

Abstract
Ime2 is a meiosis-specific protein kinase in Saccharomyces cerevisiae that is functionally related to cyclin-dependent kinase. Although Ime2 regulates multiple steps in meiosis, only a few of its substrates have been identified. Here we show that Ime2 phosphorylates Sum1, a repressor of meiotic gene transcription, on Thr-306. Ime2 protein kinase assays on Sum1 mutants and synthetic peptides define a consensus motif Arg-Pro-X-Ser/Thr that is required for efficient phosphorylation by Ime2. The carboxyl residue adjacent to the phosphoacceptor (+1 position) also influences the efficiency of Ime2 phosphorylation with alanine being a preferred residue. This information has predictive value in identifying new potential Ime2 targets as shown by the ability of Ime2 to phosphorylate Sgs1 and Gip1 in vitro, and could be important in differentiating mitotic and meiotic regulatory pathways.
Keywords: yeast, meiosis, protein kinase, Ime2, Sum1, consensus phosphorylation site
Meiosis is a specialized form of cell division that produces the haploid cells required for sexual reproduction. It is characterized by DNA replication, high levels of genetic recombination, and two sequential rounds of chromosome segregation. These processes require the choreographed expression of meiosis-specific genes in a transcriptional cascade (11, 31). In the budding yeast Saccharomyces cerevisiae, meiosis is induced by nutrient depletion and is coupled to spore formation. Starvation for key nutrients induces expression of the early gene transcriptional activator Ime1 (12). One of the genes activated by Ime1 encodes Ime2, a serine/threonine kinase that is functionally related to the Cdc28 cyclin-dependent kinase (CDK) and which shows similarity to the CMGC-group of protein kinases (10). Ime2 is required for critical events in meiosis, including the replication of DNA, the regulation of meiotic gene expression, and the meiotic divisions.
The role that Ime2 plays in regulating meiotic S phase is related to that of Cdc28 in triggering Sphase in mitosis. In both mitosis and meiosis the G1-S transition is prohibited by the Clb5,6/Cdc28 inhibitor Sic1 (24). Entry into S phase is delayed until Sic1 is phosphorylated, a reaction catalyzed in mitosis by Cdc28 in complex with the G1-phase cyclins Cln1 and Cln2 (15, 30). In meiosis, Ime2 is required for Sic1 degradation and DNA replication (7). Others have recently demonstrated that Ime2 directly phosphorylates multiple sites in Sic1 in vitro. This is insufficient to trigger meiotic Sic1 destruction, however, suggesting that Ime2 does not simply replace the function of Cln1,2-/Cdc28 in meiosis to trigger S-phase (25). Another early role for Ime2 regulation of meiosis is through its interaction with RPA1, a heterotrimeric complex required for stabilization of single stranded DNA during replication and recombination. Ime2 is required for full RPA phosphorylation in meiosis and can phosphorylate RPA when Ime2 is ectopically expressed in vegetatively growing cells (5, 6). Additionally, Ime2 directly phosphorylates the RPA subunit Rfa2 in vitro. Interestingly, Ime2 phosphoacceptor site mutants of both Sic1 and Rfa2 fail to produce obvious meiotic defects. Ime2 also autophosphorylates its activation loop to positively regulate catalysis (23). Despite the identification of phosphoacceptor residues for Ime2 in Sic1, Rfa2, and in Ime2 itself, a phosphoacceptor consensus has not been defined.
Ime2 regulates the transcriptional program of meiosis at multiple steps. In the earliest stages of meiosis Ime2 positively regulates Ime1-dependent transcription of early meiosis-specific genes (14). Ime2 also appears to promote early gene transcription in an Ime1-independent fashion. Later in the program Ime2 is also required for Ime1 destabilization and the down-regulation of early meiotic genes (8). Ime2 can phosphorylate Ime1 in vitro, although the phosphoacceptor site(s) in Ime1 have not been identified, and therefore the mechanism by which Ime2 down-regulates Ime1 is yet to be determined.
Transcription of the middle sporulation genes (MSGs) is also regulated by Ime2. MSGs contain a DNA sequence, the middle sporulation element (MSE), in their promoter regions (16). The MSE is targeted by the transcriptional activator Ndt80, which itself is expressed specifically in meiosis just prior to induction of most MSGs (4, 9). Ndt80 is phosphorylated by Ime2 in vitro, and yeast lacking Ime2 accumulate a hypophosphorylated form of Ndt80 (1, 27, 28). While these data suggest Ime2 positively regulates Ndt80 by direct phosphorylation, the phosphoacceptor site(s) in Ndt80 have not yet been identified.
During vegetative growth, MSG expression is prevented due to the lack of active Ndt80. Some MSGs, however, including NDT80 itself, have a second layer of control to ensure they are not expressed inappropriately. Genes in this subset of MSGs have MSEs that are recognized by Ndt80 during meiosis, but are also bound by the transcriptional repressor Sum1 during mitosis and early meiosis (18, 19, 33). Sum1 is transiently destabilized during meiosis, and Sum1 repression is removed during late prophase (13). Some middle genes are not expressed in ime2 mutants but are expressed in ime2 sum1 mutants (17). Although this might suggest that Sum1 is down-regulated during meiosis by Ime2 phosphorylation, the mechanism by which mutation of SUM1 alleviates the IME2 requirement for MSG expression remains uncharacterized.
Here we describe our investigation into the regulatory mechanisms of Ime2, as assayed through its interaction with the transcriptional repressor Sum1. We have identified a threonine in the NH2-terminus of Sum1 that is phosphorylated by Ime2 in vitro. The phosphoacceptor site falls within a short amino acid motif (RPXT/SA) that is required for Ime2 phosphorylation of Sum1. We also demonstrate the value of this motif in predicting other Ime2 targets, notably Gip1 and Sgs1.
MATERIALS AND METHODS
Yeast Strains and Plasmids
The yeast strains and plasmids used in this study are described in Tables 1 and 2 respectively. The SUM1-T306A yeast strains contain an ACT to GCT change in codon 306. Mutations were introduced into the chromosomal copy of SUM1 using a mutant derivative of pKMS8, an integrating URA3-SUM1 vector that was generated by subcloning the SacI/XhoI fragment from pMP208 (18) into pRS306. The pKMS8 derivatives were linearized with NruI, Ura+ transformants selected, and 5-fluoroorotic acid-resistant colonies counter-selected. Yeast mutants containing the desired substitutions in SUM1 were identified by PCR and direct sequence analysis of 5-FOA-resistant isolates.
TABLE 1.
Yeast strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| KSY187 | MATa/MATα ura3/ura3 leu2::hisG/leu2::hisG trp1ΔFA::hisG/trp1ΔFA::hisG lys2/lys2 ho::hisG/ho::hisG IME2-myc::TRP1/IME2-myc::TRP1 |
(ref. 22) |
| KSY162 | KSY187 + ime2-K97R-myc/ime2-K97R-myc | (ref. 22) |
| MSY4 | MATa/MATα ura3/ura3 leu2::hisG/leu2::hisG trp1ΔFA::hisG/trp1ΔFA::hisG lys2/lys2 arg4- NspI/ARG4 ho::hisG/ho::hisG RME1/rme1::LEU2 gip1::HIS3/ gip1::HIS3 |
A. Neiman (ref. 29) |
| MSY3 | MATa/MATα ura3-1/ura3-1 his3-11,15/his3-11,15 leu2-3,115/leu2-3,115 trp1-1/trp1-1 met15-Δ/met15-Δ sgs1::HIS3/sgs1::HIS3 can1-100/can1-100 ade2- ΔNdeI/ade2-ΔAatII |
R. Rothstein (ref. 26) |
| LNY150 | MATa/MATa ura3/ura3 leu2::hisG/leu2::hisG trp1::hisG/trp1::hisG lys2/lys2 his4-N/his4-G ho::LYS2/ho::LYS2 |
This study |
| MMY35 | LNY150 + SUM1-T306A/SUM1-T306A | This study |
| EWY88 | MATa/MATa ura3/ura3 leu2::hisG/leu2::hisG lys2/lys2 ho::LYS2/ho::LYS2 dmc1::ARG4/dmc1::ARG4 |
This study (ref. 13) |
| MMY36 | EWY88 + SUM1-T306A/SUM1-T306A | This study |
TABLE 2.
Plasmids used in this study
| Plasmid | Markers | Source |
|---|---|---|
| pMP197 | PET21b + Mbp-Sum1-523-1062 | This study |
| pMP108 | pMAL-c2 + Mbp-Sum1-1-698 | This study |
| pMP112 | pMAL-c2 + Mbp-Sum1-1-353 | This study |
| pAB221 | pMAL-c2 + Mbp-Sum1-1-340 | This study |
| pAB216 | pMAL-c2 + Mbp-Sum1-1-292 | This study |
| pAB215 | pMAL-c2 + Mbp-Sum1-1-211 | This study |
| pAB214 | pMAL-c2 + Mbp-Sum1-1-90 | This study |
| pAB257 | pMAL-c2 + Mbp-Sum1-1-353-Δ 313-348 | This study |
| pMES1 | PMAL-c2X + Mbp-Gip1-1-211 | This study |
| pMES2 | PMAL-c2X + Mbp-Sgs1-1162-1447 | This study |
| pMP208 | CEN URA3 SUM1-MYC | (ref. 18) |
| pKMS8 | CEN URA3 SUM1 | This study |
| pKMS8-T306A | CEN URA3 SUM1-T306A | This study |
| pKMS8-T306D | CEN URA3 SUM1-T306D | This study |
| pSB6 | 2μ URA3 SPO20p-HA-GIP1 | A. Neiman (ref. 29) |
| pSB6-S27A, T28A | SPO20p-GIP1-S27A,T28A | This study |
| pWJ1059 | CEN URA3 SGS1 | R. Rothstein (ref. 26) |
| pWJ1059-S1202A, T1203A |
CEN URA3 SGS1-S1202A,T1203A | This study |
To assay the consequences of mutating the region of Gip1 that is phosphorylated by Ime2, plasmid pSB6, which contains the GIP1 open reading frame driven by the SPO20 promoter, was used (29). Codons 27 and 28 were changed from TCG-ACT to GCG-GCT to alter the GIP1-encoded protein sequence from RPSTA to RPAAA and the resulting pSB6-S27A,T28A plasmid, as well as the non-mutagenized pSB6 control plasmid, were transformed into diploid strain MSY4 (made by crossing strains NY1 and NY2 from the Neiman laboratory collection) (29). Transformants were selected on minimal selective medium, grown overnight in YEPA, and transferred to sporulation medium for 36 h. Cells were assayed by microscopic inspection as well as by glusulase resistance as previously described (22). Briefly, 1 OD of cells were washed with PIPES buffer (0.1 M PIPES, pH 6.8), resuspended in glusulase diluted 1:5 in the same buffer and digested for approximately 25 minutes at 30°C. After digestion, cells were washed in PIPES buffer, serial dilutions were plated on rich medium and colony formation was scored. To assay the consequences of mutating T1203 in Sgs1 that is phosphorylated by Ime2, codons 1202 and 1203 of SGS1 in the CEN-based SGS1 plasmid pWJ1059 were changed to TCG and ACA to alter the RPST sequence to RPAA. The wild-type and mutant plasmids as well as an empty (pRS416) control vector were transformed into the sgs1-Δ/sgs1-Δ yeast strain MSY3 (made by crossing strains W3326-10B and W3326-14D from the Rothstein laboratory collection) (26). Sporulation efficiency was monitored by microscopic evaluation and by glusulase-resistance as previously described (32). Recombination at ADE2 was monitored using ade2-NdeI/ade2-AatII heteroalleles by plating serial dilutions of cells on minimal medium lacking adenine and on YAPD medium at 4 days post-induction and scoring for colony formation.
For expression of Sum1 protein fragments used in the in vitro kinase assays, PCR-generated fragments of SUM1 were cloned into maltose-binding protein (Mbp) bacterial expression vectors. Plasmid pMP197 contains the DNA coding for Sum1 residues 523-1062 in the NdeI and XhoI sites of pET21b. Plasmid pMP108 contains the DNA coding for Sum1 residues 1-698 in the BamHI and PstI sites of pMAL-C2. Plasmid pMP112 expresses the Mbp-Sum1 1-353 fusion protein and was constructed by restriction digest of a derivative of the Mbp-Sum1 fusion with NsiI and SpeI, filling in the ends with the Klenow fragment of DNA polymerase and religating to create a stop codon after residue 353 of Sum1. The Mbp-SUM1-1-340 (pAB221), Mbp-SUM1-1-292 (pAB216), Mbp-SUM1-1-211 (pAB215), and Mbp-SUM1-1-90 (pAB214) Sum1 deletion derivatives were generated by introducing a stop codon after the indicated position via site-directed mutagenesis. pAB257, which contains an Mbp-SUM1-1-353 fusion with a deletion of residues 313-348, was constructed by site-directed mutagenesis of pMP112 with primers lacking the sequence coding for residues 313-348. Plasmids expressing the T306A, T295A and T295A/T306A mutant proteins shown in Figure 1C were constructed by site-directed mutagenesis of plasmid pAB257. Plasmids expressing the K301A, E302A, R303A, P304A, S305A, T306A, N308A, and T306S mutants in the context of the Mbp-Sum1 1-353 protein fusion were constructed by site-directed mutagenesis of pMP112.
FIGURE 1.

Ime2 phosphorylates Sum1 on T306. Mbp-fusions with N-terminal portions of Sum1 were incubated with Ime2-myc isolated from sporulating cells and radiolabelled ATP. (A) Mbp-fusions to the indicated amino-terminal fragments of Sum1 (lanes 3-7) or Mbp alone (lane 2) were analyzed by electrophoresis and autoradiography (upper). The Coomassie-stained electrophoretic gel of substrates is shown below for comparison. The positions of the Ime2 autophosphorylation and Mbp-Sum1 bands are indicated. (B) Phosphoamino acid analysis of acid-hydrolyzed Mbp-Sum1 1-353 and MBP-Sum1 1-353 T306S proteins phosphorylated by Ime2-myc. The relative positions of phosphothreonine and phosphoserine are indicated. (C) Mbp-Sum1 1-353 (lane 1), and Mbp-Sum1 1-353 deleted for residues 313-348 with the indicated substitutions (lanes 2-5) were analyzed by electrophoresis and autoradiography. The Coomassie-stained gel of substrates is shown below for comparison. The positions of the Ime2 autophosphorylation and MBP-Sum1 bands are indicated.
For expression of Mbp-Gip1-1-211, an EcoRI/XbaI PCR-generated fragment containing the coding region of residues 1-211 of GIP1 was amplified from DNA from yeast of the S288C genetic background and inserted into the EcoRI and XbaI sites of pMAL-c2X to create pMES1. A mutant derivative of the Mbp-Gip1 fusion containing an ACT to GCT substitution in codon 28, that changed the GIP1-encoded protein sequence from RPST to RPSA, was constructed by site directed mutagenesis of pMES1. For expression of Mbp-Sgs1-1162-1497, an EcoRI/XbaI PCR-generated fragment containing the coding residues 1162-1497 of SGS1 was amplified from DNA from yeast of the S288C genetic background and inserted into the EcoRI and XbaI sites of pMAL-c2X to create pMES2. A mutant derivative of the Mbp1-Sgs1 fusion containing an ACA to GCA substitution in codon 1203, that changed the SGS1-encoded protein sequence from RPST to RPSA, was constructed by site-directed mutagenesis of pMES2. Oligonucleotides used in the construction of the plasmids are available on request.
Preparation of Ime2 and in vitro kinase assays
Ime2-myc was prepared from strain KSY187 and a catalytically inactive Ime2-K97R-myc version was similarly prepared from strain KSY162 (Table 1) by immunoprecipitation, as previously described (23). Briefly, frozen cell pellets were resuspended in 0.5 ml lysis buffer (50 mM Hepes, pH 7.4, 75 mM KCl, 1 mM EGTA, 1 mM MgCl2, 0.1% NP-40, 50 mM β-glycerophosphate, 1 mM sodium orthovanadate, 50 mM NaF, 100 μg/ml PMSF, 50 mM NaF, 50 mM glycerolphosphate, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 8.8 μg/ml aprotinin, 4mg/ml antipain, 0.1 μg/ml pefabloc SC, 2 μg/ml pepstatin A, 1 μg/ml chymostatin, 1 mM benzamidine, and 2 μg/ml leupeptin) and the slurry was added to an equal volume of acid-washed glass beads (Sigma). Cells were lysed with a Mini-Bead Beater (Biospec Products) 3 times for 1 min at a time, with 2 min on ice between beatings. Extracts were clarified by centrifugation at 13,000 x g for 10 minutes, and supernatants were added to 30 μl of Protein A-Agarose beads (Roche; washed 2x in PBS) along with 25 μl c-myc A14 antibody (Santa Cruz Biotechnology) and rotated for 16 h at 4° C. Immunoprecipitates were then washed two times in kinase buffer (20 mM Hepes, pH 7.4, 100 mM KCl, 10 mM MgCl2), and resuspended in kinase buffer + 50% glycerol for storage at −80° C.
For protein kinase assays of protein substrates, Ime2-myc immunoprecipitates were thawed, washed two times in kinase buffer, resuspended in kinase buffer, and added to 10 μg of either MBP or MBP-fusion protein in kinase buffer, along with 10 μM ATP and 0.2 μCi/μl [γ-32P]ATP in 25 μl final volume. Reaction mixtures were incubated at 30° C for 30 min; the reactions were terminated by boiling following the addition of an equal volume of 2X loading buffer (0.15 M Tris, pH 6.8, 24% glycerol, 12% β-mercaptoethanol, 4.85% SDS, 0.02% bromophenol blue). Reactions were resolved by 7.5% SDS-PAGE and transferred to PVDF. Incorporation of radioactive phosphate was detected by autoradiography.
Kinase reactions using peptide substrates were performed identically as for protein substrates except that the buffer concentration was increased to 100 mM HEPES, pH7.4, to eliminate pH effects that occur upon addition of high concentrations of peptide (>100μM). Peptide substrate reactions were terminated after the indicated incubations at 30°C by adding 10 μl of the reaction to 10 μl of ice cold 100 mM EDTA, pH 7.5. The mixture was spotted onto a 2 cm × 2 cm P81 phosphocellulose paper (Fisher Scientific). Spotted papers were washed 5 times for 5 min at a time in 75 mM phosphoric acid, followed by a 1 min wash in acetone. 32P incorporation was measured by scintillation spectrometry.
Phosphoamino Acid Analysis
Subsequent to autoradiography of kinase assays, bands of interest were excised from the PVDF membrane for phosphoamino acid analysis. The excised sections of membrane were wetted in methanol, rinsed with water, and submerged in 100 ¼l 6 N HCl at 110° C for 1 h. The supernatants were then transferred to a new tube, lyophilized, and resuspended in water. Samples were spotted onto a plastic-backed cellulose TLC plate (Kodak) for separation in a TLC chamber containing isobutyric acid::500 mM ammonium hydroxide (5:3), then air dried. Phosphoamino acid standards were visualized by spraying the plate with ninhydrin spray (Acros Organics) and baking it at 65° C for 15 min. Phosphoamino acids hydrolyzed from in vitro kinase substrates were visualized by autoradiography, and their migration compared to that of the phosphoamino acid standards in order to identify the amino acid species phosphorylated.
RESULTS
The Sum1 NH2 Terminus is Phosphorylated by Ime2 In Vitro
Because genetic evidence suggests that Ime2 negatively regulates Sum1, we asked whether Ime2 can phosphorylate Sum1 in vitro. For this purpose, myc-tagged Ime2immunoprecipitated from sporulating cells at 3 hours post-induction was incubated with E. coli expressed fragments of the Sum1 protein and [γ-32P]ATP. We chose to use Ime2 from this time point as it correlates with full expression of Ime2, and is subsequent to post-translational modifications required for its kinase activity (22). The protein kinase assays were performed with amino-terminal and carboxyl-terminal fragments of Sum1 (residues 1-698 and 523-1062 respectively) fused to Mbp. The amino-terminal Mbp-Sum1-1-698 fusion was phosphorylated to a significant level by Ime2; however, we were unable to detect phosphorylation of the carboxyl-terminal fragment (data not shown). Analysis of deletion derivatives of the amino-terminal Mbp-Sum1 fusion demonstrated that a fusion containing the amino-terminal 353 residues of Sum1 was able to act as a phosphoacceptor in the Ime2 assay (Figure 1A, lane 3 and data not shown).
An Mbp-Sum1 fusion expressing the first 340 residues of Sum1 was phosphorylated to a similar extent as the first 353 residues, while a fusion protein expressing only the first 292 amino acids was less extensively modified (Figure 1A, lanes 4 and 5). These data indicate that a major site of phosphorylation lies between residues 292 and 340. These results also suggest that 1 or more additional minor sites exist in the first 292 residues of Sum1 (see Discussion). Phosphoamino acid analysis of Mbp-Sum1-1-353 phosphorylated by Ime2 demonstrated that only threonine was detectably phosphorylated (Figure 1B, left lane). There are five threonines in the Sum1 292-340 interval. We identified the phosphorylated threonine by changing select threonines to non-phosphorylatable alanines, along with a strategic deletion in the Mbp-Sum1-1-353 background. Expressing a version of Mbp-Sum1-1-353 lacking residues 313-348 eliminated all but two of the potential target threonines. This deletion mutant was phosphorylated even more efficiently than Mbp-Sum1-1-353 (Figure 1C, lanes 1 and 2), indicating that the phosphorylation site was not contained in the 313-348 deleted region. Mutating one of the remaining threonines (T295) to alanine had no effect on the in vitro reaction, while substituting the threonine at position 306 with alanine reduced radiolabel incorporation to background levels (Figure 1C, lanes 3 and 4). These data suggest that T306 is the major Ime2 phosphoacceptor site in Sum1.
In order to confirm that T306 is the major site of Ime2 phosphorylation on Sum1, we performed another phosphoamino acid analysis experiment, this time on an Mbp-Sum1-1-353 fusion protein in which T306 had been replaced with serine. With this mutant we were able to demonstrate a switch in the incorporation of radiolabel into serine rather than threonine (Fig. 1B, right lane). These results unambiguously identify T306 as the residue in Sum1 that is phosphorylated by Ime2.
Ime2 Recognizes Sum1 as a Suitable Substrate Due to the Presence of a Short Phosphoacceptor Motif
We next examined whether there are any amino acids in the immediate vicinity of T306 that are required for Ime2 recognition of Sum1 as a kinase substrate. By changing nearby amino acids individually to alanine we found two residues, in addition to the phosphoacceptor site itself, that were required for incorporation of radiolabel into Sum1 in the in vitro kinase reaction. One of these substitutions, R303A, reduced the efficiency of the phosphorylation reaction to nearly background levels (Figure 2, lane 5). Alanine substitution also demonstrated that the proline at position 304 is required for the phosphorylation of Sum1 by Ime2 (Figure 2, lane 6). The data indicate that the arginine at -3 and the proline at -2, relative to the phosphoacceptor site, are required for efficient phosphorylation of Sum1. A third residue, a serine at the -1 position, contributed slightly to the overall efficiency of the reaction, but is much less critical for Ime2 recognition than R303 or P304 (Figure 2, lane 7). We carried our alanine substitution out to the -5 position and found that neither the lysine at -5 or the glutamate at -4 were required for phosphorylation. Likewise, an asparagine at the +2 position does not appear to contribute to Ime2 recognition. Ime2 was able to modify the serine in a T306S mutant just as well as the wild type substrate (Figure 2, lane 10). These data indicate that Ime2 recognizes Sum1 as a kinase substrate due to the motif RPXT/S.
FIGURE 2.

Ime2 phosphorylation of Sum1 requires an RPXT consensus. Mbp (lane 1) or Mbp-Sum1-1-353 (lanes 2-10) substrates containing the indicated substitutions were incubated with Ime2-myc and radiolabelled ATP. The Coomassie-stained gel of substrates is shown below for comparison.
The KERPSTAN peptide is sufficient to direct Ime2 phosphorylation
To further characterize the requirements of Ime2 phosphorylation, synthetic peptides containing the Sum1 phosphoacceptor site were tested as substrates in an Ime2 in vitro phosphorylation assay. The peptides used in these experiments correspond to the Sum1 301-308 sequence, KERPSTAN, plus an additional amino-terminal arginine for assay purposes (see Materials and Methods). Ime2 phosphorylated this peptide in a robust fashion, while a peptide containing an alanine in place of the threonine was not detectably phosphorylated (Figure 3). Control experiments showed that incorporation of 32P into the KERPSTAN peptide was undetectable when the catalytically inactive Ime2-K97R mutant protein was used in place of the wild-type kinase (data not shown). Incorporation of phosphate was linear for at least 90 minutes (the longest time tested, Figure 3A), and followed Michaelis-Menten kinetics where the Km for the Sum1 peptide was found to be approximately 20μM. (Figure 3B).To further characterize residues required for Ime2 phosphorylation, a series of peptides containing single amino acid substitutions were tested. Consistent with the data on Ime2 phosphorylation of Mbp-Sum1, changing either the -3 arginine or the -2 proline in the synthetic peptides to alanine reduced phosphorylation to background levels. Changing the -3 arginine to a lysine also eliminated phosphorylation of the peptide, indicating that the requirement for arginine involves more than just its basic charge. Also consistent with Mbp-Sum1, the −1 serine was found not to be required for efficient phosphorylation of the synthetic peptide. In fact, peptides containing an alanine at this position appeared to have an increased Vmax (Figure 4). The requirement for an alanine at the +1 position was also assayed in these experiments by replacing alanine with either glycine or valine, which strongly reduced phosphorylation. These experiments demonstrate that the +1 position can also play a role in substrate/enzyme interaction. Finally, a peptide in which the phosphoacceptor site was changed to serine was also examined. The Km's for the serine and threonine phosphoacceptor peptides were identical, suggesting that, at least in vitro, Ime2 does not discriminate between these residues. These results are consistent with the analysis of the mutant forms of Mbp-Sum1 fusion proteins described above and indicate that the +1 position can also play a role in substrate selection by Ime2 with alanine being a preferred residue.
FIGURE 3.

Ime2 phosphorylates a peptide containing the Sum1-phosphorylation consensus. A peptide RKERPSTAN (■), which contains the sequence 301-308 of Sum1, or the same peptide lacking the phosphoacceptor, RKERPSAAN (□), were incubated with Ime2 prepared from sporulating cells. (A) Time course of phosphorylation using 10 μM peptide (n=3). (B) Michaelis-Menten plot of peptide phosphorylation (n=3).
FIGURE 4.

Efficient phosphorylation by Ime2 requires an RPXS/TA motif. Peptides analyzed contain the sequence RKERPSTAN (■), or the same peptide containing a substitution of the RPSTA sequence for RPATA (*), RPSSA (■), RPSTG (+), RPSTV (−), RASTA (×), KPSTA (▲) and APSTA (Δ). The data shown represents the averages of 3 independent experiments.
The consensus can be used to identify Ime2 targets
There are 59 annotated open reading frames in budding yeast that contain the sequence RPXS/TA. To determine whether the consensus can be phosphorylated in the context of other proteins, we tested Gip1, which contains an exact match to this sequence. Gip1 is a targeting subunit of the protein phosphatase type 1 in yeast that is required for spore formation and is expressed exclusively during meiosis (29). Mbp-Gip1-1-211 purified from E. coli was phosphorylated in a robust fashion, while a mutant form of this fusion protein in which the threonine in the RPSTA consensus was substituted with alanine (T28A) was not detectably phosphorylated (Figure 5). This result demonstrates that RPSTA can be phosphorylated in the context of proteins other than Sum1 and suggests that the consensus can be used to discover new substrates, and to map the phosphoacceptor sites in these proteins.
FIGURE 5.
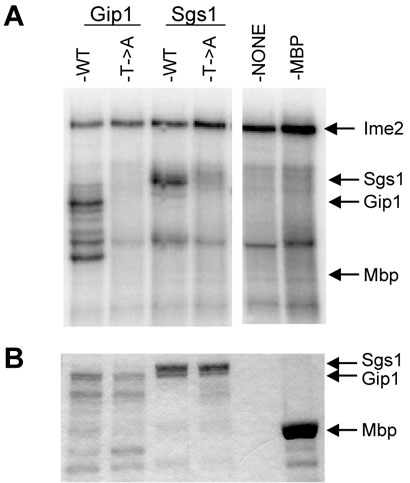
Ime2 can phosphorylate proteins other than Sum1 that contain the RPST motif. Mbp fused to residues 1-211 of Gip1, or residues 1162-1447 of Sgs1, or the same proteins containing alanine (A) in place of the threonine (T) in the RPST motif as indicated, were incubated with Ime2 in the presence of radiolabeled ATP. (A) Autoradiograph of the reactions analyzed by electrophoresis. The migration of Mbp, Mbp-Gip1-1-211, Mbp-Sgs1-1162-1447 and of the Ime2 autophosphorylation product are indicated. (B) Coomassiestained gel of the protein substrates analyzed in parallel. Analysis of the unstable Mbp-Gip1-1-211 fusion protein demonstrates that all of the observable Coomassie-stainable protein bands contain Mbp as assayed by Western immunoblot analysis (data not shown).
We also examined whether Sgs1, a RecQ helicase that regulates meiosis-specific interchromosomal interactions (21), can be phosphorylated by Ime2. Sgs1 contains the sequence RPST but contains a serine at the +1 position instead of the alanine found in Sum1. An Mbp-Sgs1-1162-1447 fusion was phosphorylated to a extent comparable to that of Mbp-Gip1, while the Mbp-Sgs1-T1203A derivative, in which the threonine in the RPST-motif was replaced with an alanine, was not detectably phosphorylated (Figure 5). These data demonstrate the predictive value of the Ime2 consensus motif derived from our analysis of Sum1. These results also indicate that the +1 alanine is not required for efficient phosphorylation in all substrates (see Discussion).
T306 in Sum1 is not required for meiosis
We constructed diploid strains of yeast in which T306 of Sum1 had been changed to alanine. The kinetics and extent of meiosis and spore formation of these sum1-T306A/sum1-T306A diploids was indistinguishable from wild-type strains. The pachytene-checkpoint block to meiosis seen in a dmc1/dmc1 strain requires SUM1 (13) (dmc1/dmc1 strains complete less than 0.5% meiosis and dmc1/dmc1 sum1/sum1 strains complete meiosis to 35% under the conditions tested). The SUM1-T306A mutant is functional, based on the absence of meiosis (less than 0.5%) observed in the dmc1/dmc1 sum1-T306A/ sum1-T306A background. Similarly, we find that a plasmid expressing a mutant form of Gip1 in which both the threonine that appears to be phosphorylated in vitro by Ime2, as well as the adjacent serine, have been replaced with alanines (pSB6-S27A,T28A) complements the sporulation defect of a gip1-Δ/gip1-Δ diploid indistinguishably from the corresponding wild-type plasmid (pSB6). A mutant version of Sgs1, with alanine substituted for both the threonine that is phosphorylated by Ime2 in vitro as well as the adjacent serine (pWJ1059-S1202A,T1203A), complements the sporulation defect of a sgs1-Δ/sgs1-Δ diploid as well as the corresponding wild-type plasmid (pJW1059). Thus, all of the Ime2 phosphoacceptor-site mutants that have been described here and in other recent studies (Sic1, Rfa2, Sum1, Gip1, and Sgs1) complete meiosis and spore formation in a manner that is apparently indistinguishable from wild-type (6, 25).
DISCUSSION
Genetic studies have shown that Ime2 regulates multiple steps in meiotic development, including pre-meiotic S-phase and entry into the meiotic divisions, as well as the transcriptional program of meiosis at the early and middle phases (10). In addition, it has been suggested that Ime2 can regulate exit from MI (23). Despite these regulatory connections, we understand relatively little about how Ime2 controls these key steps in meiotic development at the molecular level. As is the case with most other protein kinases, major impediments to our understanding of the mechanism of regulation by Ime2 include an incomplete list of substrates and an inability to predict phosphoacceptor sites within targets once they have been identified. In this study we have identified T306 within Sum1 as a major phosphoacceptor site in vitro. Analyses of the amino acids surrounding T306, using mutant forms of Sum1 as well as a panel of synthetic peptides, have identified the peptide sequence RPXS/T as being necessary for efficient phosphorylation by Ime2. We also demonstrated that the +1 position can influence the reaction with alanine being a preferred residue at this position. The RPXS/T consensus has predictive value in identifying potential Ime2 substrates and mapping phosphoacceptor sites as demonstrated by our identification of T28 in Gip1 and T1203 in Sgs1 as residues that can be phosphorylated by Ime2 in vitro. As such, the consensus motif should lead to an increased understanding of how meiosis is regulated by Ime2.
During the course of this study, phosphoacceptor sites in three in vivo targets of Ime2 were reported. The first to be reported was S27 in the Rfa2 subunit of RPA (6). This residue lies within the sequence RPGSG and conforms to the consensus. Notable is the glycine at the +1 position. Changing the +1 residue in the Sum1 peptide from alanine to glycine dramatically increased the apparent Km (Figure 4), yet introducing an additional serine to glycine substitution at the −1 position (generating the RPGSG Rfa2 sequence, flanked by Sum1 residues) decreased the Km back to approximately 20μM and increased the apparent Vmax to a level that was modestly higher than that seen with the Sum1 control-peptide (data not shown). It is also worth noting that while the Gip1 phosphoacceptor site identified in this study contains an alanine at the +1 site, the Sgs1 phosphoacceptor site, which is phosphorylated to a similar extent in vitro, contains a serine at this position. These results show that there can be significant variation in the +1 position effect and that at least some of this variation can be related to the identity of the −1position (X in the consensus). The second in vitro Ime2 substrate to be characterized is Sic1, which was assayed by mass spectrometry (25). One of the sites identified in this study (S46) is in an RPTSA sequence that conforms to the consensus and contains the preferred alanine at the +1 position. The remaining high-confidence sites identified by mass spectrometry lack the −3 arginine. However, it should be pointed out that the relative efficiency of phosphorylation of these Sic1 sites was not reported and that at least one Sic1 site appears to be preferentially phosphorylated based on the non-normal distribution of isoforms as measured by isoelectric focusing. It is worth noting that Sic1 is controlled by multi-site Cln1,-2/Cdc28 phosphorylation in mitosis and that the Sic1 sites that fail to conform to the low Km Ime2 consensus as defined in this study are imbedded in CDK consensus phosphorylation sites (S/T P-sites). Thus, as pointed out in the Sedgwick study, Ime2 may in some circumstances interact with CDK sites (25). It is unknown whether phosphorylation of CDK sites can affect the Km/Vmax of overlapping Ime2 sites or vice versa to provide a mechanism for cross-communication between Cdc28 and Ime2. This could help explain the lack of overt meiotic phenotypes seen in the Ime2 phosphosite mutants described here and elsewhere (6, 25). The third substrate to be characterized is Ime2 itself (23). Ime2 autophosphorylates T242 in its activation loop to up-regulate catalysis. T242 is contained within the low Km consensus defined in this study, except for an asparagine instead of arginine at the −3 position. This result demonstrates that suboptimal sites can be efficiently phosphorylated under certain conditions.
While Ime2 specifically phosphorylates T306 of Sum1 in vitro, we have thus far been unable to prove that Ime2 phosphorylates T306 of Sum1 in vivo. A related concern is the apparent lack of a sum1-T306A meiotic phenotype. Deletion derivatives of the amino-terminal Mbp-Sum1-1-353 that lack the region containing T306 are phosphorylated at a reduced but significant level that is reproducibly higher than Mbp alone (Figure 1A compare lanes 5 and 6 with lanes 4 and 2). In addition, while the T306A substitution reduces the extent of phosphorylation of Mbp-Sum1-1-353, this derivative is also phosphosphorylated more extensively than Mbp alone (Figure 2 compare lanes 8 and 1). Indeed, there are 12 motifs in the Sum1 protein that conform to the RPXS/T consensus except for the R at the −3 position; 5 of these are clustered in the 1-353 region. It is possible that the weak in vitro sites in Sum1 are utilized in vivo and that these additional sites are functionally redundant with the T306 site for control of Sum1 by Ime2.
We propose that the low Km Ime2 consensus identified in this study can provide a starting point for building an understanding of Ime2 substrate selection. Ime2 regulates multiple steps in meiosis, yet little is known about how this occurs. It is likely that residues phosphorylated by Ime2 in yeast cells are imbedded in motifs that have a range of inherent Km values compared to the optimal RPXS/TA motif defined in this study. For example, the Cdh1 targeting subunit of the anaphase-promoting complex/cyclosome (APC/C), which has been implicated as an Ime2 substrate (2), contains 4 motifs (RPISS, RPSTV, RPSTR, and RPSSN) that conform to the low Km consensus. Ndt80, the key checkpoint-regulated transcription factor that promotes exit from prophase and entry into MI by activating the transcription of CLB1 and other MSGs, is upregulated by Ime2 during meiosis, and Ime2 phosphorylates Ndt80 in vitro. Ndt80 contains variant sites that diverge from the low Km consensus site (there are 9 sites in Ndt80 that differ from the consensus by the −3 arginine). These considerations raise the possibility that certain sub-optimal phosphoacceptor sites are phosphorylated only at distinct steps in meiotic development due to regulated changes in Ime2. Ime2 activity fluctuates significantly during meiosis, peaking first around S-phase and then increasing to a higher level around prophase exit/MI (1, 23). This is the stage in the meiotic pathway when Ndt80 is activated, suggesting that changes in total Ime2 activity could play a role in promoting the phosphorylation of sub-optimal sites. Other mechanisms that could promote phosphorylation of suboptimal sites include regulated changes in cellular localization and association with adaptor/targeting proteins. Thus, Ime2 target-site selection might best be understood as a range of low to high Km sites, with potentially different regulatory properties and requirements.
We suggest that potential Ime2 targets and phosphoacceptor sites within known or predicted targets can be identified based on the data described here. In particular, we propose that a hierarchical ordering of proteins starting with those containing the full consensus followed by those lacking the−3 and -2 site residues will enable the prediction of novel Ime2 target proteins, when viewed in context with genomic/proteomic data sets such as the transcriptional program of meiosis (3, 20). Similar considerations should prove useful in identifying specific phosphoacceptor sites within known or suspected Ime2 targets.
ACKNOWLEDGEMENTS
We thank Michael Pierce, Aaron Neiman, Robert Reid, and Rodney Rothstein for yeast strains and plasmids. We also thank Kristin Servent for technical assistance with this project. This research was supported by grants from the National Institute of Health (GM 58762 to AKV and GM061817 to EW).
Footnotes
This work was supported by grants from the National Institutes of Health to Andrew Vershon (GM 58762) and Edward Winter GM (GM061817).
Abbreviations: RPA, replication protein A; MSG, middle sporulation gene; MSE, middle sporulation element
REFERENCES
- 1.Benjamin KR, Z. C, Shokat KM, Herskowitz I. Control of landmark events in meiosis by the CDK Cdc28 and the meiosis-specific kinase Ime2. Genes and Development. 2003;17:1524–1539. doi: 10.1101/gad.1101503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolte M, Steigemann P, Braus GH, Irniger S. Inhibition of APC-mediated proteolysis by the meiosis-specific protein kinase Ime2. Proc Natl Acad Sci U S A. 2002;99:4385–90. doi: 10.1073/pnas.072385099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. The transcriptional program of sporulation in budding yeast [published erratum appears in Science 1998 Nov 20;282(5393):1421] Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 4.Chu S, Herskowitz I. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Molecular Cell. 1998;1:685–696. doi: 10.1016/s1097-2765(00)80068-4. [DOI] [PubMed] [Google Scholar]
- 5.Clifford DM, Marinco SM, Brush GS. The meiosis-specific protein kinase Ime2 directs phosphorylation of replication protein A. J Biol Chem. 2004;279:6163–70. doi: 10.1074/jbc.M306943200. [DOI] [PubMed] [Google Scholar]
- 6.Clifford DM, Stark KE, Gardner KE, Hoffmann-Benning S, Brush GS. Mechanistic insight into the Cdc28-related protein kinase Ime2 through analysis of replication protein A phosphorylation. Cell Cycle. 2005;4:1826–33. doi: 10.4161/cc.4.12.2214. [DOI] [PubMed] [Google Scholar]
- 7.Dirick L, Bohm T, Nasmyth K. Roles and regulation of Cln-Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae. Embo J. 1995;14:4803–13. doi: 10.1002/j.1460-2075.1995.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guttmann-Raviv N, Martin S, Kassir Y. Ime2 a meiosis-specific kinase in yeast, is required for destabilization of its transcriptional activator, Ime1. Mol Cell Biol. 2002;22:2047–56. doi: 10.1128/MCB.22.7.2047-2056.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hepworth SR, Friesen H, Segall J. NDT80 and the meiotic recombination checkpoint regulate expression of middle sporulation-specific genes in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:5750–61. doi: 10.1128/mcb.18.10.5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honigberg SM. Ime2p and Cdc28p: co-pilots driving meiotic development. J Cell Biochem. 2004;92:1025–33. doi: 10.1002/jcb.20131. [DOI] [PubMed] [Google Scholar]
- 11.Kassir Y, Adir N, Boger-Nadjar E, Raviv NG, Rubin-Bejerano I, Sagee S, Shenhar G. Transcriptional regulation of meiosis in budding yeast. Int Rev Cytol. 2003;224:111–71. doi: 10.1016/s0074-7696(05)24004-4. [DOI] [PubMed] [Google Scholar]
- 12.Kupiec M, Byers B, Esposito R, Mitchell A. In: The Molecular and Cellular Biology of the Yeast Saccharomyces. Pringle J, Broach J, Jones E, editors. Cold Spring Harbor Press; Cold Spring Harbor, NY: 1997. pp. 889–1036. [Google Scholar]
- 13.Lindgren A, Bungard D, Pierce M, Xie J, Vershon A, Winter E. The pachytene checkpoint in Saccharomyces cerevisiae requires the Sum1 transcriptional repressor. Embo J. 2000;19:6489–97. doi: 10.1093/emboj/19.23.6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell AP, Driscoll SE, Smith HE. Positive control of sporulation-specific genes by the IME1 and IME2 products in Saccharomyces cerevisiae. Mol Cell Biol. 1990;10:2104–10. doi: 10.1128/mcb.10.5.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nash P, Tang X, Orlicky S, Chen Q, Gertler F, Mendenhall M, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 16.Ozsarac N, Straffon MJ, Dalton HE, Dawes IW. Regulation of gene expression during meiosis in Saccharomyces cerevisiae: SPR3 is controlled by both ABFI and a new sporulation control element. Molecular & Cellular Biology. 1997;17:1152–9. doi: 10.1128/mcb.17.3.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pak J, Segall J. Regulation of the premiddle and middle phases of expression of the NDT80 gene during sporulation of Saccharomyces cerevisiae. Mol Cell Biol. 2002;22:6417–6429. doi: 10.1128/MCB.22.18.6417-6429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierce M, Benjamin KR, Montano SP, Georgiadis MM, Winter E, Vershon AK. Sum1 and Ndt80 proteins compete for binding to middle sporulation element sequences that control meiotic gene expression. Mol Cell Biol. 2003;23:4814–25. doi: 10.1128/MCB.23.14.4814-4825.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pierce M, Wagner M, Xie J, Gailus-Durner V, Six J, Vershon AK, Winter E. Transcriptional regulation of the SMK1 mitogen-activated protein kinase gene during meiotic development in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:5970–80. doi: 10.1128/mcb.18.10.5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Primig M, Williams R, Winzeler E, Tevzadze G, Conway A, Hwang S, Davis R, Esposito R. The core meiotic transcriptome in budding yeasts. Nature Genetics. 2000;26:415–423. doi: 10.1038/82539. [DOI] [PubMed] [Google Scholar]
- 21.Rockmill B, Fung JC, Branda SS, Roeder GS. The Sgs1 helicase regulates chromosome synapsis and meiotic crossing over. Curr Biol. 2003;13:1954–62. doi: 10.1016/j.cub.2003.10.059. [DOI] [PubMed] [Google Scholar]
- 22.Schindler K, Benjamin KR, Martin A, Boglioli A, Herskowitz I, Winter E. The Cdk-activating kinase Cak1p promotes meiotic S phase through Ime2p. Mol Cell Biol. 2003;23:8718–28. doi: 10.1128/MCB.23.23.8718-8728.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schindler K, Winter E. Phosphorylation of Ime2 regulates meiotic progression in Saccharomyces cerevisiae. J Biol Chem. 2006;281:18307–16. doi: 10.1074/jbc.M602349200. [DOI] [PubMed] [Google Scholar]
- 24.Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae [see comments] [published erratum appears in Cell 1996 Jan 12;84(1):following 174] Cell. 1994;79:233–44. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 25.Sedgwick C, Rawluk M, Decesare J, Raithatha SA, Wohlschlegel J, Semchuk P, Ellison M, Yates Iii JR, Stuart DT. Saccharomyces cerevisiae Ime2 phosphorylates Sic1 at multiple PXS/T sites but is insufficient to trigger Sic1 degradation. Biochem J. 2006. [DOI] [PMC free article] [PubMed]
- 26.Shor E, Gangloff S, Wagner M, Weinstein J, Price G, Rothstein R. Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics. 2002;162:647–62. doi: 10.1093/genetics/162.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shubassi G, Luca N, Pak J, Segall J. Activity of phosphoforms and truncated versions of Ndt80, a checkpoint-regulated sporulation-specific transcription factor of Saccharomyces cerevisiae. Mol Genet Genomics. 2003;270:324–36. doi: 10.1007/s00438-003-0922-3. [DOI] [PubMed] [Google Scholar]
- 28.Sopko R, Raithatha S, Stuart D. Phosphorylation and maximal activity of Saccharomyces cerevisiae meiosis-specific transcription factor Ndt80 is dependent on Ime2. Mol Cell Biol. 2002;22:7024–40. doi: 10.1128/MCB.22.20.7024-7040.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tachikawa H, Bloecher A, Tatchell K, Neiman AM. A Gip1p-Glc7p phosphatase complex regulates septin organization and spore wall formation. J Cell Biol. 2001;155:797–808. doi: 10.1083/jcb.200107008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–60. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- 31.Vershon AK, Pierce M. Transcriptional regulation of meiosis in yeast. Curr Opin Cell Biol. 2000;12:334–9. doi: 10.1016/s0955-0674(00)00104-6. [DOI] [PubMed] [Google Scholar]
- 32.Wagner M, Briza P, Pierce M, Winter E. Distinct steps in yeast spore morphogenesis require distinct SMK1 MAP kinase thresholds. Genetics. 1999;151:1327–40. doi: 10.1093/genetics/151.4.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie J, Pierce M, Gailus-Durner V, Wagner M, Winter E, Vershon AK. Sum1 and Hst1 repress middle sporulation-specific gene expression during mitosis in Saccharomyces cerevisiae. Embo J. 1999;18:6448–54. doi: 10.1093/emboj/18.22.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]