

Abstract
Side-chain functionalized lactide analogues have been synthesized from commercially available amino acids and polymerized using stannous octoate as a catalyst. The synthetic strategy presented allows for the incorporation of any protected amino acid for the preparation of functionalized diastereomerically pure lactide monomers. The resulting functionalized cyclic monomers can be homopolymerized, and copolymerized with lactides, then quantitatively deprotected forming new functional poly(lactide)-based materials. This strategy allows for the introduction of functional groups along a poly(lactide) (PLA) backbone that after deprotection can be viewed as chemical handles for further functionalization of PLA, yielding improved biomaterials for a variety of applications.
Introduction
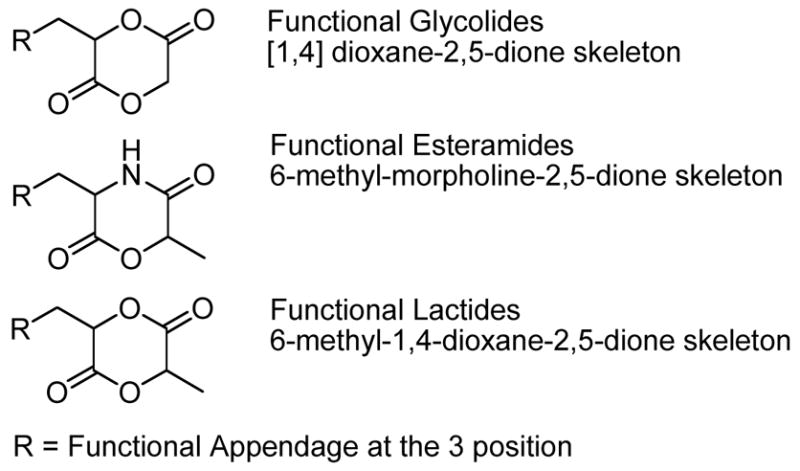
The prominence and development of biodegradable and renewable polyesters has risen dramatically over the last 30 years.1 Poly(lactide) (PLA), a poly(2-hydroxypropionic acid), used in fibers and packaging,2 is a versatile biorenewable polymeric material,3 which is also both biocompatible and bioresorbable. These traits give PLA an important role as a biomaterial in the medical field with uses in implantation devices, sutures, tissue replacement, and as a delivery vehicle.3–6 However, a major shortcoming of PLA is its lack of functional group diversity along the polymer backbone. Pendant functionalities incorporated as side-chains of a PLA would allow for greater control of its material properties such as degradation rate, hydrophilicity, and mechanical strength.7–9 Furthermore, it may allow for easy post-polymerization functionalization with a wide variety of biological moieties thereby providing a novel route towards functionalized biomaterials. While a number of these strategies have taken advantage of glycolide and morpholine-2,5-dione analogs,10–17 there is a conspicuous absence of functionally diverse lactides. The development of diverse applications for PLAs and the potential to prepare bioconjugate materials based on the lactide backbone has prompted us to develop a general synthetic and polymerization methodology towards a wide range of functional lactides.
Since Kimura’s first report on the synthesis of a glycolide-based poly(ester) with pendant carboxylic acids10 several groups have expanded on this poly(glycolide) functionalization scheme.11, 12 A significant simplification in the synthesis of functional glycolides relative to analogous lactides is the presence of only one stereocenter, which precludes the generation of diastereomers. While easier to synthesize, poly(glycolide) analogs are more hydrophilic18 and less crystalline.19 Therefore they are often poorly suited for some medical applications due to their generally faster hydrolytic degradation rates in vivo.18,19
In an analogous fashion, Feijen has reported on the development of morpholine-2,5-dione derivatives, that are subject to ring-opening polymerizations to give poly(ester-amides),13 and several other important reports describing variations of the Feijen strategy have appeared in the literature (Figure 1).14, 15 Again though, differences in the polymerization behavior of these monomers compared to their lactide analogs is observed due to the presence of the amide groups, which directs the opening of the monomer to the ester thereby retarding the rate of polymerization20 and effecting the rate of in vivo hydrolysis and enzymatic degradation rates.19 Therefore the need for a synthetic route towards functionally diverse lactides is evident.
Figure 1.
Functional lactide analogues.
In this contribution, we define a functional lactide as a 6-methyl-1,4-dioxane-2,5-dione framework with a functional group-bearing substituent at the 3 position that is amenable to further derivatization. Previous alterations to the lactide skeleton have mainly involved substitution of the 3-methyl group with other alkyl groups, phenyl, or alkenyl groups; with seminal work reported by Baker6, 16, 17 and Möller.9 Both groups have described efficient strategies towards the preparation of alkyl substituted lactides as well as their incorporation into homo- and co-polymers. In addition, there are several interesting reports on extending the substitution strategy from the mono-functionalized monomers to bi-functionalized analogs,21, 22 but there are only two reports in the literature of asymmetric functional lactide monomer analogues containing heteroatomic functionalities, amenable to further post-polymerization functionalization. The first incorporates a protected sugar, gluconic acid.11 However, the homopolymerization of this functional lactide resulted in extremely low conversions and degrees of polymerization. The most recent example describes an alternate synthetic route towards cyclic lactide monomer 1, but the authors do not report any attempts at the ring-opening polymerization.8 While pertinent, the scope of these reports is limited and the synthesis and polymerization of functional lactides remains a challenge for the development of a broadly applicable synthetic scheme towards functional lactides that will provide access to a new class of renewable and biodegradable functional materials. In this contribution, we demonstrate such a synthetic strategy by describing a synthetic pathway towards functional lactides starting from commercially available protected amino acids. It is important to note that any amino acid can be utilized in our modular strategy allowing for the synthesis of a library of functional lactides. Furthermore, we present preliminary homo- and co-polymerization results thereby demonstrating that our new monomers can be polymerized, successfully incorporated into PLA copolymers, and deprotected.
Results and Discussion
Important to the design of our synthetic strategy is the preservation of the biorenewable character of our functional monomers whereby we make use of commercially available side-chain protected L-amino acids (Figure 2). We rationalized that readily available amino acids provide a large diversity of terminal functional groups in their side-chains and that they are available as single enantiomers. To demonstrate the modular character of our functional PLA synthesis based on amino acids, we present the synthesis of three new functional lactide monomers, 1–3, containing protected amines, carboxylic acids, and alcohols in their side-chains (Figure 2) as well as their homopolymerization and copolymerization.
Figure 2.

Functional lactide monomers investigated.
Our synthetic strategy is based on the simple conversion of amino acids to their corresponding α-hydroxy acids. These are then coupled with either (S) 2-bromopropionyl chloride or (S) 2-bromopropionic acid and cyclized to provide the monofunctional lactide (Scheme 1).
Scheme 1.
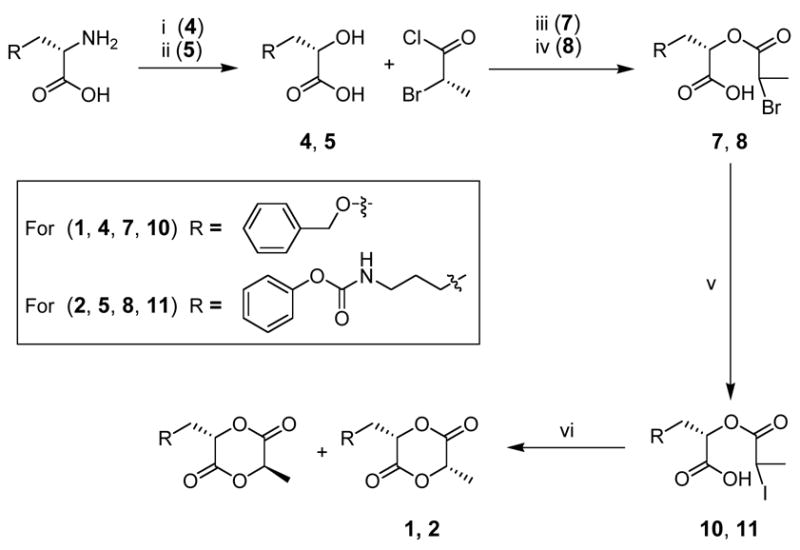
Synthesis of cyclic monomers 1 and 2 using an acid chloride coupling. Conditions: (i) NaNO2, TFA, H2O, 25 °C, 6 h; (ii) NaNO2, aq. KHSO4, 0–25 °C, 3 h; (iii) 75 °C, 6 h; (iv) DIEA, Et2O, 0–40 °C, 12 h; (v) KI, acetone, 65 °C, 12 h; (vi) DIEA, acetone, 75 °C, 10 h.
Our synthetic strategy commences with the diazotization of the amino acid to the corresponding α-hydroxy acid, with full retention of the stereochemistry.23, 24 The transformation requires sodium nitrite addition to the amino acid in an aqueous acid solution. As reported by others,23 the yields of the diazotization vary widely based on the rate of sodium nitrite addition, choice of acid, and the reaction temperature. The yields shown in Schemes 1 and 2 range from 60% for 5 to 72% for 6. Since 2-bromopropionyl chloride is only commercially available in the racemic form, we synthesized (S) 2-bromopropionyl chloride from L-alanine by diazotization in HBr to provide (S) 2-bromopropionic acid, followed by the conversion to the acid chloride with thionyl chloride.20 The conditions for the cyclization of each functionalized α-hydroxy acid with lactic acid to provide the functional cyclic diester varied depending on the lability of each protecting group (Schemes 1 and 2).
Scheme 2.
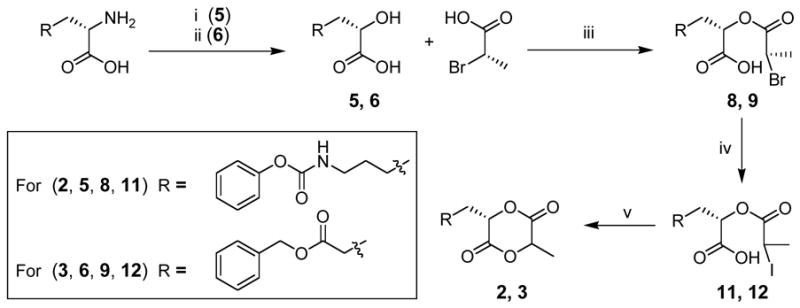
Synthesis of cyclic monomers 2 and 3. Conditions: (i) NaNO2, aq. KHSO4, 0–25 °C, 3 h; (ii) NaNO2, 1:1 AcOH/H2O (v/v), 0–25 °C, 3 h; (iii) DCC, HOBt, CH2Cl2, 25 °C, 12 h; (iv) KI, acetone, 65 °C, 12 h; (v) DIEA, acetone, 75 °C, 10 h.
The robustness of the benzyl ether protecting group made the synthesis of monomer 1 the most convenient with limited side-reactions throughout the synthetic pathway. Ester 7 was synthesized by treating α-hydroxy-acid 4 with (S) 2-bromopropionyl chloride. Racemization of the enolizable acid chloride α-proton in these acidic conditions was expected, and the diastereomeric mixture subjected to halide exchange by a Finkelstein reaction followed by cyclization without separation. The synthesis of 8, with an acid labile urethane linkage, required the use of lower temperatures and the addition of N,N-diisopropylethylamine (DIEA) to scavenge the HCl. However, these conditions were still sufficiently harsh generating persistent byproducts, which were spectroscopically and functionally quite similar to 8, and therefore difficult to remove. Because the final step is an intramolecular reaction, these unidentified byproducts posed no threat during the cyclization, and the crude monomer precursor was subjected to halide exchange and cyclized, but only low yields of product were obtained. We therefore chose to develop an alternative synthesis of monomer 2 that was also successfully employed for monomer 3.
Monomers 2 and 3 were prepared by the condensation of functionalized α-hydroxy acids with (S) 2-bromopropionic acid using dicyclohexylcarbodiimide (DCC) and 1-hydroxybenzotriazole (HOBt) to provide 8 and 9. This method circumvented racemization at the new stereocenter through: 1) milder conditions and 2) coupling of an acid instead of the more readily enolizable acid chloride. These couplings were again followed by a halogen exchange and then cyclization, as outlined in Scheme 2.
The main advantage of the coupling step of α-hydroxy acids 5 or 6 with (S) 2-bromopropionic acid is that the yields of the final cyclization are higher due to a clean conversion to the cyclic monomer precursors 8 and 9 versus monomer precursors of 7 and 8 (with persistent byproducts) generated from the acid chloride coupling method of Scheme 1.
Cyclizations of the functional monomer precursors were carried out by slowly adding uncyclized α-iodocarboxylic acids 10–12 to a dilute solution of DIEA in acetone. Linear oligomers, the primary side-products of this procedure, were easily removed by filtration of a solution of the crude product through a short plug of silica. The cyclization yield of monomer 1 was 43% with an overall yield 30%. Diastereomers of 1 from the acid chloride method were separated by column chromatography (we found that premium grade silica gel led to significantly higher recovery of product compared to standard grade, which led to decomposition of the cyclized monomers), followed by fractional crystallization from EtOAc/hexanes. The configuration of the stereocenters of the crystalline diastereomer of 1 was shown to be (S,S) as determined by X-ray structural analysis (Figure 3). The absolute configuration of cyclic monomer 1 was determined knowing that the stereocenter of the α-hydroxy acid half of the molecule retains the (S) configuration of the protected amino acid precursor under the diazotization conditions.
Figure 3.

Crystal structure of monomer 1.
As mentioned earlier the DCC/HOBt method eliminated the problem of racemization during the preparation of 8 and 9 thereby allowing for the cyclization to 2 and 3 with a > 95% diastereomeric excess (as shown by 1H-NMR), in yields unprecedented for the cyclization step of any cyclic lactide analogue. These single diastereomers were purified by column chromatography on silica gel, 3 was isolated with a 73% yield as an oil, overall yield of 48%. Monomer 2 was recrystallized and isolated with a 75% yield, overall yield of 42%, as a white crystalline solid. With the configuration of the stereocenter of monomer 2 contributed from the protected amino acid half known to be (S), the absolute configuration of the stereocenters of 2 was also shown to be (S,S) as determined by X-ray structural analysis (Figure 4).
Figure 4.
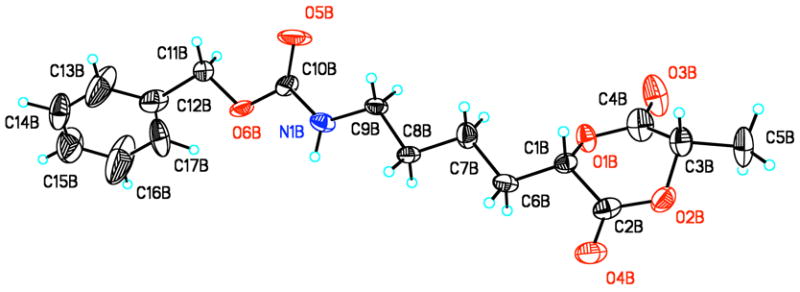
One unique structure in the asymmetric unit cell of monomer 2.
It is important to note that our new synthetic methodology allows for the retention of the stereochemistry throughout the synthesis of the functional lactides. The properties of poly(lactide)s depends strongly on the tacticity of the final polymers.2 Over the past decade, a variety of groups have developed metal catalysts that allow for the control of the tacticity during the polymerization with the appropriate lactide stereoisomer(s).25, 26 Therefore, while the scope of this study was not to investigate the stereoregular polymerization of our monomers using these catalysts, our synthetic methodology allows for the generation or separation of diastereomers that can then be employed in a stereoregular homo- or co-polymerization using these metal catalysts. This allows for complete control over polymer tacticity and thereby the properties of the resulting material.
To determine if our functionalized monomers could be polymerized, preliminary homo- and co-polymerizations were performed. For this study we restricted our investigation to the use of stannous octoate (SnOct2) as the catalyst. SnOct2 has been approved by the FDA as an indirect food additive3, 19, 27 and is among the most active and widely used catalyst for the polymerization of lactides and related monomers.22
All polymerizations were carried out in bulk. The monomers and catalyst were added to the reaction vessels in a nitrogen-filled glove box, along with a solubilizing amount of dry benzene (triply distilled). The reaction vessels were removed from the glove box, the solutions frozen, lyophilized and dried overnight. They were then heated to 140 °C under an atmosphere argon starting the polymerization. Consumption of all monomers occurred within 24 hours for the homopolymers (1H NMR). For the copolymers conversion plateaued at eight hours, with stirring ceasing for CP1 and CP2 after 3–5 hours (Scheme 3). Longer polymerization times (48 hours) and lower catalyst ratios (0.1%) did not yield higher molecular weight polymers; these same polymerization conditions gave poly(DL lactide) with a Mn of 65,000. Upon cooling, the polymers were purified by precipitation from CH2Cl2 into cold methanol (ice-water bath). Yields were based on the mass of each polymer isolated after precipitations. All polymers were characterized by 1H NMR, 13C NMR, GPC, and DSC (homopolymers Table 1; copolymers Table 2).
Scheme 3.

Homo- and co-polymerizations of 1, 2, and 3.
Table 1.
Homopolymer characterization data.
GPC in CH2Cl2 with poly(styrene) standards;
identical for either diastereomer.
Table 2.
Copolymer characterization data.
| Polymer | Mn (10−3)a | PDI | Tg (°C) | % copolyb | Yield (%) |
|---|---|---|---|---|---|
| CP1c | 31 | 2.0 | 27 | 27 % | 89 |
| CP2 | 24 | 2.1 | 25 | 25 % | 72 |
| CP3 | 5 | 1.4 | 5 | 14 % | 48 |
GPC in CH2Cl2 with poly(styrene) standards;
percentage of functional monomer incorporated into the copolymer was determined by 1H NMR after precipitation;
identical for either diastereomer.
Proton NMR showed broad signals characteristic of a polymer, and gel-permeation chromatography (GPC) confirmed the conversion to high molecular weight polymers for homopolymer P1 and copolymers CP128 and CP2. The lower molecular weights of all of these monomers versus lactide is not unexpected owing to the steric bulk of the side-chains, which is known to decrease the ΔG of the polymerization.29 However, the low molecular weight of copolymer CP3 may indicate that the side-chain of monomer 3 has other interactions with the catalyst retarding the polymerization of itself and the lactide component. All of these results are also in agreement with prior reports of the polymerization of other lactide analogs16 and substituted morpholine-2,5-dione,20 and the very low molecular weights of all the polymers derived from monomer 3 were also observed in analogs that contain similar functionalities.21 Based on the low molecular weights of most of these polymers, the near quantitative conversion of all monomers as measured by 1H NMR presumably led to low molecular weight oligomers owing to low polymerization rates and/or possible trans esterification reactions.6 However, the incorporation of the functional monomers into their copolymers is in close agreement with the stoichiometric ratio of the monomer:lactide reaction mixtures for CP1 and CP2. Differential-scanning calorimetry of each polymer showed glass-transition temperatures ranging from 5 to 27 °C, which is below the value for poly(lactide) itself. This can be attributed to the presence of the flexible side-chains, which act as internal plasticizers.16
These preliminary polymerization results clearly show the potential of monomers 1 and 2 as new functional poly(lactide) analogues. The unsatisfactory results obtained for monomer 3 may be attributed to retardation of the catalyst by coordination of the benzyl glutamate side-chain. Also, monomer 3 being an oil was harder to purify and dry in contrast to the crystalline monomers, the possible presence of scant amounts of adventitious water, despite rigorous drying procedures, may act as an undesirable initiator limiting molecular weights. Through the screening of an array of catalyst/initiator we are optimistic that higher molecular weights can be obtained for monomers 1 and 2.
Deprotection of homo- and co-polymers P1 and CP1 derived from serine, and homo- and co-polymers P3 and CP3 derived from glutamic acid was accomplished by hydrogenolysis using Pd(OH)2 as the hydrogenation catalyst revealing the hydroxyl and carboxylic acid functional groups respectively (Scheme 4A). Clean and quantitative removal of all benzyl ether and ester protecting groups from the homo- and co-polymers was confirmed by the disappearance of the aromatic and benzylic signals from the 1H NMR and 13C NMR spectra. This process did not result in the degradation of the polyester backbone, as shown by the lack of signals between 4.2 – 4.4 ppm associated with the α-hydroxy proton that would be apparent upon the cleavage of the backbone (Figure 5), this was also observed in the 1H NMR spectra of all of the copolymers.
Scheme 4.

Deprotection strategies for protected polymers: (A) Hydrogenolysis of benzyl ethers and esters; (B) Acidolysis of benzyl carbamates.
Figure 5.
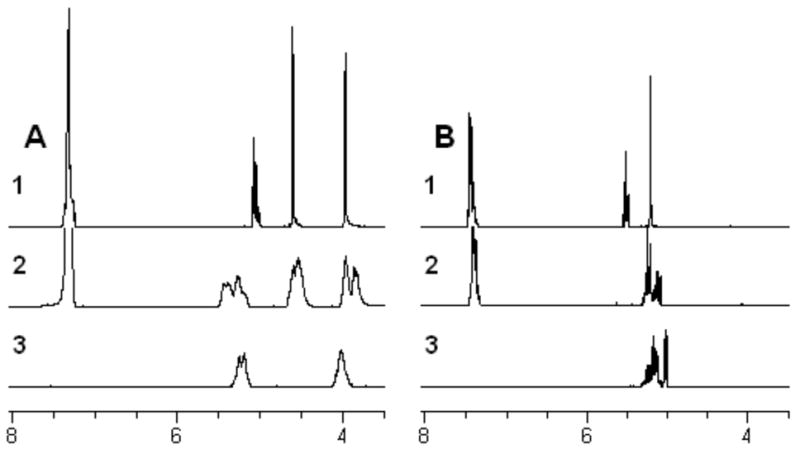
(A) 1H NMR spectra of serine derivatives in d6-acetone: 1) monomer 1; 2) homopolymer P1; 3) deprotected homopolymer P1a; (B) 1H NMR spectra of glutamic acid derivatives in d6-acetone: 1) monomer 3; 2) homopolymer P3; 3) deprotected homopolymer P3a.
However, homo- and co-polymers P2 and CP2 were recalcitrant towards hydrogenolysis and were deprotected using HBr (33%)/AcOH for two hours (Scheme 4B).
Clean and quantitative removal of the benzyl carbamate without backbone degradation was again confirmed by 1H NMR and 13C NMR, by observing the disappearance of the aromatic and benzylic signals and the absence of a signal in the region of the α-hydroxy proton in the 1H NMR spectrum (Figure 6). Unfortunately, the newly revealed functional groups of all but one of the new homopolymers precluded their characterization by GPC owing to solubility problems of the deprotected homopolymers in CH2Cl2 and the different hydrodynamic volumes of the deprotected copolymers. GPC analysis of the deprotected copolymer CP1a showed a decrease in molecular weight of approximately 35% corresponding to full cleavage of the benzyl protecting group and no cleavage of the polymer backbone. Also the GPC trace of poly(DL lactide) with a Mn of 30,000 had the same Mn and peak shape before and after being exposed to each of the deprotection conditions.
Figure 6.
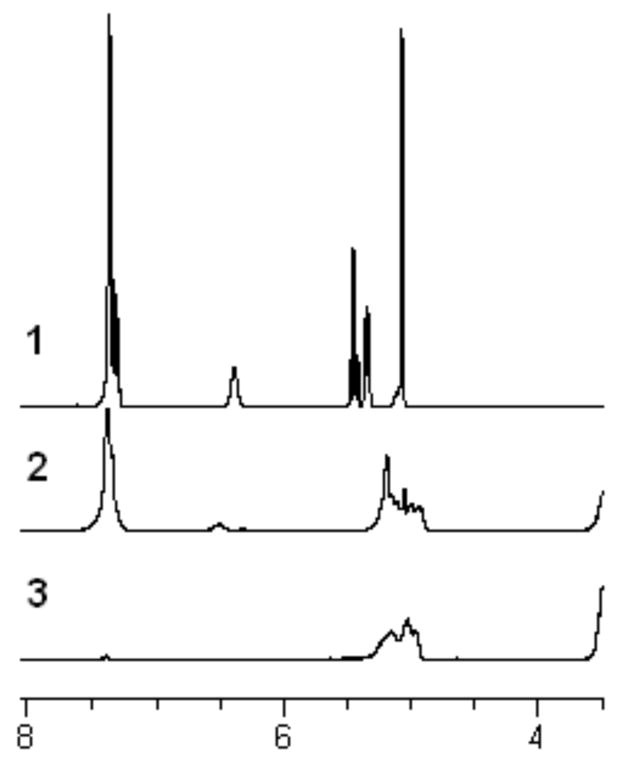
1H NMR spectra of lysine derivatives in d6-acetone: 1) monomer 2; 2) homopolymer P2; 3) deprotected homopolymer CP2a.
Conclusion
In conclusion, we have developed a modular strategy towards the synthesis and polymerization of functional lactides from commercially available protected amino acids. To demonstrate this new strategy, we have synthesized three new functional lactide monomers containing protected amine, alcohol, and carboxylic acid functionalities, we then investigated the homopolymerization behavior using the metal catalyst stannous octoate and then successfully copolymerized each monomer with lactide. Removal of the protecting groups from every polymer was quantitative with no scission of the backbone observed. Monomers 1 and 2 had the most promising polymerization results, upon deprotection; these new polymers yielded highly functional, biorenewable materials with a multitude of potential applications in the medical field. Optimization of the polymerizations for monomers 1 and 2 will be further investigated, specifically looking at an array of catalysts/initiators. Future work will focus on the development of strategies for post-polymerization modification and decomposition studies of these new functional materials.
Experimental Section
Materials and Methods
H-Ser (Benzyl)-OH was purchased from Indofine Chemical Company. H-Glu(O-Benzyl)-OH was purchased from 3B Medical Systems. H-Lys(Z)-OH was purchased from Fluka. DL lactide was purchased form Aldrich and recrystallized (2x) from dry EtOAc, dissolved in dry benzene, frozen, lyophilized and stored in a nitrogen filled glove box prior to use. N,N-Diisopropylethyl amine (DIEA), Et2O and benzene were distilled from sodium benzophenone ketyl solutions, EtOAc from CaH2, acetone from 4Å molecular sieves, and CH2Cl2 was dried via passage through Cu2O and alumina columns. All anhydrous liquids brought into the nitrogen-filled glove box were first degassed with three freeze-pump-thaw cycles using liquid nitrogen at 50 mmHg. Chromatography was performed with Sorbent Tech Premium Grade silica: porosity 60 Å, particle size 40–75 μm (200×400 mesh), surface area 450–550 m2/g, pH range 6–8., decomposition of cyclic monomer 1 was observed during chromatography using Sorbent Tech Standard Grade silica: porosity 60 Å, particle size 32–63 μm (230_450 mesh), surface area 500–600 m2/g, pH range 6.5–7.5. Compounds were analyzed by use of UV light (254 nm), I2, or a 5% solution of ammonium molybdate in 2 M sulfuric acid. IR spectra were recorded on a Schimadzu FT-IR-8400S. Melting points were determined with a Mel-Temp II apparatus fitted with a Fluke 51K/J digital thermometer and are uncorrected. Molecular weight data were collected on a Schimadzu GPC system consisting of a SCL-10Avp system controller, an LC-10ADvp pump, and a SPD-M10Avp diode array detector. The eluant was methylene chloride, and the columns were American Polymer Standards Corp. AM GPC Gel 10 μm. The molecular weights were determined relative to narrow molecular weight (polydispersity index ≤ 1.05) poly(styrene) standards from American Polymer Standards Corp. Thermal transition data were collected with a Mettler DSC 822e. The sample size ranged from 6 – 8 mg, and each sample was subjected to two cool-heat-cool cycles from 25 to −50 to 60 °C with a rate of 10 °C/min. NMR spectra were recorded at 298 K on a Varian Mercury spectrometer (300 MHz). Chemical shifts are reported in parts per million (ppm), using residual solvent as an internal standard. Mass spectral analyses were provided by the Georgia Institute of Technology Mass Spectrometry Facility. Elemental analyses were conducted at Atlantic Microlab, Inc. in Norcross, GA.
3-Benzyloxy-2-hydroxypropionic acid (4)
H-Ser(benzyl)-OH (20.0 g, 102.5 mmol) was dissolved in aq. TFA (0.7 M, 200 mL) and NaNO2 (10.6 g, 153.7 mmol) in deionized water (100 mL) was added over a period of three hours using a syringe pump at 25 °C. The reaction mixture was stirred for an additional three hours at 25 °C. NaCl (20.0 g) was added to the reaction mixture and the crude product was extracted several times with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, the solvent removed under reduced pressure, and dried in vacuo yielding a yellow oil. The product was further purified by flash column chromatography (silica gel, eluant: 97:2:1 CH2Cl2:MeOH:AcOH) and the product was dried in vacuo to give a waxy pale yellow solid (15.2 g, 76%). 1H NMR (300 MHz, CDCl3)δ: 7.25–7.35 (m, 5H), 4.5 (s, 1H), 4.3 (t, J = 3.7, 1H), 3.7–3.8 (m, 2H); 13C NMR (300 MHz, CDCl3) δ: 173.4, 138.8, 128.4, 127.7, 127.6, 73.1, 72.3, 70.8; IR (thin film) ν: 3400, 3028, 2918, 2864, 1716, 1203, 1095, 735, 696 cm−1; MS (EI) m/z (relative intensity): 196.1 (M+, 29), 107.1 (32), 91.1 (100); HRMS (EI) calcd for C10H12O4: 196.07356, found: 196.07363.
6-(Benzyloxycarbonylamino)-2-hydroxyhexanoic acid (5)
A 1 M solution of aq. KHSO4/CH3CN was prepared by dissolving KHSO4 (136.0 g, 1 mole) in deionized water (700 mL) and adding CH3CN (300 mL). H-Lys(Z)-OH (10.0 g, 35.7 mmol) was dissolved in the solution and it was then cooled to 0 °C and an aqueous solution (100 mL) of NaNO2 (37.0 g, 535.7 mmol) was added over a period 20 min using a syringe pump. The reaction mixture was kept at 0 °C and stirred for an additional three hours. NaCl (20.0 g) was added to the reaction mixture and the crude product was extracted several times with EtOAc. The combined organic layers were washed with water and brine and dried over MgSO4. The solvent was removed under reduced pressure and the residue dried in vacuo to give a waxy yellow solid. The product was further purified by flash column chromatography (silica gel, eluant: 97:2:1 CH2Cl2:MeOH:AcOH) and the product was dried in vacuo to give a pale yellow solid (6.0 g, 60%). 1H NMR (300 MHz, CDCl3) δ:7.30–7.50 (m, 5H), 5.5 (s, 2H), 4.2 (dd, J = 4.2, 7.5, 1H), 3.7 (m, 2H), 1.85-1.75 (m, 1H), 1.70-1.60 (m, 1H), 1.55-1.30 (m, 4H); 13C NMR (300 MHz, CDCl3) δ: 178.1, 154.0, 132.1, 128.8, 128.7, 128.5, 69.9, 69.8, 40.3, 33.7, 26.2, 22.1; IR (thin film) ν: 3345, 2948, 2936, 1731, 1690, 1609, 1583, 1544, 1272, 748, 731 cm−1; MS (ES+): 282.2 (M+1).
2-Hydroxypentanedioic acid 5-benzyl ester (6)
H-Glu(O-Benzyl)-OH (15.0 g, 63.3 mmol) was dissolved in a solution of 1:1 deionized water:acetic acid (350 mL) and cooled to 0 °C. NaNO2 (34.5 g, 500 mmol) in deionized water (50 mL) was added over 20 min via a syringe pump and stirred for one hour at 0 °C. The reaction was allowed to warm up to 25 °C and was stirred for an additional two hours. The crude product was extracted several times with EtOAc. The combined organic layers were washed with water and brine, and dried over MgSO4. The solvent was removed under reduced pressure and dried in vacuo to give a light brown oil (10.9 g, 72%). 1H NMR (300 MHz, CDCl3) δ: 7.3 (m, 5H), 5.1 (s, 2H), 4.3 (dd, J = 4.1, 3.8, 1H), 2.55 (m, 2H), 2.2 (m, 1H), 2.0 (m, 1H); 13C NMR (300 MHz, CDCl3) δ: 177.9, 173.5, 135.3, 128.4, 128.2, 128.1, 69.4, 66.7, 29.9, 28.9; IR (thin film) ν: 3439, 3034, 2945, 1728, 1713, 1259, 1211, 1170, 1103, 739, 698 cm−1; MS (EI) m/z (relative intensity): 238.1 (M+, 8), 210.1 (28), 108.1 (88), 107 (65), 91.1 (54), 85 (100), 79 (64), 51 (14); HRMS (EI) calcd for C12H14O5: 238.08412, found: 238.08146.
3-(Benzyloxy)-2-(2-bromopropanoyloxy)propanoic acid (7)
(S) 2-Bromopropionyl chloride (6.3 mL, 61.2 mmol) was added to 4 (10.0 g, 51 mmol) and the mixture was heated at 70 °C under an argon atmosphere in a three-neck round bottom flask equipped with a NaOH trap. Once the reaction was completed as shown by thin-layer chromatography (TLC) (approx. six hours), the dark brown oil was placed in a Kugelrohr and distilled at 60 °C and 50 mmHg overnight to remove excess 2-bromopropionic acid. Crude 7 was further purified by flash column chromatography (silica gel, eluant: 98:1.5:0.5 CH2Cl2:MeOH:AcOH). The solvent was removed in vacuo to give a brown oil in 94% yield. Compound 7 was carried on to the halogen exchange step without further purification.
6-Benzyloxycarbonylamino-2-(2-bromopropionyloxy)hexanoic acid (8)
(S) 2-Bromopropionic acid (0.242 mL, 2.7 mmol) and 1-hydroxybenzotriazole (HOBt) (0.32 g, 2.4 mmol) were dissolved in dry CH2Cl2 (10 mL). Once homogeneous, the solution was cooled to 0 °C and dicyclohexylcarbodiimide (DCC) (0.42 g, 2.0 mmol) was added in one portion. The reaction was stirred in an ice bath for 5 min and then stirred for an additional 20 min at 25 °C. Dicyclohexylurea (DCU) started to precipitate out instantly upon DCC addition. After a 25 min incubation period, a solution of 5 (0.37 g, 1.3 mmol) in CH2Cl2 (15 mL) was added dropwise over 20 min via an addition funnel. The reaction mixture was stirred for twelve hours becoming a dark brown color. It was then filtered through celite to remove DCU. The solvent was removed under reduced pressure and the brown oil redissolved in EtOAc and cooled in a freezer to precipitate out the unreacted DCC which was then filtered off through celite, and washed several times with water to remove most of the HOBt. The solvent was removed under reduced pressure and the dark brown oil was placed in a Kugelrohr and distilled at 60 °C and 50 mmHg overnight to remove excess 2-bromopropionic acid. Crude 8 was further purified by flash column chromatography (silica gel, eluant: 98:1.5:0.5 CH2Cl2:MeOH:AcOH). The solvent was removed under reduced pressure and the residue dried in vacuo to give a dark brown oil (0.51 g, 93%). Compound 8 was carried on to the halogen exchange step without further purification.
2-(2-Bromo-propionyloxy)pentanedioic acid 5-benzyl ester (9)
(S) 2-Bromo-propionic acid (0.24 mL, 2.7 mmol) and 1-hydroxybenzotriazole (HOBt) (0.32 g, 2.4 mmol) were dissolved in dry CH2Cl2 (10 mL). The solution was cooled to 0 °C and dicyclohexylcarbodiimide (DCC) (0.42 g, 2.0 mmol) was added in one portion. The reaction was stirred in an ice bath for 5 min and then stirred for an additional 20 min at 25 °C. Dicyclohexylurea (DCU) started to precipitate out instantly upon DCC addition. After a 25 min incubation period, a solution of 6 (0.32 g, 1.3 mmol) in CH2Cl2 (15 mL) was added dropwise over 20 min via an addition funnel. The reaction mixture was stirred for twelve hours becoming a light brown color. It was then filtered through celite to remove the DCU. The solvent was removed under reduced pressure and the brown oil redissolved in EtOAc and cooled in a freezer to precipitate out the unreacted DCC, which was then filtered off through celite, and then washed several times with water to remove most of the HOBt. The solvent was removed under reduced pressure and the dark brown oil was placed in a Kugelrohr and distilled at 60 °C and 50 mmHg overnight to remove excess 2-bromopropionic acid. Crude 9 was further purified by flash column chromatography (silica gel, eluant: 98:1.5:0.5 CH2Cl2:MeOH:AcOH). The solvent was removed under reduced pressure and the residue dried in vacuo to give a brown oil (0.46 g, 91%). Compound 9 was carried on to the halogen exchange step without further purification.
General procedure for the halide exchange
A solution of the α-bromocarboxylic acid (7–9) in dry acetone (100 mL) was heated to reflux with a large excess of KI (approx. 10 equivalents) under an argon atmosphere. After twelve hours (quantitative conversion by TLC) the reaction mixture was filtered through celite and the solvent was removed under reduced pressure. The residual dark red oil was redissolved in EtOAc and washed several times with a 2 M aq. Na2S2O3 solution. After the first wash, the dark red color changed to a pale yellow. The organic layer was dried over MgSO4, the solvent was removed under reduced pressure, and the residue was dried in vacuo. The α-iodocarboxylic acids (10–12) were used without further purification.
3-(Benzyloxymethyl)-6-methyl-1,4-dioxane-2,5-dione (1)
A solution of 10 (5.2 g, 13.9 mmol) in dry CH2Cl2 (100 mL) was added slowly over a period of eight hours to a refluxing solution of DIEA (4.6 mL, 27.7 mmol) in dry acetone (1 L) via a syringe pump. After the complete addition of 10 the reaction was stirred for an additional hour at reflux and the solvent was removed under reduced pressure. Et2O (100 mL) was added to the light brown oil to precipitate out ammonium iodide, which was filtered off through celite and the solvent was removed under reduced pressure. The linear oligomers were removed by passage through a short column of premium grade silica gel (3:1 v/v hexanes:EtOAc) to give the crude diastereomeric mixture as a pale brown oil (1.5 g, 43%). The diastereomers were separated by flash column chromatography (premium grade silica gel, eluant: 80:20 hexanes:EtOAc). The first diastereomer to elute was isolated as a pale brown oil (0.53 g, 36% of mixture). 1H NMR (300 MHz, CDCl3) δ:7.2–7.4 (m, 5H), 5.2 (q, J = 7.0 Hz, 1H), 5.0 (dd, J = 2.1, 2.5 Hz, 1H), 4.5 (s, 2H), 4.0 (dd, J = 2.0, 10.5 Hz, 1H), 3.9 (dd, J = 2.6, 10.5 Hz, 1H),1.6 (d, J = 7.0 Hz, 3H). The second diastereomer was recrystallized from Et2O to give a white crystalline solid (0.93 g, 64% of mixture). 1H NMR (300 MHz, CDCl3) δ: 7.2-7.1 (m, 5H), 5.07 (t, J = 3.4 Hz, 1H), 5.0 (q, J = 6.9 Hz, 1H), 4.6 (s, 2H), 4.0 (d, J = 3.5 Hz, 2H), 1.6 (d, J = 6.9 Hz, 3H); 13C NMR (300 MHz, CDCl3) δ: 166.0, 164.1, 136.4, 128.4, 128.1, 127.9, 76.0, 74.0, 73.1, 68.6, 17.8; IR (thin film) ν: 3032, 1770, 1749, 1250, 1093 cm−1; MS (EI) m/z (relative intensity): 250.1 (M+, 12), 144 (34), 107 (36), 91.1 (100), 65.1 (11); HRMS (EI) calcd for C13H14O5: 250.08384, found: 250.08412; Elemental analysis: calcd for C13H14O5: C, 62.39; H, 5.64; O, 31.97; found: C, 62.36; H, 5.75; O, 32.00; M.P. 85.4–89.8 °C.
Benzyl 4-(5-methyl-3,6-dioxo-1,4-dioxan-2-yl)butylcarbamate (2)
A solution of 11 (5.8 g, 12.5 mmol) in dry CH2Cl2 (100 mL) was added slowly over a period of eight hours to a refluxing solution of DIEA (4.1 mL, 24.9 mmol) in dry acetone (1 L) via a syringe pump. After the complete addition of 11, the reaction was stirred for an additional hour at reflux and the solvent was removed under reduced pressure. Et2O (100 mL) was added to the light brown oil to precipitate out ammonium iodide, which was filtered off through celite and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography (premium grade silica gel, eluant: 80:20 hexanes:EtOAc) to give a clear oil which was recrystallized from EtOAc/hexanes to give a white crystalline solid (3.19 g, 75%). 1H NMR (300 MHz, CDCl3) δ: 7.4-7.2 (m, 6H), 5.1 (s, 2H), 4.98 (q, J = 5 Hz, 1H), 4.89 (dd, J = 4.3, 2.8 Hz, 1H), 4.8 (br. s, 1H), 3.22 (q, J = 5.7 Hz, 2H), 2.1 (m, 2H), 1.95 (m, 2H), 1.62 (d, J = 6.7 Hz, 3H), 1.6-1.4 (br. s, 4H); 13C NMR (300 MHz, CDCl3) δ: 167.2, 166.5, 155.2, 136.4, 128.4, 128.0, 127.95, 75.5, 72.3, 66.6, 40.6, 29.55, 29.5, 21.5, 15.9; IR (thin film) ν: 3331, 2943, 2874, 1782, 1749, 1693, 1688, 1537, 1281, 1265, 1227, 696 cm−1; MS (FAB+) m/z (relative intensity): 336.1 (M+1, 100), 292.1 (42), 248.1 (12); HRMS (FAB+) calcd for C17H22NO6: 336.1447, found: 336.14352; Elemental analysis: calcd for C17H21NO6: C, 60.89; H, 6.31; N, 4.18; O, 28.63; found: C, 60.87; H, 6.33; N, 4.16, O, 28.64; M.P. 94.0–98.6 °C.
Benzyl 3-(5-methyl-3,6-dioxo-1,4-dioxan-2-yl)propanoate (3)
A solution of 12 (5.5 g, 13.2 mmol) in dry CH2Cl2 (100 mL) was added slowly over a period of eight hours to a refluxing solution of DIEA (4.3 mL, 26.3 mmol) in dry acetone (1 L) via a syringe pump. After the complete addition of 12, the reaction was stirred for an additional hour at reflux and the solvent was removed under reduced pressure. Et2O (100 mL) was added to the light brown oil to precipitate out ammonium iodide, which was filtered off through celite and the solvent was removed under reduced pressure. Crude 3 was purified by flash column chromatography (silica gel, eluant: 80:20 hexanes:EtOAc) to give a light brown oil (2.8 g, 73%). 1H NMR (300 MHz, CDCl3) δ: 7.34 (m, 5H), 5.12 (s, 2H), 5.09 (t, J = 3.8 Hz, 1H), 4.95 (q, J = 6.7 Hz, 1H), 2.63 (q, 2H), 2.48 (m, 1H), 2.21 (m, 1H), 1.64 (d, J = 6.6 Hz, 3H); 13C NMR (300 MHz, CDCl3) δ: 171.9, 167.0, 166.5, 135.3, 128.31, 128.07, 127.90, 74.0, 72.1, 66.4, 28.3, 25.0, 15.5; IR ν: (thin film) 3034, 2999, 2947, 1764, 1732, 1246, 1171, 752, 700 cm−1; HRMS (ESI+) calcd for C15H17O6: 293.106425, found: 293.101965; Elemental analysis: calcd for C15H16O6: C, 61.64; H, 5.52; O, 32.84. found: C, 61.49; H, 5.59; O, 32.92.
SnOct2 Catalyst Stock Solution
In a nitrogen-filled glove box, SnOct2 (0.32 g, 0.8 mmol)) was added to a 10 mL volumetric flask that was then filled to the graduation mark with dry, degassed benzene making a 0.8 M solution, this was transferred to a Schlenk flask and stored inside the glove box.
General Procedure for Homopolymerizations
Crystalline monomers 1 and 2 were recrystallized (2x) from anhydrous Et2O/EtOAc mixtures and monomer 3 was columned repeatedly (4x) to ensure purity. Each monomer was then frozen in benzene (triply distilled from sodium benzophenone ketyl solutions), lyophilized and stored in a nitrogen filled glove box prior to polymerization. In the nitrogen-filled glove box, the monomer of choice (0.4 mmol), SnOct2 (2 mol%, 50 μL from the stock solution), and a solubilizing amount of benzene (approximately 0.75 mL) were added to a reaction vessel with a stir bar and sealed with a side arm equipped glass stopper, the flask was removed from the glove box, the mixture was frozen, lyophilized, and dried in vacuo for 24 hours at 25 °C at 50 mmHg and was heated to 140 °C while stirring. After 24 hours the mixture was cooled, the crude polymer was dissolved in CH2Cl2 and precipitated by the dropwise addition of the polymer solution into cold MeOH at 0 °C, the precipitate was filtered off and dried in vacuo. Molecular weights and polydispersities were determined by gel-permeation chromatography and the glass-transition temperatures were determined by differential-scanning calorimetry.
Homopolymer P1
1H NMR (300 MHz, CDCl3) δ: 7.2–7.4 (s, 5H), 5.1–5.5 (m, 2H), 4.4–4.6 (m, 2H), 3.9–4.0 (m, 1H), 3.7–3.9 (m, 1H), 1.4–1.7 (m, 3H); 13C NMR (300 MHz, CDCl3) δ: 169, 166, 164, 138, 129, 128, 73, 72, 69, 68, 67, 32, 17.
Homopolymer P2
1H NMR (300 MHz, CDCl3) δ: 7.2–7.4 (m, 5H), 4.8–5.4 (m, 4H), 3.4–3.6 (m, 1H), 3.0–3.2 (m, 1H), 1.4–2.1 (m, 9H)); 13C NMR (300 MHz, CDCl3) δ: 169, 168, 156, 137, 129, 128, 72, 69, 66, 40, 30, 29, 22, 16.
Homopolymer P3
1H NMR (300 MHz, CDCl3) δ: 7.1–7.4 (m, 5H), 4.9–5.3 (m, 4H), 2.1–2.7 (m, 4H), 1.2–1.6 (m, 3H); 13C NMR (300 MHz, CDCl3) δ: 176, 172, 169, 168, 136, 135, 129, 128, 75, 72, 69, 67, 66, 31, 29, 27, 26, 16.
General Procedure for Copolymerizations
Crystalline monomers 1, 2 were recrystallized (2x) from anhydrous Et2O/EtOAc mixtures, and monomer 3 was columned repeatedly (4x) to ensure purity. Each monomer was then frozen in benzene (triply distilled from sodium benzophenone ketyl solutions), lyophilized and stored in a nitrogen filled glove box prior to polymerization. In the nitrogen-filled glove box, the monomer of choice (0.2 mmol), DL lactide (0.087 g, 0.6 mmol), SnOct2 (2 mol%, 100 μL from the stock solution), and a solubilizing amount of benzene (approximately 0.75 mL) were added to a reaction vessel with a stir bar and sealed with a side arm equipped glass stopper, the flask was removed from the glove box, the mixture was frozen, lyophilized, and dried in vacuo for 24 hours at 25 °C at 50 mmHg and heated to 140 °C while stirring. After eight hours the reaction was cooled, the crude copolymer was dissolved in CH2Cl2 and precipitated by the dropwise addition of the polymer solution into cold MeOH at 0 °C, the precipitate was filtered off and dried in vacuo. Molecular weights and polydispersities were determined by gel-permeation chromatography and the glass-transition temperatures were determined by differential-scanning calorimetry.
Copolymer CP1
1H NMR (300 MHz, CDCl3) δ: 7.2–7.4 (m, 5H), 5.1–5.4 (m, 7H), 4.4–4.60 (m, 2H), 3.8–4.0 (m, 2H), 1.4–1.7 (m, 18H), ; 13C NMR (300 MHz, CDCl3) δ: 170, 167, 166, 164, 137, 129, 128, 73, 72, 69, 68, 67, 66, 31, 17.
Copolymer CP2
1H NMR (300 MHz, CDCl3) δ: 7.2–7.4 (m, 5H), 4.8–5.3 (m, 18H), 3.4–3.5 (m, 1H), 3.0–3.2 (m, 2H), 1.1–2.1 (m, 50H); 13C NMR (300 MHz, CDCl3) δ: 170,169, 168, 167, 156, 137, 129, 128, 78, 72, 69, 66, 40, 39, 30, 29, 22, 20, 16.
Copolymer CP3
1H NMR (300 MHz, CDCl3) δ: 7.2–7.4 (m, 5H), 4.9–5.3 (m, 10H), 2.2–2.6 (m, 4H), 1.4–1.8 (m, 22H); 13C NMR (300 MHz, CDCl3) δ176, 172, 170, 169, 168, 167, 137, 136, 135, 129, 128, 78, 77, 75, 70, 69, 68, 66, 30, 29, 26, 25, 16.
General Procedure for Hydrogenolysis Deprotections
A mixture of polymer, reagent grade EtOAc (25 mL), and a catalytic amount of wet 20% Pd(OH)2 on carbon was sealed in a pressure flask on a hydrogenator and charged to 50 PSI with H2 at 25 °C and agitated for four hours. The solution was filtered through celite, the solvent removed under reduced pressure and the product was dried in vacuo.
General Procedure for Acidolysis Deprotections of Benzyl Carbamates
The polymer was dissolved in 33% w/w HBr in AcOH under anhydrous conditions and stirred in an inert atmosphere, after two hours the deprotected homopolymers were precipitated by the addition of Et2O and dried in vacuo. For the deprotected copolymer HBr and AcOH were removed in vacuo. All deprotected polymers were redissolved in acetone then dried. This was repeated two times to ensure complete removal of all acidic components, after which all polymers were again dried in vacuo.
Homopolymer P1a
1H NMR (300 MHz, d6-acetone) δ: 5.1–5.4 (m, 2H), 3.9–4.1 (m, 1H), 3.7–3.9 (m, 1H), 3.0–3.2 (m, 1H), 1.8–2.0 (m, 1H), 1.4–1.8 (m, 3H); 13C NMR (300 MHz, d6-acetone) δ: 169, 166, 164, 75, 73, 69, 68, 67, 62, 32, 23, 16.
Homopolymer P2a
1H NMR (300 MHz, d6-acetone) δ: 4.8–5.3 (m, 2H), 3.4–3.5 (m, 1H), 3.0–3.3 (m, 1H), 1.8–2.1 (m, 2H), 1.3–1.8 (m, 7H); 13C NMR (300 MHz, d6-acetone) δ: 172, 169, 72, 69, 40, 29, 28, 26, 20, 16.
Homopolymer P3a
1H NMR (300 MHz, d6-acetone) δ: 5.1–5.4 (m, 1H), 5.0–5.1 (m, 1H), 2.4–2.8 (m, 2H), 2.2–2.4 (m, 2H), 1.3–1.6 (m, 3H); 13C NMR (300 MHz, d6-acetone) δ: 177, 176, 173, 172, 171, 77, 76, 71, 70, 69, 62, 56, 31, 28, 27, 26, 18.
Copolymer CP1a
1H NMR (300 MHz, CDCl3) δ: 5.1–5.3 (m, 7H), 3.7–4.0 (m, 2H), 1.3–1.8 (m, 18H); 13C NMR (300 MHz, CDCl3) δ: 170, 167, 166, 164, 74, 73, 69, 68, 67, 32, 22, 17, 16.
Copolymer CP2a
1H NMR (300 MHz, CDCl3) δ: 4.9–5.3 (m, 16H), 3.4–3.5 (m, 1H), 3.0–3.2 (m, 1H), 1.1–2.1 (m, 50H); 13C NMR (300 MHz, CDCl3) δ: 170, 169, 75, 69, 68, 41, 40, 31, 23, 17.
Copolymer CP3a
1 H NMR (300 MHz, CDCl3) δ: 5.0–5.3 (m, 8H), 2.4–2.6 (m, 2H), 2.2–2.4 (m, 2H), 1.4–1.7 (22H); 13C NMR (300 MHz, CDCl3) δ: 180, 174, 173, 172, 170, 169, 167, 72, 70, 69, 68, 66, 30, 29, 26, 25, 16.
Supplementary Material
General experimental details and crystallographic data as a .cif file. This material is available free of charge on the Internet at http://pubs.acs.org.
Acknowledgments
Financial support has been provided by the National Science Foundation (ChE-0239385) and the National Institute of Health. MW gratefully acknowledges a 3M Untenured Faculty Award, a DuPont Young Professor Award, an Alfred P. Sloan Fellowship, a Camille Dreyfus Teacher Scholar Award, and the GIT Blanchard Assistant Professorship. Initial support of our program in functional lactide research is supported by an award from the Whitaker Foundation to AG and DMC and by the NSF ERC in Tissue Engineering at Georgia Tech and the Georgia Tech/Emory NSF ERC on the Engineering of Living Tissues (EEC-9731643).
References
- 1.Albertsson AC, Varma IK. Biomacromolecules. 2003;4:1466–1486. doi: 10.1021/bm034247a. [DOI] [PubMed] [Google Scholar]
- 2.Drumright RE, Gruber PR, Henton DE. Adv Mater. 2000;12:1841–1846. [Google Scholar]
- 3.Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Chem Rev. 2004;104:6147–6176. doi: 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- 4.Holy CE, Fialkov JA, Davies JE, Shoichet MS. J Biomed Mater Res, Part A. 2003;65A:447–453. doi: 10.1002/jbm.a.10453. [DOI] [PubMed] [Google Scholar]
- 5.Jin S, Gonsalves KE. J Mater Sci: Mater Med. 1999;10:363–368. doi: 10.1023/a:1026425824686. [DOI] [PubMed] [Google Scholar]
- 6.Simmons TL, Baker GL. Biomacromolecules. 2001;2:658–663. doi: 10.1021/bm005639+. [DOI] [PubMed] [Google Scholar]
- 7.Kimura Y, Shirotani K, Yamane H, Kitao T. Polymer. 1993;34:1741–1748. [Google Scholar]
- 8.Leemhuis M, van Steenis JH, van Uxem MJ, van Nostrum CF, Hennink WE. Eur J Org Chem. 2003:3344–3349. [Google Scholar]
- 9.Trimaille T, Möller M, Gurny R. J Polym Sci, Part A: Polym Chem. 2004;42:4379–4391. [Google Scholar]
- 10.Kimura Y, Shirotani K, Yamane H, Kitao T. Macromolecules. 1988;21:3338–3340. [Google Scholar]
- 11.Marcincinova-Benabdillah K, Boustta M, Coudane J, Vert M. Biomacromolecules. 2001;2:1279–1284. doi: 10.1021/bm015585j. [DOI] [PubMed] [Google Scholar]
- 12.Yang J-y, Yu J, Pan H-z, Gu Z-w, Cao W-x, Feng X-d. Chin J Polym Sci. 2001;19:509–516. [Google Scholar]
- 13.in't Veld PJA, Dijkstra PJ, Feijen J. Makromol Chem. 1992;193:2713–2730. [Google Scholar]
- 14.Barrera DA, Zylstra E, Lansbury PT, Jr, Langer R. J Am Chem Soc. 1993;115:11010–11011. [Google Scholar]
- 15.Feng Y, Klee D, Höcker H. Macromol Chem Phys. 2002;203:819–824. [Google Scholar]
- 16.Yin M, Baker GL. Macromolecules. 1999;32:7711–7718. [Google Scholar]
- 17.Vogeley NJ, Baker GL, Smith MR., III Polym Prepr (Am Chem Soc, Div Polym Chem) 2005;46:336. [Google Scholar]
- 18.Jeong SI, Kim BS, Lee YM, Ihn KJ, Kim SH, Kim YH. Biomacromolecules. 2004;5:1303–1309. doi: 10.1021/bm049921i. [DOI] [PubMed] [Google Scholar]
- 19.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Chem Rev. 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 20.Barrera DA, Zylstra E, Lansbury PT, Langer R. Macromolecules. 1995;28:425–432. [Google Scholar]
- 21.Ouchi T, Fujino A. Makromol Chem. 1989;190:1523–1530. [Google Scholar]
- 22.Radano CP, Baker GL, Smith MR., III Polym Prepr (Am Chem Soc, Div Polym Chem) 2002;43:727–728. [Google Scholar]
- 23.Deechongkit S, You SL, Kelly JW. Org Lett. 2004;6:497–500. doi: 10.1021/ol036102m. [DOI] [PubMed] [Google Scholar]
- 24.Winitz M, Bloch-Frankenthal L, Izumiya N, Birnbaum SM, Baker CG, Greenstein JP. J Am Chem Soc. 1956;78:2423–2430. [Google Scholar]
- 25.Chamberlain BM, Cheng M, Moore DR, Ovitt TM, Lobkovsky EB, Coates GW. J Am Chem Soc. 2001;123:3229–3238. doi: 10.1021/ja003851f. [DOI] [PubMed] [Google Scholar]
- 26.O'Keefe BJ, Hillmyer MA, Tolman WB. J Chem Soc, Dalton Trans. 2001:2215–2224. [Google Scholar]
- 27.US Food and Drug Administration. The List of “Indirect” Additives Used in Food Contact Substances. http://www.cfsan.fda.gov/~dms/opa-indt.html.
- 28.Yang J-y, Yu J, Li M, Gu Z-w, Feng X-d. Chin J Polym Sci. 2002;20:413–417. [Google Scholar]
- 29.Johns DB, Lenz RW, Luecke A. Ring-Opening Polymerization. Vol. 1. Elsevier Applied Science Publisher; London: 1984. pp. 461–521. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental details and crystallographic data as a .cif file. This material is available free of charge on the Internet at http://pubs.acs.org.