

Abstract
Cross-linked polyelectrolyte multilayer films (CL PEM) have an increased rigidity and are mechanically more resistant than native (e.g. uncrosslinked) films. However, they are still biodegradable, which make them interesting candidates for biomedical applications. In this study, CL PEM films have been explored for their multifunctional properties as i) mechanically resistant ii) biodegradable and iii) bioactive films. Toward this end, we investigated drug loading into CL chitosan/hyaluronan (CHI/HA) and poly(L-lysine)/hyaluronan (PLL/HA) films by simple diffusion of the drugs. Sodium diclofenac and paclitaxel were chosen as model drugs and were successfully loaded into the films. The effect of varying the number of layers in the (CHI/HA) films as well as the cross-linker concentration on diclofenac loading were studied. Diclofenac was released from the film in about ten hours. Paclitaxel was also found to diffuse within CL films. Its activity was maintained after loading in the CL films and cellular viability could be reduced by about 55% over three days. Such simple approach may be applied to other types of cross-linked films and to other drugs. These results prove that it is possible to design multifunctional multilayer films that combine mechanical resistance, biodegradability and bioactivity properties into a single PEM architecture.
Introduction
Local drug delivery at a specific implantation site is particularly interesting in many biomedical and tissue engineering applications as it may reduce toxicity and increase the efficiency of the drug. Various approaches include embedding of drugs in biodegradable polymers 1,2, chemical grafting of a drug to a natural or polymeric matrix and its subsequent release upon hydrolysis 3,4. Recently, a new class of polymeric surface coatings, the polyelectrolyte multilayer films (PEM) has attracted great attention due to its large potentialities 5-9. As PEM films can be coated on any type of implant materials 10-12 with virtually any shape 13,14, they appear as interesting candidates for the localized delivery of bioactive molecules including peptides 15, drugs 16 or DNA 17,18. Bioactive molecules can be incorporated into such films by different strategies : simple adsorption of the bioactive molecules at a certain step during the film buildup 19, 15, 20,21 22 16, simple adsorption by dipping the preformed film in a drug containing solution 23 24, which is also called the post-diffusion method or more sophisticated strategies. Theses more elaborated strategies make use of either cyclodextrins as hydrophobic drug or plasmid carriers 18,25, of a prodrug approach 26 or rely on the preparation of films with polysaccharides grafted with alkyl chains, which are amphiphilic and allow the formation of hydrophobic nanocavities in the films 27. Bioactive molecules may also be released from degradable PEM or hollow capsules via a diffusion mechanism or via degradation of the film using hydrolytically degradable polycations 28,29, enzymatically degradable polyelectrolytes 12,30, or via sensitivity to pH or sodium chloride solutions 31-34. Layer-by-layer films were also recently used to provide a diffusion barrier to encapsulated glucose oxydase inside alginate microspheres 35. Diffusion of dyes is also the subject of intense studies in order to better understand the interaction of small molecules with polymer multilayers 36,37.
A usual requirement for the design of bioactive films is that the cell type, which is targeted by the bioactive molecules, has to come in contact and adhere to the film to be in contact with the drug, unless the function of the film would precisely to be non-adhesive or cell resistant 38. Very often, thick and hydrated films such as those that can be designed as drug “reservoirs” are often unfavourable for primary cell adhesion possibly due to their high hydration or high softness 38-40. Several strategies have therefore been developed to enhance cell adhesion such as increasing cell stiffness by embedding of nanoparticules 41, chemically cross-linking the films by heating 39 or using carbodiimide chemistry 42, grafting cell adhesive peptides 43,44 or sugar molecules 45 and more recently capping the film with few layers of synthetic polyelectrolytes 24. All these strategies have consequences on the film chemistry, on its mechanical properties, or on both 46.
Very interestingly, soft PEM films made of polysaccharides and polypeptides initially exhibiting a poor cell adhesion were found to favour, once cross-linked, cell adhesion and proliferation of various primary cell types 44,47. Furthermore, it is possible to take benefit of the biodegradability of films made of natural polyelectrolytes, such as polysaccharides, which can be degraded by various enzymes, and to tune this biodegradability by controlling the concentration of the cross-linking agent 48. So far, cross-linked films have always been used without any bioactive molecule inserted in the film bulk. It is known that the cross-linking procedure modifies the relative abundance of chemical groups present in the films 42,48, as amide bonds are created upon cross-linking whereas the number of free carboxylic groups is decreased. The charge balance within the films is thus expected to be greatly modified.
Therefore, an important question that remains so far unanswered is whether the cross-linked films can also be functionalized by insertion of bioactive molecules, which would allow both a control of film mechanical properties and of their bioactivity. In addition, if these films are biodegradable, one could envision combining all these properties within a single multifunctional PEM architecture. In this study, we will show for the first time that two model drugs can be embedded in CL PEM films. The nonstereoidal anti-inflammatory drug diclofenac 49-51 and the anti-cancer drug paclitaxel (taxol) 52, which has efficient antiproliferative and antimitotic properties, were studied as model drugs (Figure 1). These drugs have been mostly loaded in gels 53 54, in particles 55 or grafted to polysaccharides 53 or polypeptides 56 but only few studies focused on their embedding and bioactivity in films 24,26. Two different types of films made either of chitosan (CHI) or poly(L-lysine) (PLL) as polycations and hyaluronan (HA) as polyanion will be investigated. We will demonstrate that it is possible to combine advantages of cross-linked films, e.g. enhanced cellular adhesion, enhanced mechanical resistance and biodegradability properties with the possibility to render film bioactive.
FIGURE 1.
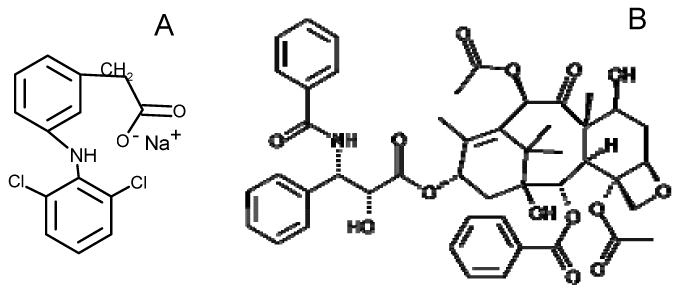
Chemical structures of (A) sodium diclofenac (B) paclitaxel (ta)
Experimental
Polyelectrolyte solutions
Film preparation and cross-linking
HA (sodium hyaluronate, 4×105 g/mol) was purchased from Bioiberica (Spain), CHI (5×103 g/mol) was purchased from Medipol (Switzerland) and PLL (3×104 g/mol) was purchased from Sigma (France). CHI and HA were dissolved at 1 mg/mL in 0.15 M NaCl in water at pH 4.5 for CHI/HA films and PLL and HA were dissolved at pH 6.5 for PLL/HA films. (CHI/HA)i and (PLL/HA)i films were prepared with an automatic dipping machine on 14 mm glass slides as previously described (Dipping Robot DR3, Kirstein and Riegler GmbH, Germany) 42 12 (VWR Scientific, France) cleaned with 10 mM SDS and 0.1 N HCl and extensively rinsed. For film cross-linking, we followed the previously published protocol using the water soluble carbodiimide, 1-Ethyl-3-(3-Dimethylamino-propyl)Carbodiimide (EDC) and N-Hydrosulfosuccinimide (sulfo-NHS) (both purchased from Sigma) 42. Briefly, EDC and sulfo-NHS were prepared at 70 mg/mL and 22 mg/mL respectively in a 0.15 M NaCl solution at pH 4.5. 1 mL of the mixed EDC/Sulfo-NHS solution (v/v) at pH 4.5 (for CHI/HA films and at pH 5 for PLL/HA films) was deposited in the wells containing the film coated glass slides and left for 18 hours at 4°C. All the experiments were performed at this cross-linker concentration unless otherwise noted.
Atomic Force Microscopy
Atomic force images were obtained in contact mode in air with the Multimode Nanoscope IV from Veeco (Santa Barbara, CA, USA). Cantilevers with a spring constant of 0.03 N/m and with silicon nitride tips were used (Model MLCT-AUHW Park Scientific, Sunnyvale, CA, USA). Deflection and height mode images were scanned simultaneously at a fixed scan rate (between two and four Hz) with a resolution of 512 × 512 pixels. The mean roughness of the films was calculated according to : where zij is the height of a given pixel, zmean is the average height of the pixels, and Nx and Ny are the number of pixels in the x and y directions.
Determination of the film mechanical properties by force spectroscopy
The measurements aimed at the determination of the elasticity of the films were performed on (CHI/HA)24 films using a homemade AFM working in the indenter-type mode (IT-AFM) 57. This number of layer pairs was chosen as these films can be easily visualized by confocal microscopy and their thickness be measured (for input in the curve fitting procedure) 40. The probe was constituted by a borosilicate sphere of 5 μm in diameter fixed to the cantilever whose spring constant was either 0.06 or 0.38 N m−1 as indicated by the manufacturer (BioForce, Nanosciences Inc., Ames, IA) and confirmed by the thermal fluctuation technique 58. All experiments were performed in liquid environment, i.e. the films were immersed in a drop of 0.15M NaCl aqueous solution and the curves were fitted to extract the Young's modulus of the film E0 using a modified Hertz model that was previously detailed 59.
Diclofenac deposition and measurement of adsorbed amount
Cross-linked (CHI/HA)i (i=12, 24, 48) films were equilibrated for one hour in a 0.15M NaCl solution at pH 7.4. 1 mL of 2mg/mL diclofenac solution (D899, Sigma, in NaCl 0.15M pH=7.4) was deposited and let adsorbed for two hours. The supernatants were collected and replaced by 1 mL of 0.01M SDS to extract the diclofenac from the film. After three hours, the SDS supernatants were collected. Measurements of the sodium diclofenac concentration after diclofenac insertion and after its extraction were performed by UV spectrometry at 276 nm (Beckman Coulter, DU640 Spectrophotometer). The absorbance values were translated to concentration values by comparing to standard solutions of known concentrations that were used to plot the calibration curve (data not shown). This calibration curve followed the Beer-Lambert law. The drug loading was estimated for various numbers of layer pairs i (i= 12, 24 and 48).
For the drug release experiments, cross-linked films (CHI/HA)24 charged with diclofenac were put in 1 mL of PBS (Gibco). After 2, 4, 6, 24, 30 and 48 hours, 200 μL of the supernatant were collected and replaced by 200 μL of fresch PBS.
Adsorption of paclitaxel
Paclitaxel, commercially known as taxol, was a generous gift from the Hôpitaux Universitaires de Strasbourg, France. For cell experiments, it was adsorbed overnight at 20 μg/mL on native and cross-linked (PLL/HA)12 films. Films were then rinsed three times in 0.15M NaCl (pH 6.5). For confocal laser scanning microscopy (CLSM) observations, paclitaxel Oregon Green 488 labeled (paclitaxelGreen 488, Molecular Probes, Oregon, USA) was first dissolved in ethanol/cremophor solution (v/v at 500 μg/mL) and then adjusted to 20 μg/mL in a 0.15 M NaCl solution at pH 6.5.
HT 29 Cell culture
Human colonic adenocarcinoma cells HT29 were kindly provided by M. Kedinger (INSERM UMR-S682, Strasbourg, France)60. They were routinely cultured in 25 cm2 culture flasks in Glutamax I DMEM 25 mM glucose (Invitrogen, France), without sodium pyruvate and supplemented with 10% v/v heat-inactivated fetal calf serum, penicillin (100 U/ml) and streptomycin 0.1 mg/ml (Invitrogen, France). Cultures were incubated at 37 °C in humidified air atmosphere (95%)-CO2 (5%). Medium was changed every other day. For subcultures, cells were incubated in a mixture of 0.05% trypsin and 0.53M EDTA and then harvested. The cells were distributed into 24-well plates containing the film coated glass slides (104/well/slide) in 1 mL of medium. Viability measurements were performed after 24h, 48h and 72h in culture. Experiments were performed in triplicate.
Cell viability
The measurement of viability by the acid phosphatase method makes it possible to quantify the number of living cells with a greater sensitivity than the XTT test 61. The principle relies on the titration of the enzyme whose activity increases proportionally with the number of viable cells. The culture medium was first replaced with 300 μl PBS. The buffer containing 0.1 M sodium acetate (pH 5.5), 0.1% Triton X-100 and 10 mM p-nitrophenyl phosphate (pNPP, Sigma) was added in each well. The plates were placed for 3h at 37 °C in the cell culture incubator. The reaction was stopped by addition of 40 μl of 1N sodium hydroxide and the absorbance of the solution (yellow color) was measured at 405 nm using a spectrophotometer (Labsystems iEMS Reader MF, Gibco). Statistical analysis were performed using Sigma-stat software.
Results and discussion
Mechanical and biodegradability properties of (CHI/HA) and (PLL/HA) cross-linked films
The comparison between both film types in terms of roughness, Young's modulus and biodegradability properties is presented in Table 1. Mechanical properties of native and cross-linked (PLL/HA) films have already been characterized 40 and their biodegradability in vivo in contact with hyaluronidase and macrophages have been evaluated 62. Cross-linked films are much more rigid than native ones and less susceptible to biodegradation. The (CHI/HA) films appear to be more susceptible to rapid degradation in vivo in contact with various enzymes 12 and to be rapidly degraded by macrophages in vitro and in vivo 48. This more rapid biodegradation may be explained by the fact that CHI is a polysaccharide whereas PLL is a synthetic polypeptide. The mechanical properties of native and cross-linked (CHI/HA) films were measured using the AFM-nano indentation technique (Table 1). They were found to be qualitatively similar to that of (PLL/HA) films : whereas native films are very soft, cross-linked films are about ten times stiffer, which may explain their enhanced resistance in vivo 48. Consequences of film cross-linking appear thus to lead to common features that are i) increased roughness ii) increased mechanical properties iii) increased resistance to biodegradation.
Table 1.
Summary of the physico-chemical and biodegradability properties of native and cross-linked (PLL/HA)24 and (CHI/HA)24 films.
| (PLL/HA) films | (CHI/HA) films | |||
|---|---|---|---|---|
| Parameter | Native | CL | Native | CL |
| Film roughness (nm) | 1.1 ± 0.4 | 5.8 ± 0.5 | 7.3 ± 1 nm | 39.6 ± 5.3 |
| E0 (kPa) | 20 ± 31 | 250 ± 301 | 15 ± 4 | 159 ± 40 |
| In vitro Enzymatic degradation | Yes2 (in about 10-15 hours) | Not visible over A 48h time period2 | Yes3 rapid (few hours) | Yes3 but much slower (only superficial attack is visible) |
| In vivo Biodegradability | NA | NA | Yes3 rapid (few hours) | Yes3, but slower (several days/weeks) |
Incorporation of diclofenac into cross-linked (CHI/HA) films
A first strategy was tested. It consisted in first adsorbing sodium diclofenac on a native film and then cross-linking the film at pH 5. However, in this case, we noticed that the film became white when the cross-linking solution was added. This may be due to the precipitation of diclofenac in the film. Therefore, a second strategy was investigated. It consisted in first equilibrating the cross-linked film in a NaCl 0.15 M solution at pH 7.4 and then putting the film in contact with diclofenac solution for two hours. UV spectrometry was used to assess that diclofenac effectively adsorbed in the film by measuring the amount of diclofenac extracted from the films using a surfactant (SDS at 0.01 M). By comparing the diclofenac concentration in the supernatant and in the film (after its extraction), it was possible to check that all the drug is either in the film or in the supernatant and that all the drug could be extracted upon addition of the detergent (data not shown).
We then examined the influence of the diclofenac solution concentration on diclofenac loading by varying its concentration over the range 0.2 to 2 mg/mL. A maximum loading was reached for concentrations greater than 1 mg/mL concentration (data not shown). For the highest diclofenac solution concentrations (1 and 2 mg/mL respectively), the maximum incorporated amount was of the order of 23 to 12 % respectively. In addition, for a given diclofenac concentration (2 mg/mL), drug loading can be modulated by varying the number of layers in the film. We observed that the amount of diclofenac incorporated in the film is directly related to the number of layers in the film, for films containing between 12 and 48 layer pairs (Figure 2). Thus, once some parameters such as the adsorption time 27 and the concentration of the drug solution in contact with the film 24 have been fixed, it is still possible to modulate the capacity of the drug reservoir by varying the number of layer pairs in the film for a given diclofenac solution concentration. Our results are qualitatively similar to that obtained recently by Rubner et al. for nanoporous films 23. These authors showed that the total amount of drug released increased as the number of porous layer pairs increased. This indirectly indicates that the adsorbed amounts, although not directly measured in the aforementioned study were also related to the number of layer pairs. The results suggest that the diclofenac is diffusing through the whole film and that the film behaves as a drug reservoir.
FIGURE 2.

Influence of the number of layer pairs in the films on the diclofenac loaded concentration in cross-linked (CHI/HA)i films (with i=12, 24 and 48). The diclofenac solution concentration in contact with the film was fixed at 1 mg/mL. (Error bars are SD of three PEM coated slides).
As diclofenac is negatively charged at neutral pH (pKa = 4) and also slightly hydrophobic, it may associate with positive charges in the film or with hydrophobic domains. We investigated whether the diclofenac adsorbed amount depends on the extent of film cross-linking (Figure 3). We found that the adsorbed amount did not depend on cross-linker concentration, which rather suggests that diclofenac preferentially interacts with hydrophobic film regions as the number of free carboxylic and amine groups is decreased upon cross-linking 42. HA three dimensional structure has been shown to be stabilized by hydrogen bonds and molecular modeling suggested that hydrophobic interactions help to stabilize these structures 63. In a recent work, HA has been shown to interact with graphite, a hydrophobic surface. The authors concluded that HA molecules may interact with graphite through hydrophobic patches along its surface 64. Such hydrophobic cavities may also be naturally present in various film types such as poly(acrylic acid)/poly(allyl amine hydrochloride), which have also been found to incorporate hydrophobic drugs such as cytochalasin D and Ketoprofen 23. In addition, the number or size of the hydrophobic nanodomains could be recently adjusted by using chemically modified polysaccharides (such as amphiphilic polymers) 27,65, which were shown to increase the loading capacity of the films 27.
FIGURE 3.

Influence of the cross-linker concentration EDC on the diclofenac loaded concentration in (CHI/HA)24 films. The initial diclofenac concentration was the same as for Figure 2. (Error bars are SD of three PEM coated slides).
Release of diclofenac
Diclofenac loaded films were then set in contact with PBS to investigate the release behaviour of the films. The release appeared to be in two phases with a first burst effect within about half an hour followed by a slower release phase (Figure 4). This kind of two phase behaviour has already been observed for various drug loaded hydrogels or nanoparticules 66,67, for which various parameters such as pH and gel of composite nanoparticules formulation were indeed found to influence the release 68. For chitosan nanoparticles, the molecular weight of polysaccharide used for preparing the particles was also found to play a role 66. In our case, about 90% of the loaded amount is released within about 20 hours. This is a relatively high release rate as compared to films containing hydrophobic nanodomains for which 60% of release can be obtained at best in non physiologic conditions 27. For other dye containing films, release was strongly dependent on pH of the release solution and occurred typically in about 15 to 50 min at physiological pH depending on the dye and on film type 37 36. In addition, the fraction of dye released never reach 100% and varied from 15% to 40% at most 37 36. For (CHI/HA) films containing the drug paclitaxel coupled to HA, half-life of paclitaxel was about 3 hours 26 whereas nanoporous thin films could release drugs over several days or weeks 23. It thus seems that release kinetics of PEM films strongly depends on film architecture and on dye/film or drug/film interactions. The exact mechanisms that lead to entrapment and subsequent release of dyes and drugs are not very well understood. In our case, as cross-linked films are stable in PBS, we hypothesize that the release is due to a passive diffusion out of the film as the drug is not covalently bound to the film and as it may be entrapped in hydrophobic domains.
FIGURE 4.

Diclofenac release curve in PBS from a (CHI/HA)24 film. Results are expressed in terms of percentage of the total diclofenac concentration loaded in the film. (error bars are SD of three measurements but are not visible).
Incorporation of Paclitaxel in (PLL/HA) films and cell activity
To further validate the concept of drug loading into CL films, another type of drug, paclitaxel was loaded in another film architecture composed of cross-linked (PLL/HA) films. Paclitaxel was recently coupled to hyaluronan (HA-Pac) and was found to remain active on macrophages when inserted in (chitosan/HA-Pac) films 26. It was also loaded in native (PLL/HA) films capped with (PSS/PAH) synthetic layers that were needed for improving cell adhesion 24. In the present study, we worked at constant paclitaxel concentration for loading and did not investigate the influence of its concentration. We first verified the incorporation of paclitaxel in the film by observing CL (PLL/HA)24 loaded with fluorescently labeled paclitaxel by CLSM (Figure 5). The visualization of a green band indicates that Paclitaxel is diffusing throughout the CL film similarly to native (PLL/HA) films 24. The low fluorescence intensity is due to the low fluorescent paclitaxel concentration. This diffusion is analogous to what occurs for diclofenac in (CHI/HA) films.
FIGURE 5.
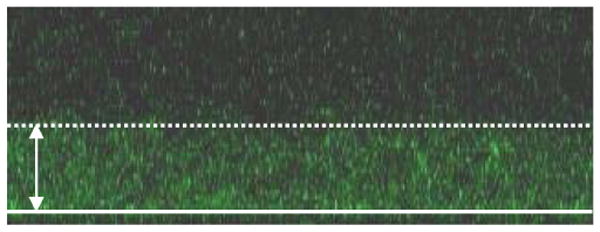
Confocal laser scanning (CLSM) image of a CL (PLL/HA)24 film loaded with fluorescent paclitaxel (paclitaxelGreen 4888) at 20 μg/mL. The white line corresponds to the glass slide and the white arrow to the film thickness (approximately 4 μm).
Human colonic adenocarcinoma cells (HT29) cell activity was then evaluated onto CL films that contained paclitaxel. First, we verified that HT29 cells initial activity was significantly increased on CL films as compared to native ones (Figure 6). A good cell adhesion is effectively an important prerequisite for testing the subsequent action of paclitaxel on cells. Only few HT29 cells adhered and remained viable on native films, which is similar to results obtained for other cells types such as chondrosarcomas, primary chondrocytes, and primary motoneurons 12,42. Even if the high thickness and gel like character of native (PLL/HA) films make them a good reservoir for drugs, such films are clearly not appropriate when cells have first to come into contact and adhere onto films before being able to “sense” bioactive molecules. We first investigated paclitaxel activity when paclitaxel was loaded in (PLL/HA) films prior to film cross-linking. However, in this case, no paclitaxel activity was observed (data not shown), which is probably due to a loss of activity of paclitaxel upon cross-linking during the formation of amide bonds. As a consequence, paclitaxel was loaded into CL films after cross-linking. Cell viability was followed over a three days period and the number of viable cells was counted everyday (Figure 7). Figure 7A are representing the same data but in a different manner. In Figure 7A, cell viability was set at 100% for the control CL films at 24H. Cell viability is clearly increasing on the CL films, similarly to what was already found for chondrosarcomas cells and primary chondrocytes onto such CL (PLL/HA) films 47,62. In comparison, cell viability was steadily decreasing on the paclitaxel loaded CL films. In Figure 7B, CL films without drug were taken as reference (cell viability being set at 100% for comparison) for each time period in culture. After 24 hours in culture, there was already a significant difference between paclitaxel loaded CL films and controls. The differences were even increasing over the three days culture period. Paclitaxel had a dramatic effect on HT29 viability over this period as less than 46% of the initial cells remained viable. When cell number on drug loaded films is compared to the number of cells on control CL films, this percentage falls down to 10% over the 72H time period. The differences between both film types were significantly different at each time period. The activity of the drug incorporated into CL films was thus fully conserved. The mechanism of paclitaxel action requires its internalization by the cells. Such internalization has indeed recently been shown on adherent cells anchoring to films capped with PSS/PH layers that contained fluorescently labeled paclitaxel loaded at high concentration (100 μg/mL) 69. It is however difficult to know whether the molecules are diffusing toward the cell membrane through the film or whether the cells would be able, via membrane protrusions (pseudopodia or filopodia) to detect the presence of the molecules in the film. In any case, if paclitaxel were diffusing out of the films, its diffusion would probably occur within hours or even days as the effect of paclitaxel was still visible three days after cell plating even if the medium was changed every two days.
FIGURE 6.

Acid phosphatase (AP) activity for HT29 cells cultured on native or cross-linked (PLL/HA)12 films after 24 hours. The error bars represent the standard deviation. The value of 100% has been arbitrary attributed to CL films (*** p<0.001).
FIGURE 7.

Acid phosphatase (AP) activity for HT29 cells cultured on cross-linked (PLL/HA)12 films loaded or not with paclitaxel, after different time periods of 24H, 48H and 72H in culture. The error bars represent the standard deviation. Two different representations are shown: (A) The value of 100% has been arbitrary attributed to CL films at 24H; (** p<0.01; *** p<0.001 versus control at 24H for control films or versus paclitaxel at 24H for paclitaxel loaded films). (B) The value of 100% has been arbitrary set at 100% for CL films at each time period (* p<0.05; ** p<0.01; *** p<0.001 versus controls, which are the CL films at time 24H, 48H, and 72H respectively.
The optimal duration of release in vivo would strongly depend on the type of biomedical application. For instance, in vascular therapy, it has been suggested that a sustained release of the drug for at least three weeks after stent deployment is required to present the cascade of biological events that lead to restenosis 70. In the case of solid tumor however (like metastatic breast tumors), one has to notice that the effect of drugs even on few days is interesting as one day is the time by which the tumors in normal conditions have almost doubled in volume 71.
Conclusion
We have developed multifunctional polyelectrolyte multilayer films that are i) mechanically resistant, ii) biodegradable and iii) bioactive. The films made of polypeptides and polysaccharides are mechanically resistant as they are cross-linked and are also cell adhesive. Two types of film architectures made (PLL/HA) and (CHI/HA) were chosen as they were already well characterized. They are biodegradable as they can be degraded in vitro and in vivo. In addition, they are bioactive and can function as drug carriers. To render the film bioactive, the model drugs sodium diclofenac and paclitaxel were loaded after film cross-linking using the simple post-diffusion method. The amount of drug could be controlled by changing the number of layer in the film. Paclitaxel was found to remain active when loaded in CL (PLL/HA) films leading to a dramatic decrease in cell viability over a three days period. All together, these results prove that it is possible to prepare multifunctional films with defined mechanical properties, biodegradability properties and bioactivity. These films will now be employed to investigate the respective roles of film bioactivity versus film mechanical properties on cell adhesion and differentiation.
Acknowledgments
This work has been partly supported by the Action Concertée Incitative “Nanosciences” (ACI NR204) from the Ministère de la Jeunesse, de l'Education Nationale et de la Recherche, by the NIH (R21 grant) via a subcontract to CP (n°544168A) and by the “Association pour la Recherche sur le Cancer” (CP, grant n°7918). AS is indebted to the « Region Alsace » and GF is indebted to the “Faculté de Chirurgie Dentaire” for financial support. We thank Dr M. Kédinger (INSERM UMR-S 682, Strasbourg) for kindly providing the HT29 cells.
References
- 1.Seal BL, Otero TC, Panitch A. Mat Sci Eng C. 2001;34:147–230. [Google Scholar]
- 2.Whittlesey KJ, Shea LD. Experimental Neuroogyl. 2004;190:1–16. doi: 10.1016/j.expneurol.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 3.Luo Y, Prestwich GD. Curr Cancer Drug Targets. 2002;2:209–226. doi: 10.2174/1568009023333836. [DOI] [PubMed] [Google Scholar]
- 4.Prestwich GD, Marecak DM, Marecek JF, Vercruysse KP, Ziebell MR. J Control Release. 1998;53:93–103. doi: 10.1016/s0168-3659(97)00242-3. [DOI] [PubMed] [Google Scholar]
- 5.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 6.Hiller J, Mendelsohn JD, Rubner MF. Nat Mater. 2002;1:59–63. doi: 10.1038/nmat719. [DOI] [PubMed] [Google Scholar]
- 7.Tang Z, Kotov NA, Magonov S, Ozturk B. Nat Mater. 2003;2:413–418. doi: 10.1038/nmat906. [DOI] [PubMed] [Google Scholar]
- 8.Decher G, Hong JD, Schmitt J. Thin Solid Films. 1992;210-211:831–835. [Google Scholar]
- 9.Lvov L, Haas H, Decher G, Moehwald H, Mikhailov A, Mtchedlishvily B, Morgunova E, Vainshtein B. Langmuir. 1994;10:4232–4236. [Google Scholar]
- 10.Zhu Y, Gao C, He T, Liu X, Shen J. Biomacromolecules. 2003;4:446–452. doi: 10.1021/bm025723k. [DOI] [PubMed] [Google Scholar]
- 11.Tan Q, Ji J, Barbosa MA, Fonseca C, Shen J. Biomaterials. 2003;24:4699–4705. doi: 10.1016/s0142-9612(03)00363-6. [DOI] [PubMed] [Google Scholar]
- 12.Etienne O, Schneider A, Taddei C, Richert L, Schaaf P, Voegel JC, Egles C, Picart C. Biomacromolecules. 2005;6:726–733. doi: 10.1021/bm049425u. [DOI] [PubMed] [Google Scholar]
- 13.Schultz P, Vautier D, Richert L, Jessel N, Haikel Y, Schaaf P, Voegel JC, Ogier J, Debry C. Biomaterials. 2005;26:2621–2630. doi: 10.1016/j.biomaterials.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 14.Gao C, Leporatti S, Moya S, Donath E, Mohwald H. Langmuir. 2001;17:3491–3495. [Google Scholar]
- 15.Etienne O, Picart C, Taddei C, Haikel Y, Dimarcq JL, Schaaf F, Voegel JC, Ogier JA, Egles C. Antimicrob Agents Chem. 2004;48:3662–3669. doi: 10.1128/AAC.48.10.3662-3669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thierry B, Winnik FM, Merhi Y, Silver J, Tabrizian M. Biomacromolecules. 2003;4:1564–1571. doi: 10.1021/bm0341834. [DOI] [PubMed] [Google Scholar]
- 17.Jewell CM, Zhang J, Fredin NJ, Lynn DM. J Control Release. 2005;106:214–223. doi: 10.1016/j.jconrel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 18.Jessel N, Oulad-Abdelghani M, Meyer F, Lavalle P, Haikel Y, Schaaf P, Voegel JC. Proc Natl Acad Sci U S A. 2006;103:8618–8621. doi: 10.1073/pnas.0508246103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ai H, Jones SA, Lvov YM. Cell Biochem Biophys. 2003;39:23–43. doi: 10.1385/CBB:39:1:23. [DOI] [PubMed] [Google Scholar]
- 20.Etienne O, Gasnier C, Taddei C, Voegel JC, Aunis D, Schaaf P, Metz-Boutigue MH, Bolcato-Bellemin AL, Egles C. Biomaterials. 2005;26:6704–6712. doi: 10.1016/j.biomaterials.2005.04.068. [DOI] [PubMed] [Google Scholar]
- 21.van den Beucken JJ, Walboomers XF, Boerman OC, Vos MR, Sommerdijk NA, Hayakawa T, Fukushima T, Okahata Y, Nolte RJ, Jansen JA. Journal of Controlled Release. 2006;113:63–72. doi: 10.1016/j.jconrel.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Vodouhe C, Schmittbuhl M, Boulmedais F, Bagnard D, Vautier D, Schaaf P, Egles C, Voegel JC, Ogier J. Biomaterials. 2005;26:545–554. doi: 10.1016/j.biomaterials.2004.02.057. [DOI] [PubMed] [Google Scholar]
- 23.Berg MC, Zhai L, Cohen RE, Rubner MF. Biomacromolecules. 2006;7:357–364. doi: 10.1021/bm050174e. [DOI] [PubMed] [Google Scholar]
- 24.Vodouhe C, Guen EL, Garza JM, Francius G, Dejugnat C, Ogier J, Schaaf P, Voegel JC, Lavalle P. Biomaterials. 2006;27:4149–4156. doi: 10.1016/j.biomaterials.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 25.Jessel N, Schwinté P, Falvey P, Darcy R, Haïkel Y, Schaaf P, Voegel JC, Ogier J. Adv Funct Mater. 2004;14:174–182. [Google Scholar]
- 26.Thierry B, Kujawa P, Tkaczyk C, Winnik FM, Bilodeau L, Tabrizian M. J Am Chem Soc. 2005;127:1626–1627. doi: 10.1021/ja045077s. [DOI] [PubMed] [Google Scholar]
- 27.Guyomard A, Nysten B, Muller G, Glinel K. Langmuir. 2006;22:2281–2287. doi: 10.1021/la052871y. [DOI] [PubMed] [Google Scholar]
- 28.Vazquez E, Dewitt DM, Hammond PT, Lynn DM. J Am Chem Soc. 2002;124:13992–13993. doi: 10.1021/ja026405w. [DOI] [PubMed] [Google Scholar]
- 29.Wood KC, Boedicker JQ, Lynn DM, Hammond PT. Langmuir. 2005;21:1603–1609. doi: 10.1021/la0476480. [DOI] [PubMed] [Google Scholar]
- 30.Serizawa T, Yamaguchi M, Akashi M. Angew Chem Int Ed. 2003;42:1115–1118. doi: 10.1002/anie.200390293. [DOI] [PubMed] [Google Scholar]
- 31.Schüler C, Caruso F. Biomacromolecules. 2001;2:921–926. doi: 10.1021/bm010052w. [DOI] [PubMed] [Google Scholar]
- 32.Antipov AA, Sukhorukov GB. Adv Colloid Interface Sci. 2004;111:49–61. doi: 10.1016/j.cis.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 33.Balabushevich NG, Tiourina OP, Volodkin DV, Larionova NI, Sukhorukov GB. Biomacromolecules. 2003;4:1191–1197. doi: 10.1021/bm0340321. [DOI] [PubMed] [Google Scholar]
- 34.Sukhishvili S, Granick S. Macromolecules. 2002;35:301–310. [Google Scholar]
- 35.Srivastava R, Brown JQ, Zhu H, McShane MJ. Macromolecular Bioscience. 2005;5:717–727. doi: 10.1002/mabi.200500061. [DOI] [PubMed] [Google Scholar]
- 36.Burke SE, Barrett CJ. Macromolecules. 2004;37:5375–5384. [Google Scholar]
- 37.Kharlampieva E, Sukhishvili SA. Langmuir. 2004;20:9677–9685. doi: 10.1021/la048763d. [DOI] [PubMed] [Google Scholar]
- 38.Mendelsohn JD, Yang SY, Hiller J, Hochbaum AI, Rubner MF. Biomacromolecules. 2003;4:96–106. doi: 10.1021/bm0256101. [DOI] [PubMed] [Google Scholar]
- 39.Yang SY, Mendelsohn JD, Rubner MF. Biomacromolecules. 2003;4:987–994. doi: 10.1021/bm034035d. [DOI] [PubMed] [Google Scholar]
- 40.Richert L, Engler AJ, Discher DE, Picart C. Biomacromolecules. 2004;5:1908–1916. doi: 10.1021/bm0498023. [DOI] [PubMed] [Google Scholar]
- 41.Koktysh DS, Liang X, Yun BG, Pastoriza-Santos I, Matts RL, Giersig M, Serra-Rodríguez C, Liz-Marzán LM, Kotov NA. Adv Funct Mater. 2002;12:255–265. [Google Scholar]
- 42.Richert L, Boulmedais F, Lavalle P, Mutterer J, Ferreux E, Decher G, Schaaf P, Voegel JC, Picart C. Biomacromolecules. 2004;5:284–294. doi: 10.1021/bm0342281. [DOI] [PubMed] [Google Scholar]
- 43.Berg MC, Yang SY, Hammond PT, Rubner MF. Langmuir. 2004;20:1362–1368. doi: 10.1021/la0355489. [DOI] [PubMed] [Google Scholar]
- 44.Picart C, Elkaim R, Richert L, Audoin F, Da Silva Cardoso M, Schaaf P, Voegel JC, Frisch B. Adv Funct Mater. 2005;15:83–94. [Google Scholar]
- 45.Schneider A, Bolcato-Bellemin AL, Francius G, Jedrzejwska J, Schaaf P, Voegel JC, Frisch B, Picart C. Biomacromolecules. 2006 doi: 10.1021/bm0605208. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson MT, Berg MC, Tobias IS, Lichter JA, Rubner MF, Van Vliet KJ. Biomacromolecules. 2006;7:1990–1995. doi: 10.1021/bm060146b. [DOI] [PubMed] [Google Scholar]
- 47.Schneider A, Francius G, Obeid R, Schwinté P, Frisch B, Schaaf P, Voegel JC, Senger B, Picart C. Langmuir. 2006;22:1193–1200. doi: 10.1021/la0521802. [DOI] [PubMed] [Google Scholar]
- 48.Picart C, Schneider A, Etienne O, Mutterer J, Egles C, Jessel N, Voegel JC. Adv Funct Mater. 2005;15:1771–1780. [Google Scholar]
- 49.Weindl G, Schaller M, Schafer-Korting M, Korting HC. Skin Pharmacology and Physiology. 2004;17:207–213. doi: 10.1159/000080213. [DOI] [PubMed] [Google Scholar]
- 50.Mulhall KJ, Curtin WA, Given HF. Orthopedics. 2003;26:1219–1223. doi: 10.3928/0147-7447-20031201-12. [DOI] [PubMed] [Google Scholar]
- 51.Marsolais D, Cote CH, Frenette J. Lab Invest. 2003;83:991–999. doi: 10.1097/01.lab.0000078688.07696.ac. [DOI] [PubMed] [Google Scholar]
- 52.Schiff P, Fant J, Horwith S. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 53.Luo Y, Ziebell MR, Prestwich GD. Biomacromolecules. 2000;1:208–218. doi: 10.1021/bm000283n. [DOI] [PubMed] [Google Scholar]
- 54.Ruan G, Feng SS. Biomaterials. 2003;24:5037–5044. doi: 10.1016/s0142-9612(03)00419-8. [DOI] [PubMed] [Google Scholar]
- 55.Sipahigil O, Gursoy A, Cakalagaoglu F, Okar I. Int J Pharm. 2006;311:130–138. doi: 10.1016/j.ijpharm.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 56.Milas L, Mason KA, Hunter N, Li C, Wallace S. Int J Radiat Oncol Biol Phys. 2003;55:707–712. doi: 10.1016/s0360-3016(02)04153-6. [DOI] [PubMed] [Google Scholar]
- 57.Hemmerle J, Altmann SM, Maaloum M, Horber JK, Heinrich L, Voegel JC, Schaaf P. Proc Natl Acad Sci. 1999;96:6705–6710. doi: 10.1073/pnas.96.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lévy R, Maaloum M. Nanotechnology. 2002;13:33–37. [Google Scholar]
- 59.Francius G, Hemmerle J, Ohayon J, Schaaf P, Voegel JC, Picart C, Senger B. Micros Res Techniq. 2006;69:84–92. doi: 10.1002/jemt.20275. [DOI] [PubMed] [Google Scholar]
- 60.Simon-Assmann P, Leberquier C, Molto N, Uezato T, Bouziges F, Kedinger M. J Cell Sci. 1994;107(Pt 3):577–587. doi: 10.1242/jcs.107.3.577. [DOI] [PubMed] [Google Scholar]
- 61.Ueda Y, Walsh E, Nakanishi H, Yoshida K. Neurosci Lett. 1994;165:203–207. doi: 10.1016/0304-3940(94)90745-5. [DOI] [PubMed] [Google Scholar]
- 62.Richert L, Schneider A, Vautier D, Jessel N, Payan E, Schaaf P, Voegel JC, Picart C. Cell Biochem Biophys. 2006;44:273–276. doi: 10.1385/CBB:44:2:273. [DOI] [PubMed] [Google Scholar]
- 63.Scott JE, Heatley F. Biomacromolecules. 2002;3:547–553. doi: 10.1021/bm010170j. [DOI] [PubMed] [Google Scholar]
- 64.Spagnoli C, Korniakov A, Ulman A, Balazs EA, Lyubchenko YL, Cowman MK. Carbohyd Res. 2005;340:929–941. doi: 10.1016/j.carres.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 65.Rinaudo M, Auzely R, Vallin C, Mullagaliev I. Biomacromolecules. 2005;6:2396–2407. doi: 10.1021/bm0580025. [DOI] [PubMed] [Google Scholar]
- 66.Fernandez-Urrusuno R, Calvo P, Remunan-Lopez C, Vila-Jato JL, Alonso MJ. Pharm Res. 1999;16:1576–1581. doi: 10.1023/a:1018908705446. [DOI] [PubMed] [Google Scholar]
- 67.Boonsongrit Y, Mitrevej A, Mueller BW. Eur J Pharm Biopharm. 2006;62:267–274. doi: 10.1016/j.ejpb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 68.Fitzpatrick D, Corish J. Int J Pharm. 2005;301:226–236. doi: 10.1016/j.ijpharm.2005.05.040. [DOI] [PubMed] [Google Scholar]
- 69.Vodouhê C, Guen EL, Garza JM, Francius G, Dejugnat C, Ogier J, Schaaf P, Voegel JC, Lavalle P. Biomaterials. 2006;27:4149–4156. doi: 10.1016/j.biomaterials.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 70.Kamath KR, Barry JJ, Miller KM. Adv Drug Deliv Rev. 2006;58:412–436. doi: 10.1016/j.addr.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 71.Ahmed F, Pakunlu FI, Brannan A, Bates F, Minko T, Discher DE. Journal of Controlled Release. 2006 doi: 10.1016/j.jconrel.2006.07.012. in press, July 30th 2006. [DOI] [PubMed] [Google Scholar]