to preparative scale applications.
Supplementary Material
Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
Funding for this work was provided by the National Institutes of Health (National Institute of General Medical Sciences, GM068443). Research support from Eli Lilly, Amgen, and GSK is gratefully acknowledged. X-ray crystallography was performed by Dr. Peter White. J.S.J. is an Alfred P. Sloan Fellow and a Camille-Dreyfus Teacher Scholar.
References
- 1.Sardina FJ, Rapoport H. Chem Rev. 1996;96:1825–1872. doi: 10.1021/cr9300348. [DOI] [PubMed] [Google Scholar]
- 2.Padwa A, Dharan M, Smolanoff J, Wetmore JSI. J Am Chem Soc. 1973;95:1954–1961. [Google Scholar]
- 3.Kakuuchi A, Taguchi T, Hanzawa Y. Tetrahedron Lett. 2001;42:1547–1549. [Google Scholar]
- 4.Sato A, Ito H, Okada M, Nakamura Y, Taguchi T. Tetrahedron Lett. 2005;46:8381–8383. [Google Scholar]
- 5.Davis FA, Ramachandar T, Lui H. Org Lett. 2004;6:3393–3395. doi: 10.1021/ol0485971. [DOI] [PubMed] [Google Scholar]
- 6.Murry JA, Frantz DE, Soheili A, Tillyer R, Grabowski EJJ, Reider PJ. J Am Chem Soc. 2001;123:9696–9697. doi: 10.1021/ja0165943.For related examples, see: Mattson AE, Scheidt KA. Org Lett. 2004;6:4363–4366. doi: 10.1021/ol0481129.Li GQ, Dai LX, You SL. Chem Commun. 2007:852–854. doi: 10.1039/b611646a. In.
- 7.Mennen SM, Gipson JD, Kim YR, Miller SJ. J Am Chem Soc. 2005;127:1654–1655. doi: 10.1021/ja042650z. [DOI] [PubMed] [Google Scholar]
- 8.Enders D, Tedeschi L, Bats JW. Angew Chem Int Ed. 2000;39:4605–4607. [PubMed] [Google Scholar]
- 9.Reich HJ, Holtan RC, Bolm C. J Am Chem Soc. 1990;112:5609–5617. [Google Scholar]
- 10.Linghu X, Potnick JR, Johnson JS. J Am Chem Soc. 2004;126:3070–3071. doi: 10.1021/ja0496468. [DOI] [PubMed] [Google Scholar]
- 11.Nahm MR, Linghu X, Potnick JR, Yates CM, White PS, Johnson JS. Angew Chem Int Ed. 2005;44:2377–2379. doi: 10.1002/anie.200462795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nahm MR, Potnick JR, White PS, Johnson JS. J Am Chem Soc. 2006;128:2751–2756. doi: 10.1021/ja056018x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walsh R. Acc Chem Res. 1981;14:246–252. [Google Scholar]
- 14.Lombardo M, Trombini C. Synthesis. 2000:759–774. [Google Scholar]
- 15.Kirby GW, McLean D. J Chem Soc Perkin Trans 1. 1985:1443–5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the internet at http://pubs.acs.org.