

Abstract
High levels of interstrand cross-link damage in mammalian cells cause chromatid breaks and radial formations recognizable by cytogenetic examination. The mechanism of radial formation observed following DNA damage has yet to be determined. Due to recent findings linking homologous recombination and non-homologous end-joining to the action of the Fanconi anemia pathway, we speculated that radials might be the result of defects in either of the pathways of DNA repair. To test this hypothesis, we have investigated the role of homologous recombination proteins RAD51 and RAD52, non-homologous end-joining proteins Ku70 and LIG4, and protein MRE11 in radial formation and cell survival following interstrand crosslink damage with mitomycin C. For the studies we used small inhibitory RNA to deplete the proteins from cells, allowing for evaluation of radial formation and cell survival. In transformed normal human fibroblasts, depletion of these proteins increased interstrand crosslink sensitivity as manifest by decreased cell survival and increased radial formation. These results demonstrate that inactivation of proteins from either of the two separate DNA repair pathways increases cellular sensitivity to interstrand crosslinks, indicating each pathway plays a role in the normal response to interstrand crosslink damage. We can also conclude that homologous recombination or non-homologous end-joining are not required for radial formation, since radials occur with depletion of these pathways.
Introduction
Interstrand cross-link (ICL) damage poses a unique DNA repair problem. It occurs when a covalent bond forms between nucleotides on the two strands of DNA, precluding the availability of an undamaged complementary strand as a template for the process of DNA repair (Metzler 1986). When treated with high concentrations of ICL agents such as mitomycin C (MMC) or diepoxybutane (DEB), normal mammalian cells show chromosomal abnormalities comprising radial formations and chromatid breaks (Newell et al. 2004). Mammalian cells particularly sensitive to the damaging effects of ICLs, such as Fanconi anemia (FA) or Bloom syndrome (BS) cells, show a much more marked phenotype, displaying radials and breaks at much lower concentrations of treatment (Sasaki and Tonomura 1973; Sasaki 1975; German 1997). Indeed, the cellular phenotype of radials and breaks following MMC or DEB exposure is the standard for diagnosis of FA (Auerbach et al. 1981; Cervenka et al. 1981; reviewed in D’Andrea and Grompe 2003). For both normal cells and those from FA or BS patients, radial formation correlates with decreased cell survival; that is, cellular toxicity of ICL agents correlates with radial formation. Thus, it appears that radials reflect a failure in DNA damage processing which is associated with decreased cell survival.
While used for the diagnosis of FA, radial formations can be observed in non-FA cells following DNA damage by ICL agents or ionizing radiation (IR) (Bruun et al. 2003; Houghtaling et al. 2005). Radial formations can take several forms and comprise the association of chromatids, whether two or more. However, the mechanism of radial formation is not yet known. Radials may form during normal DNA repair, but be resolved through the processing of DNA damage, or they may form and persist through failure of repair. Radials are thought to be formed secondarily to DNA strand breaks, although direct evidence for this is lacking. It is known that radials form exclusively between non-homologous chromosomes or non-homologous regions of chromosomes and it has also been shown in FA cells that while the full length of all autosomes appears to be involved, there are no preferential chromosome pairings found in radials (Newell et al. 2004). It has been an open question as to whether radial formation is dependent on recombination or end-joining functions.
In yeast, multiple pathways, including non-homologous end joining (NHEJ) and homologous recombination (HR), are used for repair of ICLs (De Silva et al. 2000; McHugh et al. 2000; Dronkert and Kanaar 2001; Grossmann et al. 2001). In mammalian cells, it is known that ICLs are recognized in S-phase (Akkari et al. 2000), initiating the repair response. Of known cellular DNA repair processes, two that have been implicated in mammalian ICL repair are homologous recombination repair (HRR) and NHEJ.
HR-based repair depends on the RAD51 and RAD52 proteins in mammalian cells (reviewed in Harrison and Haber 2006; reviewed in Su 2006). RAD51 is loaded onto single stranded DNA in a BRCA2 (FANCD1)-mediated manner in the early stages of HR (Davies et al. 2001). RAD51 is recruited by RAD52 to single stranded DNA (Song and Sung 2000). RAD52 then assists in pairing complementary DNA and is thought to be involved in postsynaptic annealing of one end of a double strand break (DSB) (Miyazaki et al. 2004). Foci made up of RAD51, RAD52, MRE11, BRCA1, FANCD2, and other DNA repair-involved proteins form at the sites of DNA damage following not only IR and ICL exposure, but other types of DNA damage (Garcia-Higuera et al. 2001; Nakanishi et al. 2002).
NHEJ is another repair pathway that influences the sensitivity of mammalian cells to ICL agents (Muller et al. 1998; reviewed in Riha et al. 2006). The normal ICL response appears to lead to a DSB (Rothfuss and Grompe 2004). To repair DSBs, mammalian cells appear to rely heavily on NHEJ (Valerie and Povirk 2003; reviewed in Burma et al. 2006; Chen et al. 2006). Thus a plausible mechanism for involvement of NHEJ in ICL repair would be operation on DSB intermediates. NHEJ-mediated repair is initiated by the action of DNA-dependent protein kinase (DNA-PK). The regulatory subunit of DNA-PK is the Ku70/Ku80 heterodimer, which recognizes and binds to DNA ends. Following the action of DNA-PK, the XRCC4/DNA LIG4 complex acts to reseal breaks in DNA (Li et al. 1995; Grawunder et al. 1998).
MRE11 is a protein that appears to act as a sensor for DNA damage and is involved in both NHEJ and HRR (Morales et al. 2005). An mre11 yeast mutant strain has been shown to be sensitive to ICL damage and defective in DSB repair, suggesting a role for MRE11 in ICL repair (McHugh et al. 2000). In yeast, Mre11 forms a complex with Rad50 and Xrs2, the MRX complex, which is involved in both HRR and NHEJ (Haber 1998). In mammalian cells, this complex is known as the MRE11/RAD50/NBS1 (MRN) complex. This complex is necessary for proper in vitro DNA end-joining (Zhong et al. 2002). ICL damage has been shown to activate the assembly of MRN proteins in subnuclear foci (Pichierri et al. 2002). Together, these findings indicate a role for MRE11 in mammalian ICL response.
In this study, we investigated whether HR and NHEJ are processes involved in radial formation and the cellular response to ICL damage. We used small inhibitory RNA (siRNA) depletion (Elbashir et al. 2001) of HR and NHEJ proteins in transformed normal human GM 639 cells to determine if either process serves a role in radial formation and cellular survival after ICL damage. One advantage of the technique is that it is possible to deplete proteins required for long term viability. We determined that depletion of RAD51, RAD52, MRE11, Ku70, or LIG4 results in increased ICL sensitivity, using the end points of decreased cell survival and increased ICL-induced radial formation, relative to non-depleted cells. There was good correlation between increased radial formation and decreased cell survival. Therefore, HRR, and NHEJ are required for appropriate cellular response to ICL damage, and are not required for radial formation. Indeed, since loss of these pathways increases radial formation with ICL damage, we conclude that radial formation directly reflects a failure in either pathway. These findings indicate that the NHEJ and HR pathways are each required for maintenance of genome stability following ICL damage. In addition, depletion of RAD51 induced spontaneous radial formation in the absence of MMC treatment. This indicates that RAD51 plays a key role in genome stability and prevention of radial formation both during and in the absence of ICL damage.
Materials and Methods
Cells and Media
Transformed normal human cell line GM639 was obtained from the NIHGMS Human Gene Mutant Repository. Cells were maintained in α-modified Eagle Medium (GIBCO/BRL) with 10% fetal calf serum (Hyclone), 1 × glutamine (GIBCO/BRL), and 50 μg/ml Gentamicin (GIBCO/BRL) at 37°C and 5%CO2.
siRNA Transfection
siRNA duplexes for relevant genes were acquired as SmartPool samples from Dharmacon Research (Lafayette, CO). Transfections were performed as previously described (Bruun et al. 2003). Controls were transfected with a non-functional (scrambled) siRNA duplex.
Immunoblotting
Cells were transfected with siRNA on 100 mm dishes as previously described (Bruun et al. 2003), using a 6 ml final solution volume, then washed with PBS, trypsinized, pelleted, and frozen at −80°C at the indicated time points. The controls were transfected with non-functional siRNA and harvested at 48 hours. Cell lysates were prepared as previously described (Bruun et al. 2003). 50 μg whole protein extract from each lysate was run on an 8% acrylamide gel. Gels were transferred to Immobilon-P PVDF (Millipore). Membranes were blocked overnight in TBST (TBS plus 0.1% Tween) or PBST (PBS plus 0.1% Tween) and 5% dry milk. For RAD51 and Ku70 blots, the membrane was probed with the respective mouse monoclonal antibody (Novus) at a 1:500 dilution in TBST with milk. MRE11 blots were probed with anti-MRE11 rabbit polyclonal antibody (Novus) at a 1:10,000 dilution in TBST with milk. LIG4 blots were probed with anti-LIG4 rabbit polyclonal antibody (Sigma) at a dilution of 1:1000 in TBST with milk. β-tubulin controls were probed with a rabbit polyclonal antibody (Santa Cruz) at a 1:3000 dilution in TBST. RAD51 and Ku70 blots were then incubated with goat anti-mouse IgG HRP antibody (Calbiochem) at a 1:10,000 dilution in TBST with milk. MRE11 and LIG4 blots were incubated with goat anti-rabbit IgG HRP antibody (BioRad) at a 1:10,000 dilution in TBST with milk. Detection was performed using a chemiluminescence kit (Perkin Elmer).
Radial Analysis
Twenty-four hours after siRNA transfection, flasks were treated with MMC (Table 1) alongside untreated control cells. Control cultures were transfected with a scrambled siRNA. 32 hours following MMC treatment, colcemid (0.05 μg/ml) (GIBCO/BRL) was added for a 3 hour incubation. Cells were then harvested, treated with hypotonic solution (0.075 M KCL, 5% fetal calf serum) for 10 minutes, and fixed with 3:1 methanol acetic acid. Cells were dropped onto slides for metaphase spreads and stained with Wright’s stain (Fisher Scientific) for 3.5 minutes. 50 metaphases for each sample were scored for radial content on a Nikon Eclipse E800 photoscope, and representative photographs were taken using CytoVision software from Applied Imaging.
Table 1. Radial formation in human cells depleted of HR and NHEJ proteins.
Average percentage of GM639 cells containing radials following depletion of listed proteins and MMC treatment. Average percentage represents three independent trials. “None” represents a transfection with scrambled siRNA.
| siRNA Depletion | [MMC] ng/ml | Average % cells containing radials | St. dev. (+/−) |
|---|---|---|---|
| None
None |
0
60 |
0
16 |
0
9.17 |
| RAD51
RAD51 |
0
40 |
34
70.67 |
6
3.06 |
| RAD52
RAD52 |
0
60 |
1.33
47.67 |
1.15
10.69 |
| MRE11
MRE11 |
0
60 |
0
71.33 |
0
2.31 |
| Ku70
Ku70 |
0
60 |
0.67
52 |
1.15
10 |
| LIG4
LIG4 |
0
60 |
2
63.33 |
2
9.87 |
Cell Survival Assay
Twenty-four hours after transfection with siRNA or scrambled siRNA, cells were plated on 100 mm dishes at 300 cells per dish and treated with 5, 10, 20, and 40 ng/ml MMC in duplicate. Cells were fixed in a solution of 50% MeOH, 1% new methylene blue (Sigma) after 10 days growth in MMC. Colonies were counted, and standard deviations were derived from the duplicate data sets.
Results
siRNA treatment depletes proteins of interest
To test the involvement of the HR and NHEJ proteins in ICL response, we used siRNA to deplete proteins from each of the two pathways of DNA repair. siRNAs with specificity to RAD51, RAD52, MRE11, Ku70, or LIG4 were transfected into transformed normal fibroblast cell line GM639. By immunoblot analysis, the proteins of interest were successfully depleted over the 72 hour time-period necessary for the MMC treatment and harvest prior to analysis (Fig. 1).
Figure 1. siRNA treatment depletes proteins of interest.
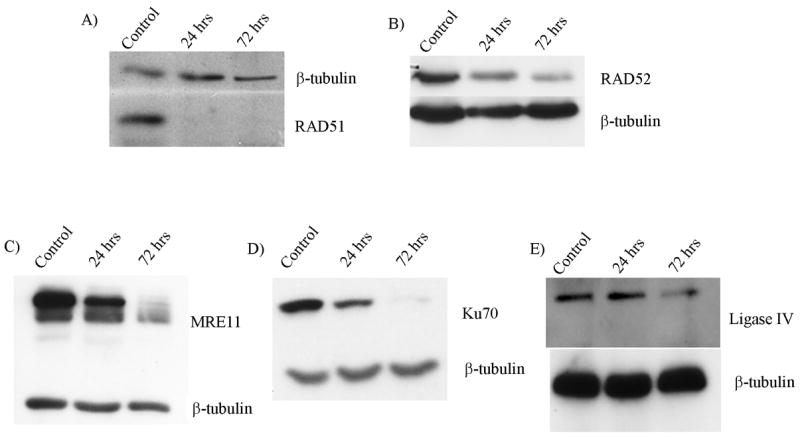
Immunoblot analysis of A) RAD51, B) RAD52, C) MRE11, D) Ku70, and E) LIG4 following siRNA treatment. Cells were treated with respective siRNAs as described and samples harvested for immunoblot analysis at time points shown. The blots display depletion over the time period of MMC treatment through harvest for cytogenetic analysis. Controls were transfected with a non-functional siRNA.
Depletion of HR proteins induces MMC sensitivity and radial formation
HRR is an important pathway in yeast ICL repair. To investigate the role of HR in mammalian processing of ICLs, we examined cells depleted of RAD51 or RAD52 proteins, as well as HR/NHEJ protein MRE11 for cytogenetic evaluation and cell survival response with ICL damage.
RAD51-depleted cells were treated with a range of concentrations of MMC in order to analyze radial formation and cell survival. Sensitivity to ICL formation was manifest by increased radial formation (Table 1 and Fig. 2). Depletion of RAD51 also increased MMC sensitivity when compared to non-depleted control cells in cell survival assays (Fig. 3a). Depletion of RAD51 also led to spontaneous radial formation in controls lacking MMC treatment (Table 1 and Fig. 2 b, d). Thus radial formation correlates well with cell survival as an index of sensitivity to ICL adducts.
Figure 2. RAD51 depleted cells form radials.
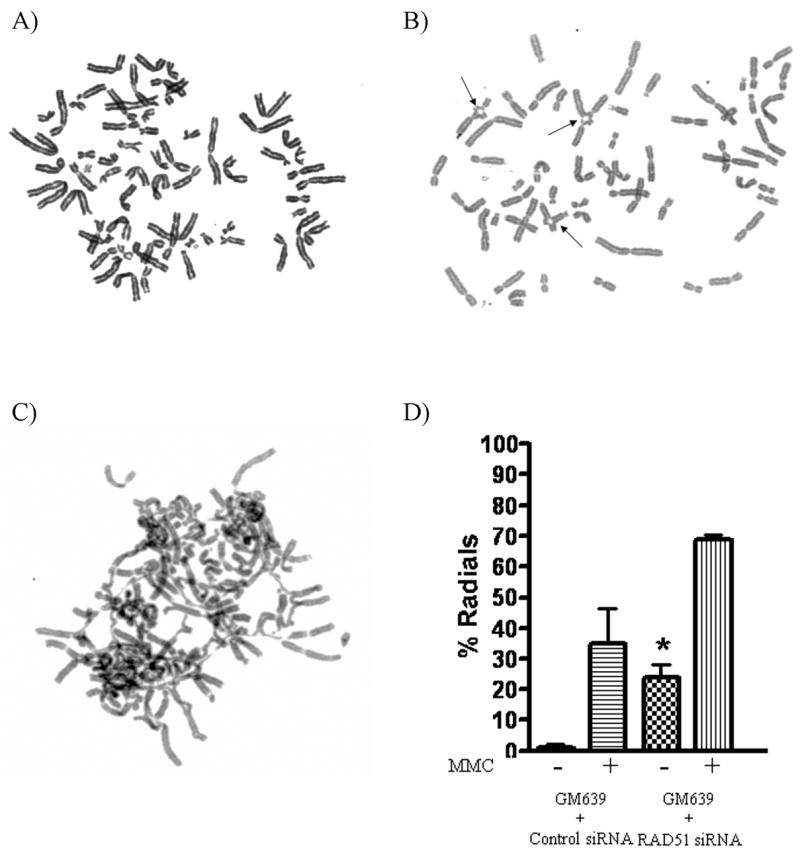
Metaphase spreads of normal GM639 cells treated with A) negative control scrambled siRNA and no MMC B) RAD51 siRNA and no MMC showing spontaneous radial formation (arrows), and C) RAD51 siRNA and 40 ng/ml of MMC. D) Depletion of RAD51 from normal GM639 cells caused increased spontaneous radials (*), with further increase following MMC treatment.
Figure 3. Cells depleted of proteins of interest are sensitive to MMC.
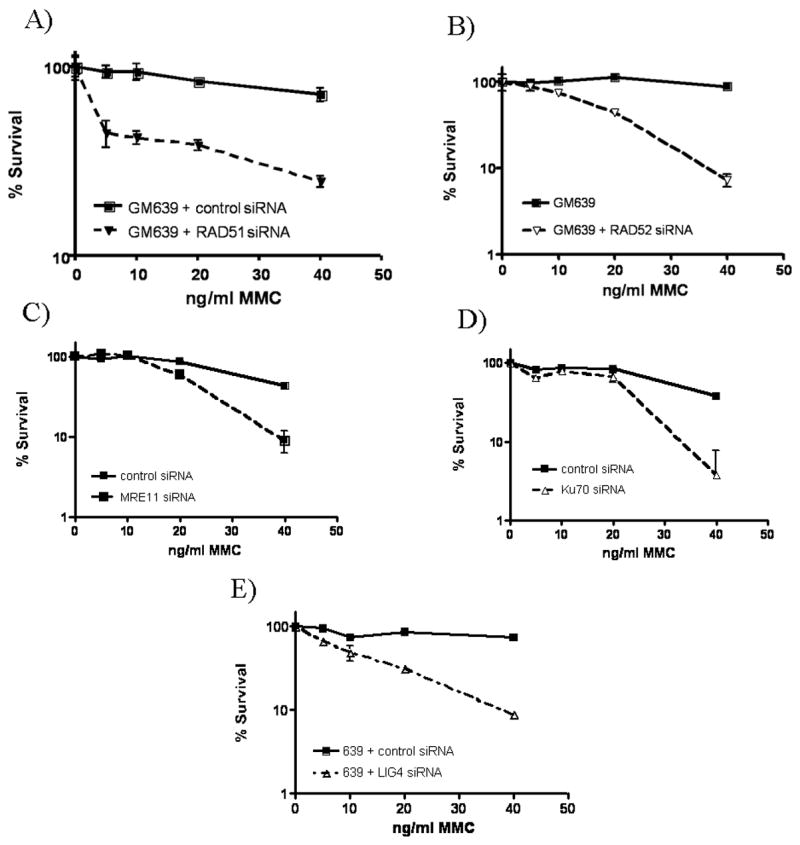
Normal GM639 cells siRNA depleted of A) RAD51, B) RAD52, C) MRE11, D) Ku70, and E) LIG4 proteins show MMC sensitivity through colony formation evaluated following 10 days of designated MMC concentration treatment.
RAD52 depletion also increased MMC sensitivity and radial formation (Table 1, Fig. 3b) in response to ICL treatment. However, unlike the RAD51-depleted cells, spontaneous radials were not noted. These data indicate that, like RAD51, RAD52 is not required for radial formation but actually functions in prevention of radial formation. This eliminates HR as a basis for radial formation.
MRE11 has been reported to act in HR repair (Haber 1998). Therefore, we depleted MRE11 from the GM639 cells. As with RAD52, depletion of MRE11 increased MMC sensitivity as detected by cell survival and radial formation (Table 1, Fig. 3c.) but did not increase spontaneous radial formation.
The increased ICL sensitivity of cells depleted of RAD51, RAD52, and MRE11, indicates a requirement for HRR in normal ICL damage repair and maintenance of genome stability. Furthermore, the radial formation seen in these cells following MMC treatment, and the spontaneous radial formation seen in RAD51 depleted cells, suggests that these proteins are required for either prevention or resolution of radial formation.
Depletion of NHEJ proteins induces MMC sensitivity and radial formation
In addition to homologous recombination, another separate repair process in the mammalian ICL response is NHEJ. To analyze the role of NHEJ in ICL damage response and radial formation, we depleted several proteins acting in NHEJ. Cells depleted of Ku70 had increased sensitivity to ICL damage, as illustrated through increased radial formation. Depleted cells exhibited increased radial formation when compared to normal cells transfected with control siRNA (Table 1). Increased ICL sensitivity was also seen in decreased cell survival following Ku70 depletion (Fig. 3d).
To further test the involvement of NHEJ in the ICL response and radial formation, cells depleted of LIG4 were tested for MMC-induced radial formation and cell survival. As with cells depleted of Ku70, LIG4-depleted cells formed increased numbers of radials following MMC treatment (Table 1) and had increased sensitivity to MMC in cell survival assays (Fig. 3e).
Overall, depletion of NHEJ proteins MRE11 (above), Ku70 and LIG4 caused an increase in ICL-induced radial formation. These results indicate that the NHEJ process is involved in preventing radial formation response and cellular processing of ICL damage. Again the correlation of radial formation with decreased cell survival is uniform.
Discussion
RAD51 and RAD52 are necessary for ICL damage response
HRR has been indicated as a possible pathway involved in human cell response to ICL damage (Niedernhofer et al. 2005). This has been difficult to investigate since null mutations in RAD51 cause both embryonic and cell lethality. However, mammalian cell lines with mutations in RAD51 paralogs, RAD51C, XRCC2, and XRCC3, are viable and therefore assessable. Survival of such mutant cells is impaired following exposure to ICL agents (Liu et al. 1998; Godthelp et al. 2002), indicating that HRR plays a role in response to ICL damage. The work reported here underscores the utility of siRNA depletion, since it allows, within the time frame used, studies with depletion of proteins for which mutant cell lines are not available or viable. RAD51 or RAD52 depletion caused human fibroblasts to have increased sensitivity to MMC as seen by radial formation and cell survival. This demonstrates that RAD51- and RAD52-dependent HRR is involved in the ICL damage response.
In addition to increased ICL-induced sensitivity and radial formation, RAD51-depleted cells displayed spontaneous radial formation even when not treated with MMC. This suggests involvement of RAD51 in more than one pathway for maintenance of genome stability since inactivation of either HR or NHEJ alone does not lead to spontaneous radial formation.
NHEJ proteins are required for appropriate processing of ICL damage
It has been suggested that excision of an ICL adduct leads to formation of a DSB (Rothfuss and Grompe 2004). In addition, IR-induced DSBs are known to lead to radial formation, with NHEJ being a major repair response in a mouse model (Houghtaling et al. 2005). In agreement with this, we find the NHEJ proteins Ku70, MRE11, and LIG4 all appear to be needed for the normal process of repair following MMC treatment. Since NHEJ acts in DSB repair, loss of NHEJ would increase radial formation, suggesting DSBs may be precursors to radials.
Radial formation is not mediated by HR or NHEJ
In this study, we found that depleting any of several HR or NHEJ proteins resulted in an FA-like phenotype of increased radial formation, and decreased cell survival following treatment with MMC. These results indicate that HR and NHEJ proteins are required for either prevention or resolution of radial formation following ICL damage as well as for normal cell survival. Thus, two distinct repair pathways are needed for a normal cellular response after ICL formation.
Radial structures have been suggested to be chromatid fusions following ICL-induced DSBs, dependent on recombination (Niedernhofer et al. 2005). The presence of ICL-induced radials in HR-deficient or NHEJ-depleted cells excludes the dominant RAD51-dependent recombination pathway as the basis for such a mechanism. In addition, the cohesion of sister chromatids does not appear to be required for radial formation, since cells depleted of SMC1, a ‘cohesin’ protein, show normal radial formation following MMC treatment (data not shown).
An increasing number of proteins functioning in separate DNA repair paths are being identified as necessary for avoidance of damage-induced radial formation (Table 2). This indicates that radials are not reflective of any single DNA repair pathway or its defects. It has also been demonstrated that more than one type of DNA damage can lead to radial formation. Since radials are noted with such an array of repair protein deficiencies, and after several types of DNA damage, they are non-specific. Radials might arise from a single structure resulting from a failure by any of several repair responses to process ICL damage normally. The nature of such a putative intermediate is unknown, but it would occur in response to ICL damage and arise with failure of any of several repair functions. As shown here, loss of NHEJ proteins or HR proteins leads to increased radials after ICL, precluding a requirement for HR or NHEJ in radial formation. Lastly, the correlation of decreased cell survival with radial formation following ICL adducts indicates that radials reflect DNA damage inadequately repaired.
Table 2. Summary of protein deficiencies leading to an increase in radial formation.
Loss of function or siRNA depletion of proteins listed here causes increased radial formation either spontaneously, following ICL-damage, or both. Five of these have been shown in the depletions in this manuscript. BLM is Bloom syndrome protein. DNA-PK is for the mouse Prkdc protein, the catalytic subunit of DNA-dependent protein kinase.
| Protein deficiency causing ICL-induced radial increase |
|---|
| FA proteins1 |
| BLM5 |
| RAD512* |
| RAD522 |
| LIG42 |
| MRE112 |
| Ku702 |
| BRCA13 |
| BRCA23 |
| SNM14 |
| ERCC16 |
| DNA-PK7 |
(Schroeder et al. 1964; reviewed in D’Andrea and Grompe 2003)
shown in this manuscript.
(German 1997).
spontaneous radials without clastogen exposure.
Acknowledgments
We thank Donald Bruun for technical advice in experiment protocol. We also thank Yumi Torimaru for technical assistance. This work was supported by an NHLBI Program Project grant PO1 HL048546, and a post-doctoral NIH Training Grant GM008796 to Y.M.N.A.
References
- Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M. DNA replication is required To elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol Cell Biol. 2000;20(21):8283–9. doi: 10.1128/mcb.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach AD, Adler B, Chaganti RS. Prenatal and postnatal diagnosis and carrier detection of Fanconi anemia by a cytogenetic method. Pediatrics. 1981;67(1):128–35. [PubMed] [Google Scholar]
- Bruun D, Folias A, Akkari Y, Cox Y, Olson S, Moses R. siRNA depletion of BRCA1, but not BRCA2, causes increased genome instability in Fanconi anemia cells. DNA Repair (Amst) 2003;2(9):1007–13. doi: 10.1016/s1568-7864(03)00112-5. [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Chen DJ. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair (Amst) 2006;5(9–10):1042–8. doi: 10.1016/j.dnarep.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Cervenka J, Arthur D, Yasis C. Mitomycin C test for diagnostic differentiation of idiopathic aplastic anemia and Fanconi anemia. Pediatrics. 1981;67(1):119–27. [PubMed] [Google Scholar]
- Chen CC, Motegi A, Hasegawa Y, Myung K, Kolodner R, D’Andrea A. Genetic analysis of ionizing radiation-induced mutagenesis in Saccharomyces cerevisiae reveals TransLesion Synthesis (TLS) independent of PCNA K164 SUMOylation and ubiquitination. DNA Repair (Amst) 2006;5(12):1475–88. doi: 10.1016/j.dnarep.2006.07.007. [DOI] [PubMed] [Google Scholar]
- D’Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3(1):23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7(2):273–82. doi: 10.1016/s1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20(21):7980–90. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486(4):217–47. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–8. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7(2):249–62. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- German J. Bloom’s syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet. 1997;93(1):100–6. doi: 10.1016/s0165-4608(96)00336-6. [DOI] [PubMed] [Google Scholar]
- Godthelp BC, Wiegant WW, van Duijn-Goedhart A, Scharer OD, van Buul PP, Kanaar R, Zdzienicka MZ. Mammalian Rad51C contributes to DNA cross-link resistance, sister chromatid cohesion and genomic stability. Nucleic Acids Res. 2002;30(10):2172–82. doi: 10.1093/nar/30.10.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA LIG4 is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. 1998;2(4):477–84. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- Grossmann KF, Ward AM, Matkovic ME, Folias AE, Moses RE. S. cerevisiae has three pathways for DNA interstrand crosslink repair. Mutat Res. 2001;487(3–4):73–83. doi: 10.1016/s0921-8777(01)00106-9. [DOI] [PubMed] [Google Scholar]
- Haber JE. The many interfaces of Mre11. Cell. 1998;95(5):583–6. doi: 10.1016/s0092-8674(00)81626-8. [DOI] [PubMed] [Google Scholar]
- Harrison JC, Haber JE. Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet. 2006;40:209–35. doi: 10.1146/annurev.genet.40.051206.105231. [DOI] [PubMed] [Google Scholar]
- Hemphill AW, Bruun D, Thrun L, Akkari Y, Torimaru Y, Hejna K, Jakobs PM, Hejna J, Jones S, Olson SB, Moses RE. Mammalian SNM1 is required for genome stability. Mol Genet Metab. 2008 doi: 10.1016/j.ymgme.2007.11.012. http://dx.doi.org/10.1016/j.ymgme.2007.11.012. [DOI] [PMC free article] [PubMed]
- Houghtaling S, Newell A, Akkari Y, Taniguchi T, Olson S, Grompe M. Fancd2 functions in a double strand break repair pathway that is distinct from non-homologous end joining. Hum Mol Genet. 2005;14(20):3027–33. doi: 10.1093/hmg/ddi334. [DOI] [PubMed] [Google Scholar]
- Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, Taccioli GE, Alt FW. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell. 1995;83(7):1079–89. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- Liu N, Lamerdin JE, Tebbs RS, Schild D, Tucker JD, Shen MR, Brookman KW, Siciliano MJ, Walter CA, Fan W, Narayana LS, Zhou ZQ, Adamson AW, Sorensen KJ, Chen DJ, Jones NJ, Thompson LH. XRCC2 and XRCC3, new human Rad51-family members, promote chromosome stability and protect against DNA cross-links and other damages. Mol Cell. 1998;1(6):783–93. doi: 10.1016/s1097-2765(00)80078-7. [DOI] [PubMed] [Google Scholar]
- McHugh PJ, Sones WR, Hartley JA. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20(10):3425–33. doi: 10.1128/mcb.20.10.3425-3433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler M. DNA adducts of medicinal drugs: some selected examples. J Cancer Res Clin Oncol. 1986;112(3):210–5. doi: 10.1007/BF00395914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Bressan DA, Shinohara M, Haber JE, Shinohara A. In vivo assembly and disassembly of Rad51 and Rad52 complexes during double-strand break repair. Embo J. 2004 doi: 10.1038/sj.emboj.7600091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Theunissen JW, Kim CF, Kitagawa R, Kastan MB, Petrini JH. The Rad50S allele promotes ATM-dependent DNA damage responses and suppresses ATM deficiency: implications for the Mre11 complex as a DNA damage sensor. Genes Dev. 2005;19(24):3043–54. doi: 10.1101/gad.1373705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller C, Christodoulopoulos G, Salles B, Panasci L. DNA-Dependent protein kinase activity correlates with clinical and in vitro sensitivity of chronic lymphocytic leukemia lymphocytes to nitrogen mustards. Blood. 1998;92(7):2213–9. [PubMed] [Google Scholar]
- Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, Mathew CG, Kastan MB, Weaver DT, D’Andrea AD. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol. 2002;4(12):913–20. doi: 10.1038/ncb879. [DOI] [PubMed] [Google Scholar]
- Newell AE, Akkari YM, Torimaru Y, Rosenthal A, Reifsteck CA, Cox B, Grompe M, Olson SB. Interstrand crosslink-induced radials form between non-homologous chromosomes, but are absent in sex chromosomes. DNA Repair (Amst) 2004;3(5):535–42. doi: 10.1016/j.dnarep.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123(7):1191–8. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24(13):5776–87. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P, Averbeck D, Rosselli F. DNA cross-link-dependent RAD50/MRE11/NBS1 subnuclear assembly requires the Fanconi anemia C protein. Hum Mol Genet. 2002;11(21):2531–46. doi: 10.1093/hmg/11.21.2531. [DOI] [PubMed] [Google Scholar]
- Riha K, Heacock ML, Shippen DE. The role of the nonhomologous end-joining DNA double-strand break repair pathway in telomere biology. Annu Rev Genet. 2006;40:237–77. doi: 10.1146/annurev.genet.39.110304.095755. [DOI] [PubMed] [Google Scholar]
- Rothfuss A, Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2004;24(1):123–34. doi: 10.1128/MCB.24.1.123-134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki MS. Is Fanconi’s anaemia defective in a process essential to the repair of DNA cross links? Nature. 1975;257(5526):501–3. doi: 10.1038/257501a0. [DOI] [PubMed] [Google Scholar]
- Sasaki MS, Tonomura A. A high susceptibility of Fanconi’s anemia to chromosome breakage by DNA cross-linking agents. Cancer Res. 1973;33(8):1829–36. [PubMed] [Google Scholar]
- Schroeder TM, Anschutz F, Knopp A. Spontaneous chromosome aberrations in familial panmyelopathy. Humangenetik. 1964;1(2):194–6. doi: 10.1007/BF00389636. [DOI] [PubMed] [Google Scholar]
- Song B, Sung P. Functional interactions among yeast Rad51 recombinase, Rad52 mediator, and replication protein A in DNA strand exchange. J Biol Chem. 2000;275(21):15895–904. doi: 10.1074/jbc.M910244199. [DOI] [PubMed] [Google Scholar]
- Su TT. Cellular responses to DNA damage: one signal, multiple choices. Annu Rev Genet. 2006;40:187–208. doi: 10.1146/annurev.genet.40.110405.090428. [DOI] [PubMed] [Google Scholar]
- Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. 2003;22(37):5792–812. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Boyer TG, Chen PL, Lee WH. Deficient nonhomologous end-joining activity in cell-free extracts from Brca1-null fibroblasts. Cancer Res. 2002;62(14):3966–70. [PubMed] [Google Scholar]