

Abstract
Background
The pregnane X receptor (PXR; NR1I2), a member of the nuclear receptor superfamily, regulates the expression of metabolic enzymes and transporters involved in the response of mammals to their chemical environment.
Objective
To summarize the functions and clinical implications of PXR.
Methods
In the current review, the clinical implications of PXR are discussed, and the use of genetically engineered PXR mouse models is highlighted.
Results/conclusion
Recent advances in mouse models, including Pxr-null and PXR-humanized mice, provide in vivo tools for evaluating the physiological functions of PXR and its role in controlling xenobiotic metabolism and transport. By using the PXR knockout and humanized mouse models, PXR was found to influence drug-drug interactions, hepatic steatosis, and the homeostasis of vitamin D, bile acids and steroid hormones. PXR was also shown to influence inflammatory bowel diseases.
Keywords: clinical implications, nuclear receptor, pregnane X receptor, PXR-humanized mouse, Pxr-null mouse
1. Introduction to the pregnane X receptor
The pregnane X receptor (PXR; NR1I2), identified in 1998 as a member of the nuclear receptor (NR) superfamily, is expressed in liver and intestine, front line organs involved in the absorption, distribution, metabolism and elimination of xenobiotics and endobiotics, in all mammalian species examined to date [1–4]. PXR, generally regarded as a sensor activated by exogenous and endogenous chemicals, regulates a large number of enzymes and transporters involved in the response of mammals to their chemical environment [5, 6]. PXR activation is ligand dependent; following ligand binding, PXR forms a heterodimer with the retinoid X receptor (RXR) that binds to PXR response elements, located in the 5′-flanking regions of PXR target genes, resulting in their transcriptional activation. PXR is mainly associated with the cellular response to xenobiotics, including induction of enzymes involved in drug oxidation and conjugation, as well as induction of xenobiotic and endobiotic transporters [7]. Metabolic enzymes and transporters induced by PXR activation affect the pharmacokinetics of both xenobiotics and endobiotics (Figure 1).
Figure 1. Pregnane X receptor (PXR) activation and its function.
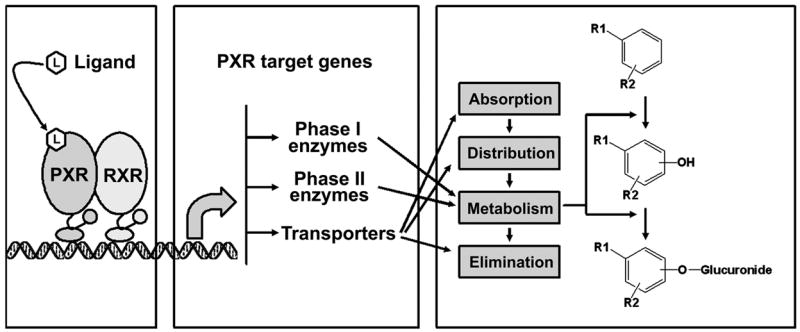
PXR activation is ligand dependent. Following ligand binding, PXR forms of a heterodimer with retinoid X receptor (RXR), and subsequently binds to PXR response elements in the 5′-flanking region of the PXR target genes resulting in the transcriptional activation. Among the target genes are those encoding including both phase I and phase II metabolic enzymes, as well as transporters which contribute to absorption, distribution, metabolism and elimination of xenobiotics and endobiotics.
Unlike other NRs such as the steroid receptors that interact selectively with their physiological ligands, PXR ligands are structurally diverse and include prescription drugs, herbal medicines, dietary supplements, environmental pollutants, and endobiotics [8–10]. Indeed, elucidation of the three-dimensional structure of the PXR ligand-binding domain (LBD) revealed that it has a large, spherical ligand binding cavity that allows it to interact with a wide range of hydrophobic chemicals [11, 12]. Many PXR ligands have been identified among prescription drugs, and include the antibiotics rifampicin, clotrimazole, and ritonavir; the antineoplastic drugs cyclophosphamide, cyproterone acetate, taxol, tamoxifen, and RU486; the anti-inflammatory agent dexamethasone; the anti-type 2 diabetes drug troglitazone; the anti-hypertensive drugs nifedipine and spironolactone; and the sedatives glutethimide and phenobarbital [9]. Commonly used herbal medicines can also activate PXR, such as St. John’s Wort, Gugulipid®, and kava kava [13]. Among dietary supplements, vitamins K2 and E have been established as weak PXR activators [14, 15]. Recently, multiple research groups reported that a number of environmental pollutants are PXR ligands, such as organochlorine pesticides, and polybrominated diphenyl ether flame retardants [16, 17]. In addition, some endobiotics were identified as PXR ligands, including certain bile acid precursors, estrogens and progestogens [2, 18–20].
PXR activation regulates a large network of genes. In rats treated with pregnenolone 16α-carbonitrile (PCN), a rodent specific PXR ligand, 138 genes were induced, while 82 genes were repressed [21]. Activation of PXR in PXR-humanized mice on a Pxr-null background revealed that 146 genes were differentially expressed, with 56 upregulated and 90 downregulated [7]. It should be noted, that most of the genes detected in these studies were not validated by direct quantification of the mRNAs and many may not be direct targets of PXR. Thus, the numbers of putative target genes are likely to be overestimated. However, among the PXR upregulated genes found in these studies were those encoding multiple metabolic enzymes and transporters, many of which are known PXR target genes with PXR regulatory elements. The most significantly induced P450s were from the CYP3A and CYP2B subfamilies [1, 22]. The human CYP3A genes located on chromosome 7q22.1, consist of four members, CYP3A4, CYP3A5, CYP3A7, and CYP3A43 [23]. Among these, CYP3A4 is among the most abundant of the P450s expressed in liver, and participates in the metabolism of more than 50% of marketed drugs, and some endogenous substrates such as steroids and bile acids [24]. CYP2B, although not as abundant as CYP3A, plays an important role in metabolism of the antidepressant bupropion, the non-nucleoside reverse transcriptase inhibitor efavirenz, and the alkylating antineoplastic drugs ifosfamide and cyclophosphamide. UDP-glucuronosyltransferases and glutathione S-transferases are the major phase 2 conjugating enzymes induced following PXR activation [25, 26]. Both oxidation and conjugation reactions generally result in the formation of more polar metabolites of the original chemicals. PXR activation also upregulates transporters, such as P-glycoprotein, multidrug resistance-related protein-3, and organic anion transporting polypeptide-2 (OATP2) [27–29]. OATP2 is an influx transporter, while both P-glycoprotein and multidrug resistance-related protein-3 are efflux transporters. P-glycoprotein is an ATP-dependent efflux pump with broad substrate specificity, and serves as a defense mechanism against potentially harmful chemicals and their metabolites. Multidrug resistance-related protein-3 is involved in the transport of biliary and intestinal excretion of organic anions. Due to the role of PXR in regulation of metabolic enzymes and transporters, PXR activation can greatly affect the fate of chemical substances, such as drugs, hormones, nutrients and toxins. The current review summarizes and discusses the role of PXR activation in drug-drug interactions, hepatic steatosis, vitamin D homeostasis, bile acids homeostasis, steroid hormones homeostasis, and inflammatory bowel diseases. The use of genetically modified PXR mouse models is highlighted.
2. Animal models used in the study of PXR
High-throughput in vitro PXR activation and binding assays have been used to identify PXR ligands [30, 31]. Primary cultures of human hepatocytes have also been employed in studies on PXR activation and PXR target gene regulation, and for prediction of drug-drug interactions [32]. However, the general problem remains one of extrapolating from in vitro findings to the clinical situation in vivo, and PXR-expressing cell lines were limited to the study of overall functions of PXR functions, such as PXR-mediated adverse drug interactions and toxicities. In this regard, animal models provide appropriate in vivo tools for evaluating the functions of PXR in a whole animal system. In this regard, animal models provide appropriate in vivo tools for evaluating the functions of PXR. Recent advances in animal models for PXR include the Pxr-null mouse and the PXR-humanized mouse models (Figure 2).
Figure 2. Recent advances in animal models on PXR studies: Pxr-null mouse model and PXR-humanized mouse model.

The Pxr-null mouse model is generally used to identify the PXR-dependent signaling pathways and the PXR-humanized mouse model is used to overcome the species differences in response to PXR ligands.
2.1 Pxr-null mouse models
Two Pxr-null mouse models were generated successfully by using similar strategies of disrupting the mouse Pxr gene by homologous recombination [33, 34]. The Pxr-null mice did not display any overt phenotypic abnormalities and extensive biochemical analysis did not reveal any significant differences in serum cholesterol, triglycerides, glucose, or liver enzyme levels. In addition, Pxr-null mice developed and reproduced normally [5, 12]. These data suggest that PXR is not essential for mouse development or physiological homeostasis. However, as expected, the Pxr-null mice did not respond to PXR ligands; hepatic PXR target genes were strongly induced in wild-type mice treated with PCN, but not induced in Pxr-null mice [33, 34]. Thus, the Pxr-null mouse was validated as a reliable model to identify PXR-dependent signaling pathways (Figure 2).
2.2 PXR-humanized mouse models
The major reason for generation of the PXR-humanized mouse model is the marked species differences in response to PXR ligands. Drugs such as rifampicin, clotrimazole, and troglitazone activate human PXR but are weak activators of rat and mouse PXR. In contrast, dexamethasone and PCN activate the rodent PXR but are weak activators of human PXR [12]. The molecular basis for species differences in response to PXR ligands is likely to be amino acid difference in the LBD or in regions of the protein that affect the structure of the LBD. Like all the other NRs, PXR has a highly conserved DNA binding domain of ~70 amino acids and a LBD of ~300 amino acids in the C-terminal portion of the protein [1, 35]. The PXR orthologs in human, rhesus monkey, rabbit, rat, and mouse, share >94% amino acid identity in the DNA binding domain. However, the amino acid differences in the LBD are more extensive; human PXR shares 82% sequence similarity with rabbit, 77% with mouse, 76% with rat, and 52% with fish in the LBD [10, 12, 36]. While the specific amino acid residues responsible for the differences in ligand affinity between species have not been elucidated, the structure of the human PXR LBD reveals an expansive ligand-binding pocket that differs between species [37, 38].
Generation of a PXR-humanized mouse model would provide a solution to the problem of species differences in ligand specificity. To this end, two PXR-humanized mouse models were generated successfully, the Alb-hPXR mice and BAC-hPXR mice [33, 39]. The Alb-SXR/hPXR mouse model was produced by use of a cDNA, while the BAC-hPXR mouse model was generated with a bacterial artificial chromosome (BAC). These PXR-humanized mouse models were developed in the Pxr-null mouse background and responded to the human-specific PXR ligand rifampicin; no significant response was found with the rodent-specific PXR ligand PCN [33, 39]. The notable differences between these two humanized models is that the human PXR cDNA is driven by heterologous promoter that yields liver-specific expression, while the BAC transgene, contains the complete PXR gene is under control of the native human PXR promoter. This distinction is quite important since PXR is also expressed in the gut where it can influence the metabolism and transport of drugs [39, 40]. A third model, in which the human PXR protein was fused to a viral VP16 coactivator, was also generated yielding ligand independent gene activation resulting in constitutive activation of PXR target genes [33, 41]. The PXR-humanized mice serve as models to surmount the species differences in response to PXR ligands (Figure 2).
3. Clinical implications of PXR
PXR activation regulates enzymes and transporters that affect the metabolism and elimination of xenobiotics and endobiotics which could result in altered clinical responses. Indeed, data from preclinical research and in vivo studies using the Pxr-null and PXR-humanized mouse models, together with clinical reports, revealed the role of PXR in drug-drug interactions, triglyceride homeostasis and hepatic steatosis, vitamin D homeostasis, metabolic bone disorders, bile acid homeostasis, steroid hormone homeostasis, and the potential benefit of PXR in inflammatory bowel diseases (Figure 3).
Figure 3. Clinical implications of PXR.

PXR is highly expressed in human liver and intestine. PXR activation results in multiple clinical responses. PXR was proposed as a risk factor contributing to drug-drug interactions, hepatic steatosis, metabolic bone disorders, and perturbation of steroid hormone homeostasis. In contrast, PXR activation might be protective for inflammatory bowel diseases, and protective for cholestasis via the regulation of bile acids homeostasis.
3.1. PXR mediated drug-drug interactions
The most common clinical implication for the activation of PXR is the occurrence of drug-drug interactions (Figure 3 and 4). Multiple-therapy regimens are the major reason for drug-drug interactions, especially involving patients with tuberculosis, cancer, HIV, cardiovascular disease, and diabetes. Drug-drug interactions have become a critical issue in health care, as serious adverse drug reactions in patients frequently require hospitalization, and some result in permanent disability or death [42]. Therefore, understanding the mechanism and predicting drug-drug interactions is an important goal for the improvement of drug safety. The identification of PXR revealed a molecular mechanism for many drug-drug interactions. When two or more drugs are combined, and one is a PXR ligand, and others are the substrates of PXR target gene encoded enzymes or transporters, drug-drug interactions can occur. PXR mediated drug-drug interactions are based on pharmacokinetics and result from the interference of the metabolic clearance of one drug by another co-administered drug. The clinical consequences of PXR mediated drug-drug interactions are generally decreased therapeutic efficacy and, occasionally, increased drug toxicity (Figure 4).
Figure 4. PXR mediated drug-drug interactions.
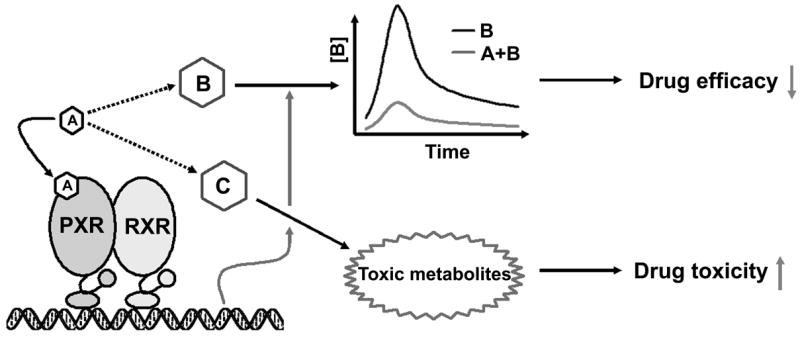
PXR mediated drug-drug interactions are based on pharmacokinetics and result from the interference in the metabolic clearance of one drug by another co-administered drug. For example, drug A is co-administered with drug B or C, and drug A is a PXR ligand that activates PXR and enhances the metabolic clearance of drug B or C. For the drug-drug interactions between drug A and B, the efficacy of drug B decreases due to the lowered area under the curve and the peak plasma concentration. For the drug-drug interactions between drug A and C, the toxicity of drug C increases because of the accumulation of toxic metabolites.
3.1.1 Decreased efficacy via PXR-enhanced metabolism
Rifampicin, a human PXR ligand used at a high dose and long-term for tuberculosis treatment, is associated with PXR-mediated drug-drug interactions in humans [43]. Rifampicin interacts with over 100 drugs, most notably drugs that are CYP3A substrates, including oral contraceptives, pre-anesthetic midazolam, and anti-HIV protease inhibitors. Drug-drug interactions between rifampicin and oral contraceptives were first reported in the early 1970s. In tuberculosis patients under chemotherapy with rifampicin, a significant high incidence of unwanted pregnancies and menstrual cycle disorders was noted in female patients using oral contraceptives [44]. Drug-drug interactions were also found with midazolam, a fast-acting benzodiazepine used for short minor surgical procedures such as dental extraction. However, midazolam is ineffective in patients treated with rifampicin. Indeed, pharmacokinetic studies on midazolam revealed a ~95% decrease in the area under the curve (AUC) and the peak plasma concentration (Cmax), ~60% decrease in the half-life (t1/2β) in healthy volunteers pretreated with rifampicin [45]. Drug-drug interactions between rifamycins and anti-HIV protease inhibitors in tuberculosis and HIV co-infected patients, can result in the loss of HIV suppression. Pretreatment with rifampicin markedly changed the pharmacokinetics of anti-HIV protease inhibitors as revealed by a decrease of the AUC: 82% for amprenavir, 92% for indinavir, 82% for nelfinavir, and 70% for saquinavir [43, 46, 47]. It is now clear that PXR is involved in these drug-drug interactions; rifampicin activates PXR and upregulates the PXR target gene CYP3A4, resulting in increased metabolic clearance of oral contraceptives, midazolam, and anti-HIV protease inhibitors, leading to decreased efficacy (Figure 4).
St John’s Wort is a herbal medicine used for centuries, dating from the early Greeks, for medicinal purposes in the treatment of mental disorders and nerve pain. Today, St John’s Wort is widely used for depression, anxiety, and sleep disorders [48]. However, the alarm regarding St John’s Wort was raised when a life-threatening adverse drug reaction was reported in two heart transplant patients treated with cyclosporine, one who had self-administered St John’s Wort and the other who was prescribed St John’s Wort by a psychiatrist [49]. Cyclosporine is an immunosuppressant used in organ transplant patients to reduce the risk of organ rejection. However, in organ transplant patients using St John’s Wort, the organ transplant failed. Further studies revealed that St John’s Wort contains multiple PXR ligands, most notably hyperforin, which was found to activate PXR with an EC50 at 21 nM [50]. Thus, St John’s Wort activates PXR, upregulates CYP3A expression, and accelerates cyclosporine metabolism. Pharmacokinetic studies confirmed the drug-drug interactions between St John’s Wort and cyclosporine, through demonstrating a marked decrease of cyclosporine blood levels in patients coadministered St John’s Wort [51, 52].
3.1.2 Increased toxicity via PXR mediated metabolic activation
In most cases, PXR activation is associated with detoxication due to the increased metabolism and elimination of xenobiotics. However, accelerated metabolism might be harmful for some drugs because of the production and accumulation of toxic metabolites (Figure 4). Acetaminophen (APAP), widely used analgesic for relief of fever and headaches, is considered safe for humans at recommended doses. However, acute overdose can result in liver damage and is the leading cause of liver failure in the United States [53]. APAP is metabolized primarily in the liver, where its major metabolites include metabolically inactive sulfate and glucuronide conjugates. The minor metabolic pathway is via hepatic P450, which is responsible for the generation of the putative toxic alkylating metabolite N-acetyl-p-benzo-quinone imine (NAPQI). At recommended doses, NAPQI is quickly detoxicated through conjugation with glutathione to produce a non-toxic derivative. However, under conditions of P450 induction, the risk of APAP toxicity increases due to excess hepatic NAPQI. Pretreatment with the CYP2E1 inducer isoniazid or the CYP1A2 inducer 3-methylcholanthrene increases APAP hepatotoxicity [54, 55]. Enhanced APAP hepatotoxicity by PXR activation was recently reported in mice pretreated with the PXR ligand PCN. Pretreatment with the PXR activator markedly enhanced APAP-induced hepatic injury in wild-type but not in Pxr-null mice, suggesting that PXR plays a critical role in APAP-induced hepatic toxicity by inducing CYP3A11 expression and hence increasing bioactivation [56]. However, the role of PXR in APAP hepatotoxicity in humans is not clear.
3.2 Vitamin D homeostasis and PXR mediated metabolic bone disorders
Vitamin D promotes bone formation and mineralization and is essential in skeleton development. Vitamin D deficiency leads to bone softening diseases, such as rickets in children and osteomalacia in adults (Figure 3). In mammals, two major forms of vitamin D exist, vitamin D2 and vitamin D3. In humans, vitamin D3 is more effective than vitamin D2 [57] while vitamin D2 is more effective than vitamin D3 in rats [58]. 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), the physiologically active form of vitamin D in humans, is synthesized from vitamin D3 by hepatic CYP27A1 and CYP2R1, and renal CYP27B1. 1,25(OH)2D3 mediates its biological effects by binding to the vitamin D receptor, and vitamin D receptor activation in the intestine, bone, and kidney leads to the maintenance of calcium and phosphorus levels in the blood and to the maintenance of bone content [59]. Thus, 1,25(OH)2D3 homeostasis is important in controlling bone metabolism. Renal CYP24 is well known to be the major enzyme contributing to the metabolism of 1,25(OH)2D3 to the inactive form 1,24,25-trihydroxyvitamin D3 (1,24,25(OH)3D3). CYP24 also contributes to the metabolism of 25(OH)D3 to 24,25-dihydroxyvitamin D3 (24,25(OH)2D3), which decreases formation of 1,25(OH)2D3 from 25(OH)D3. Recently, CYP24 was identified as a PXR target gene by both in vivo and in vitro studies, suggesting that drugs such as PXR ligands can modulate CYP24 gene expression and alter the homeostasis of 1,25(OH)2D3 [60]. The PXR target CYP3A4 catalyzes 23- and 24-hydroxylation of 1,25(OH)2D3, resulting in production of the biologically inactive metabolites 1,23R,25-trihydroxyvitamin D3 (1,23R,25(OH)3D3), 1,23S,25-trihydroxyvitamin D3 (1,23S,25(OH)3D3), and 1,24S,25-trihydroxyvitamin D3 (1,24S,25(OH)3D3) [61]. Compared to CYP24, CYP3A4 expression in humans is very abundant especially in liver. However, the affinity and efficiency of CYP3A4 in 1,25(OH)2D3 metabolism are ~10-fold lower than that of CYP24 as revealed by enzyme kinetics studies [61].
Whatever the effect of CYP24 and CYP3A4 on 1,25(OH)2D3 homeostasis, the role of PXR in metabolic bone disorders in humans remains unclear, because: (i) Although a significant proportion of the world’s population has been treated with rifampicin over the past 40 years, rifampicin-mediated osteomalacia was not reported to any significant degree [62]; (ii) There was no significant change in plasma 1,25(OH)2D3 levels during short- or long-term treatment with rifampicin [63, 64]; (iii) PXR was also proposed to be a factor that increases bone density. Thus, while it is not likely that activation of PXR alters the biological activity of vitamin D, there is a potential for drug-hormone interactions. Vitamin K2, a critical factor required for blood clotting, was identified as a weak PXR ligand [14]. Indeed, vitamin K2 supplementation increases bone density in vivo, and it is used clinically in the management of osteoporosis. In vitro, vitamin K2 was able to induce bone markers in primary osteocytes isolated from wild-type mouse calvaria but not in cells isolated from Pxr-null mice [15]. Further studies indicated that the osteoblastgenic transcription factor MSX2, a target gene for PXR, mediated the osteoprotective action of vitamin K2 [65]. Overall, the role of PXR in vitamin D homeostasis and metabolic bone disorders is not conclusive. For studies on drug-induced osteomalacia, the role of constitutive androstane receptor (CAR) should also be considered, because: (i) CAR ligands can also regulate metabolism and affect vitamin D homeostasis; (ii) CAR ligands, such as phenytoin and phenobarbital, are much more frequently associated with osteomalacia than PXR ligand rifampicin [62].
3.3 Role of PXR in bile acid homeostasis
Bile is produced and secreted by hepatocytes into the gut where it facilitates the digestion and absorption of lipids and vitamins. In addition, bile secretion is an important pathway for the elimination of large hydrophobic endobiotic and xenobiotic metabolites, including many high Mr conjugates. The major components of bile are cholesterol, lecithin, bile pigments, bile acids, and bicarbonate ions. Among the bile components, bile acids are crucial in maintaining bile flow, eliminating of cholesterol from the liver, and emulsify lipids in the gut. Bile acid levels are tightly regulated by multiple NRs, including hepatic nuclear factor 4α(HNF4α), liver X receptor (LXR), farnesoid X receptor, vitamin D receptor, constitutive androstane receptor, and PXR [34, 66–68]. Under certain pathological conditions such as cholestasis that results from impaired bile flow, bile acids are potentially toxic. Indeed, serious hepatotoxicity was noted in mice over exposed to lithocholic acid [34, 69]. PXR is involved in regulation of the biosynthesis, transport, and metabolism of bile acids (Figure 3). Bile acids are produced in the liver from cholesterol via multiple enzyme-dependent steps with the rate limiting step being 7-hydroxylation of cholesterol by cholesterol 7α-hydroxylase (CYP7A1). Since PXR activation downregulates CYP7A1, it can impact bile acid synthesis [34]. Further studies suggest that PXR activation promotes PXR interaction with HNF4αand blocks peroxisome proliferator-activated receptor (PPAR) γcoactivator 1αactivation of HNF4α, resulting in inhibition of CYP7A1 gene transcription [70]. PXR activation also regulates the expression of OATP2, sulfotransferase, and murine Cyp3a11/human CYP3A4, which may promote the metabolism and transport of bile acids [34]. OATP2 is localized on the basolateral membrane of the hepatocyte and is involved in the cellular uptake of bile acids. Its induction by PXR would presumably increase the uptake of bile acids from the sinusoidal blood into the hepatocyte where the detoxication pathways such as hydroxylation and sulfation could take place by Cyp3a11/CYP3A4 and sulfotransferase, respectively [34].
PXR activation was proposed to decrease the bile acid synthesis via downregulation of CYP7A1, and to accelerate bile acid metabolism and elimination through upregulation of metabolic enzymes and transporters [34, 70]. Clinically, the human PXR ligand rifampicin has been used in patients with primary biliary cirrhosis for its potential effect on bile acid metabolism. Primary biliary cirrhosis, a disease characterized by inflammatory destruction of the small bile ducts within the liver, presents with cholestasis, jaundice and pruritus. Rifampicin treatment decreased pruritus, and the serum concentration of total and conjugated bile acids was also strikingly reduced [71]. The benefit of rifampicin for primary biliary cirrhosis was proposed as the metabolic regulation, as shown by an increase of antipyrine clearance and the enhanced urinary excretion of 6β-hydroxycortisol [71]. However, the clinical data are controversial on the role of PXR in the biliary system. In humans with or without hepatic cirrhosis, a significant elevation of plasma bile acids was noted after two hours of rifampicin treatment [72]. Cholestatic hepatitis was reported in humans treated with rifampicin alone [73]. At the same time, significant high incidences of hepatotoxicity were reported in primary biliary cirrhosis patients with the treatment of rifampicin [74, 75]. Thus, further studies are required to determine the role of PXR in the biliary system, and the high risk of rifampicin-induced hepatotoxicity should be considered for the patients with primary biliary cirrhosis.
3.4 PXR mediated hepatic steatosis
The abnormal retention of lipids within the cells results in steatosis, which reflects an impairment of the normal processes of synthesis and breakdown of triglycerides. As the liver is the primary organ of lipid metabolism, steatosis most often occurs in this tissue (Figure 3). While patients with hepatic steatosis have few or no symptoms, infrequently, they may complain of fatigue, malaise and dull right upper quadrant abdominal discomfort. However, the danger of hepatic steatosis is the result of the sequelae, such as liver fibrosis, cirrhosis, and carcinoma. In humans, hepatic steatosis is commonly associated with alcohol abuse or metabolic syndrome (diabetes, hypertension and dyslipidemia), but may also be caused by drugs and certain toxins. Recently, PXR activators were proposed as risk factors for hepatic steatosis. Hepatic lipid accumulation was noted in PXR-humanized mice treated with the human PXR activator rifampicin [76]. Expression of an activated PXR in the livers of transgenic mice also resulted in an increased lipid accumulation that was independent of activation of the lipogenic transcriptional factor sterol regulatory element-binding protein-1c but associated with an increased expression of the free fatty acid transporter CD36, and other accessory lipogenic enzymes, such as stearoyl-CoA desaturase-1 and long-chain free fatty acid elongase [76]. It was recently shown that PXR may promote hepatic steatosis by increasing the expression of CD36 directly or indirectly through the PXR-mediated activation of PPARγ [77]. PXR-mediated gene regulation and lipid accumulation are required for the hepatic regenerative response to liver resection, and it was suggested that PXR is essential for normal progression of liver regeneration by modulating lipid homeostasis [78]. T0901317, a high-affinity ligand for both LXR and PXR, induced liver steatosis. Further studies indicated that T0901317-associated hepatic steatosis was mediated by PXR, but not by LXR [79].
The putative role for PXR in hepatic steatosis raised great concern about the safety of drugs that are also PXR ligands, such as the glucocorticoid dexamethasone, the antibiotic rifampicin, and the antimycotic clotrimazole [9]. However, there are very few clinical reports concerning drug-induced hepatic steatosis by PXR ligands. For example, rifampicin, although used by a large number of tuberculosis patients since 1970s, there are no significant clinical reports of rifampicin-induced hepatic steatosis, fibrosis, cirrhosis, or carcinoma. In addition, liver biopsies were carried out on 100 tuberculosis patients, eight cases after anti-tuberculosis treatment and 92 cases before anti-tuberculosis treatment. There was no evidence indicating rifampicin-induced hepatic steatosis [80]. However, in preclinical studies, fatty livers were noted in rats given a high dose of rifampicin [81, 82]. The physiological and pharmacological relevance of this finding to humans is questionable since supra-pharmacological doses were used; rifampicin is a human specific PXR ligand, having virtually no effect on rat PXR at human-equivalent doses [12]. Overall, the role of human PXR in lipid metabolism and hepatic steatosis warrants further investigation.
3.5 PXR and steroid hormone homeostasis
Preclinical studies support the concept of PXR as a potential endocrine disrupting factor that might have broad implications in steroid hormone homeostasis and drug-hormone interactions. Activation of PXR markedly increased plasma concentrations of corticosterone and aldosterone, and their increases were associated with increased expression of adrenal steroidogenic enzymes, including CYP11A1, CYP11B1, CYP11B2, and 3β-hydroxysteroid dehydrogenase [83]. CYP3A4, a PXR target gene, also contributes to the metabolism of steroid hormones. Indeed, recombinant CYP3A4 showed exhibited significant cortisol and testosterone metabolism [84]. Cortisol and testosterone 6β-hydroxylase activities were also used as biomarkers for CYP3A4 induction or inhibition [85, 86]. Among the major human hepatic P450s, 6β- and 16α-hydroxylation of progesterone was catalyzed most efficiently by CYP3A4 [87]. In addition, CYP3A4 catalyzes 2-, 4-, and 16-hydroxylation of estradiol [88, 89].
In a CYP3A4-transgenic mouse line expressing both human and murine CYP3A, females were found to be deficient in lactation, leading to a markedly lower pup survival. This impaired lactation phenotype was associated with significantly reduced serum estradiol levels in CYP3A4-transgenic mice, suggesting that CYP3A4 may play an important role in estradiol homeostasis [90]. This may be of relevance to the treatment of pregnant and lactating women with drugs that are PXR activators. Of note, rifampicin is contraindicated in pregnancy except in the presence of a severe disease untreatable with other drugs, such as tuberculosis, because of teratogenicity found in animal studies and case reports of malformation, death and haemorrhage in infants whose mothers were exposed to rifampicin [91]. The role of PXR in the homeostasis of steroid hormones, especially sex hormones, may provide an important clue as to the mechanism by which rifampicin compromises pregnancy.
3.6 PXR in inflammatory bowel disease
Inflammatory bowel disease (IBD) refers to a chronic inflammatory condition of the digestive tract occurring as one of two major types, ulcerative colitis and Crohn’s disease. Ulcerative colitis is limited to the colon while Crohn’s disease most commonly affects the small intestine and/or the colon, but can involve any part of the gastrointestinal tract from the mouth to the anus. However, the etiology of IBD is unknown. To date, genetic, infectious, immunologic, and psychological factors have all been implicated in influencing the development of IBD. Recently PXR was identified as a gene strongly associated with the susceptibility to IBD in humans [92]. In patients with IBD, decreased expression of PXR and PXR target genes was also noted [93, 94].
Drug treatment is the main method for relieving the symptoms of both ulcerative colitis and Crohn’s disease. Progress is being made in the development of medications for treating IBD, such as anti-inflammatory drugs and immunosuppressive agents. Thus, identification of the role of PXR in IBD might provide new stratagem for IBD therapeutics. In the dextran sulfate sodium (DSS)-induced IBD mouse acute colitis model, treatment with the PXR ligand PCN protected against DSS-induced colitis compared with vehicle-treated mice, as defined by body weight loss, diarrhea, rectal bleeding, colon length, and histology. However, this treatment did not decrease the severity of DSS-induced colitis in Pxr-null mice indicating a role for PXR in protection against IBD [95]. It has recently been reported that hepatic SCD1 is downregulated in mice with DSS-induced colitis and that this leads to elevated levels of proinflammatory saturated fatty acids and reduced levels of antiinflammatory unsaturated fatty acids [96]. It should be noted that SCD1 was upregulated in mice by PXR activation [76], and thus PXR activation should be expected to ameliorate the symptoms of DSS-induced colitis in mice having low levels of expression of SCD1 through increased production of unsaturated fatty acids.
Interestingly, budesonide, a glucocorticoid derivative frequently used as an anti-inflammatory drug for IBD, was recently identified as a PXR ligand [97]. Rifaximin, approved by the Food and Drug Administration in 2004 for the treatment of travelers’ diarrhea [98] was found to be of potential value in the treatment of chronic gastrointestinal disorders including ulcerative colitis and Crohn’s disease. Despite the differences in dose and duration, rifaximin was found to be beneficial in the treatment of active ulcerative colitis, mild-to-moderate Crohn’s disease as well as prevention of postoperative recurrence of Crohn’s disease [99–101]. The mechanism contributing to the beneficial effects of rifaximin in chronic gastrointestinal disorders are not fully understood. By using PXR-humanized, Pxr-null, and wild-type mice, rifaximin was identified as a gut-specific human PXR activator [102]. Further human studies are suggested to assess the potential role of PXR activation in therapeutics of IBD.
3.7 Miscellaneous implications
PXR in cancer and chemotherapy: Resistance to chemotherapeutic agents is the major clinical problem and cause of failure in the chemotherapy of human cancer. Understanding the molecular basis of chemoresistance will be valuable for developing more effective chemotherapy. Several molecular targets have been shown to be related to chemoresistance, which include efflux transporters, phase I and II detoxication enzymes and DNA-repair enzymes. Most of these chemoresistance related enzymes, are encoded by PXR target genes [103], such as P-glycoprotein, multidrug resistance proteins, CYP3A, UDP-glucuronosyltransferase and glutathione S-transferases. Some chemotherapeutic agents, such as cyclophosphamide, tamoxifen, and taxol, have been identified as human PXR ligands [8, 9]. Activation of PXR will induce a battery of enzymes and transporters to accelerate metabolism and elimination of chemotherapeutic agents, which may contribute to acquired resistance and multi-drug resistance in chemotherapy. Additionally, PXR and its target genes have been detected in cancerous tissues, including in prostate, breast, endometrium and colon. A localized function of PXR in chemoresistance has been proposed [104–107]. However, the function of PXR in cancerous tissues is not limited to chemoresistance, as a biological function of PXR in human breast carcinoma has been proposed [108]. In human colon cancer cells, the antiapoptotic role of PXR was reported [109].
PXR and antifibrogenesis: PXR was recently proposed as a potential target for antifibrotic therapy. In rats chronically treated with carbon tetrachloride, hepatocyte necrosis and liver fibrogenesis were produced. Interestingly, the extent of fibrosis caused by carbon tetrachloride was significantly inhibited by PCN [110]. The further study investigated the effects of human PXR activators on human hepatic stellate cell transdifferentiation to a profibrogenic phenotype. The expression of PXR was detected in primary cultured hepatic stellate cells. Short-term treatment of hepatic stellate cells with the PXR ligand rifampicin inhibited the expression of fibrosis-related genes. Long-term treatment with rifampicin reduced the proliferation and transdifferentiation of hepatic stellate cells, and all these rifampicin effects were PXR-dependent. These data suggested that PXR activators inhibit transdifferentiation and proliferation of human hepatic stellate cells, and PXR may therefore be a potential target for antifibrotic therapy [111].
PXR and oxidative stress: The role of PXR in the oxidative stress response has been reported. In this study, a heightened sensitivity to oxidative toxicant paraquat was noted both in PXR-humanized mice and wild-type mice upon PXR activation. Consistent with the in vivo study, cell lines with activated human PXR were also sensitive to paraquat, and an increased production of reactive oxygen species was observed. These data suggested that PXR activation was a risk factor for oxidative stress caused by an imbalance between the production of reactive oxygen and detoxication of the reactive intermediates [112]. Further studies are indicated on the role of PXR in oxidative stress, because of the importance of oxidative stress in drug toxicity and human disease.
4. Conclusions
The Pxr-null and PXR-humanized mouse lines serve as valuable in vivo models for investigations on PXR. New information such as the target gene network controlled by PXR, and the species differences in response to PXR ligands were obtained using these models. Due to the importance of PXR in the regulation of xenobiotic-metabolizing enzymes and transporters, PXR activation greatly affects the metabolism of drugs, hormones, nutrients and toxins. Based upon data acquired from preclinical studies, especially in vivo studies using the aforementioned mouse models, together with clinical reports, it has become clear that PXR is central to drug-drug interactions, and may have a role in vitamin D, bile acid, and steroid hormone homeostasis, hepatic steatosis, and IBD. Among these clinical implications, PXR mediated drug-drug interactions have been confirmed by both preclinical studies and clinical studies, and most cases of PXR mediated drug-drug interactions result in decreased efficacy. All the clinical implications of PXR beyond drug-drug interactions were generally based on preclinical studies, and their importance to humans is not clear; however, these preclinical results provided valuable directions for the future research on PXR.
5. Expert opinion
It should be emphasized that due to the species differences between rodents and humans, the data generated from preclinical studies should be confirmed in clinical studies (Figure 5). For preclinical studies, the use of Pxr-null and PXR-humanized mouse models is recommended. Patients treated with drugs that are PXR activators are potential subjects for clinical studies on PXR, such as the tuberculosis patients under chemotherapy with rifampicin. Additional studies are warranted to investigate on the role of PXR in vitamin D homeostasis and metabolic bone disorders, bile acid homeostasis, hepatic steatosis, steroid hormones homeostasis, and IBD. Typically, cell based PXR-reporter assays or primary cultured human hepatocytes can be used to determine if a compound is a PXR agonist. However, the identification of PXR agonists in herbal medicines remains problematic. In addition to PXR agonists, studies on PXR antagonists will also become important, as they might be useful for preventing PXR mediated drug-drug interactions or perturbations of hormone and vitamin homeostasis.
Figure 5. Brief summary of the studies on PXR.
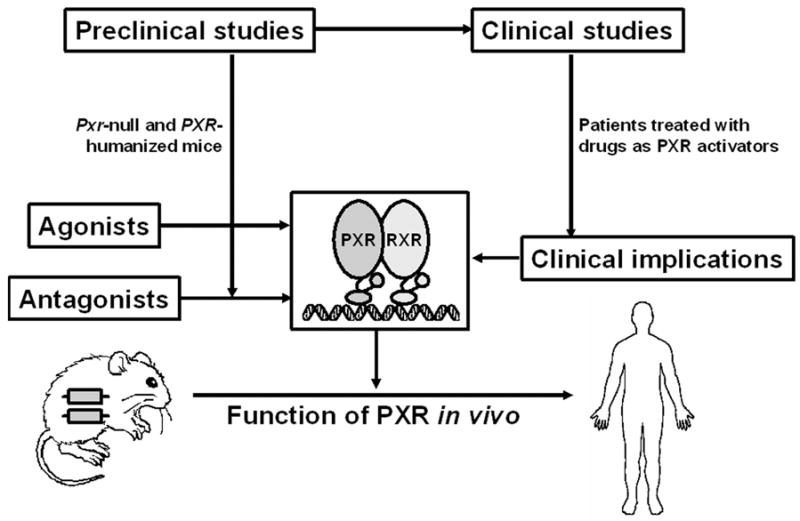
The overall goals of preclinical and clinical studies on PXR are to illustrate the function of human PXR in vivo, and to predict the risk of PXR agonists and antagonists to which humans are exposed. Preclinical studies on PXR by using Pxr-null and PXR-humanized mouse models provided valuable information on PXR. However, the preclinical results should be translated to clinical studies because of the species differences between rodents and humans. Clinical studies on PXR are suggested in patients treated with drugs as PXR activators.
Footnotes
Xiaochao Ma, MD, PhD, Postdoctoral
Fellow Jeffrey R. Idle, PhD, Professor Frank
J. Gonzalez, PhD, Laboratory Chief
Declaration of interest
The authors have no conflicts of interest to declare.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1••.KLIEWER SA, MOORE JT, WADE L, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92(1):73–82. doi: 10.1016/s0092-8674(00)80900-9. The first report on PXR. [DOI] [PubMed] [Google Scholar]
- 2••.LEHMANN JM, MCKEE DD, WATSON MA, et al. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102(5):1016–1023. doi: 10.1172/JCI3703. The first paper indicating the PXR-mediated drug-drug interactions via CYP3A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.BLUMBERG B, SABBAGH W, JR, JUGUILON H, et al. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12(20):3195–3205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MOORE LB, MAGLICH JM, MCKEE DD, et al. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol Endocrinol. 2002;16(5):977–986. doi: 10.1210/mend.16.5.0828. [DOI] [PubMed] [Google Scholar]
- 5.KLIEWER SA, WILLSON TM. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J Lipid Res. 2002;43(3):359–364. [PubMed] [Google Scholar]
- 6.KLIEWER SA. The nuclear pregnane X receptor regulates xenobiotic detoxification. J Nutr. 2003;133(7 Suppl):2444S–2447S. doi: 10.1093/jn/133.7.2444S. [DOI] [PubMed] [Google Scholar]
- 7•.ROSENFELD JM, VARGAS R, JR, XIE W, et al. Genetic profiling defines the xenobiotic gene network controlled by the nuclear receptor pregnane X receptor. Mol Endocrinol. 2003;17(7):1268–1282. doi: 10.1210/me.2002-0421. The first paper revealing the gene network controlled by PXR. [DOI] [PubMed] [Google Scholar]
- 8•.POSO A, HONKAKOSKI P. Ligand recognition by drug-activated nuclear receptors PXR and CAR: structural, site-directed mutagenesis and molecular modeling studies. Mini Rev Med Chem. 2006;6(8):937–947. doi: 10.2174/138955706777935008. An excellent review elucidating the structural properties of PXR, and the molecular determinants of its ligand and species specificities. [DOI] [PubMed] [Google Scholar]
- 9.HONKAKOSKI P, SUEYOSHI T, NEGISHI M. Drug-activated nuclear receptors CAR and PXR. Ann Med. 2003;35(3):172–182. doi: 10.1080/07853890310008224. [DOI] [PubMed] [Google Scholar]
- 10.CARNAHAN VE, REDINBO MR. Structure and function of the human nuclear xenobiotic receptor PXR. Curr Drug Metab. 2005;6(4):357–367. doi: 10.2174/1389200054633844. [DOI] [PubMed] [Google Scholar]
- 11.JACOBS MN, DICKINS M, LEWIS DF. Homology modelling of the nuclear receptors: human oestrogen receptorbeta (hERbeta), the human pregnane-X-receptor (PXR), the Ah receptor (AhR) and the constitutive androstane receptor (CAR) ligand binding domains from the human oestrogen receptor alpha (hERalpha) crystal structure, and the human peroxisome proliferator activated receptor alpha (PPARalpha) ligand binding domain from the human PPARgamma crystal structure. J Steroid Biochem Mol Biol. 2003;84(2–3):117–132. doi: 10.1016/s0960-0760(03)00021-9. [DOI] [PubMed] [Google Scholar]
- 12••.KLIEWER SA, GOODWIN B, WILLSON TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23(5):687–702. doi: 10.1210/er.2001-0038. An excellent review on the role of PXR in metabolic regulation. [DOI] [PubMed] [Google Scholar]
- 13••.STAUDINGER JL, DING X, LICHTI K. Pregnane X receptor and natural products: beyond drug-drug interactions. Expert Opin Drug Metab Toxicol. 2006;2(6):847–857. doi: 10.1517/17425255.2.6.847. An excellent review focuses on PXR activation by natural products. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14•.LANDES N, BIRRINGER M, BRIGELIUS-FLOHE R. Homologous metabolic and gene activating routes for vitamins E and K. Mol Aspects Med. 2003;24(6):337–344. doi: 10.1016/s0098-2997(03)00029-3. This paper provides the evidence that vitamins E and K are PXR activators. [DOI] [PubMed] [Google Scholar]
- 15•.TABB MM, SUN A, ZHOU C, et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J Biol Chem. 2003;278(45):43919–43927. doi: 10.1074/jbc.M303136200. The first study indicated that vitamin K2 regulates bone homeostasis via PXR. [DOI] [PubMed] [Google Scholar]
- 16.PACYNIAK EK, CHENG X, CUNNINGHAM ML, et al. The flame retardants, polybrominated diphenyl ethers, are pregnane X receptor activators. Toxicol Sci. 2007;97(1):94–102. doi: 10.1093/toxsci/kfm025. [DOI] [PubMed] [Google Scholar]
- 17.COUMOUL X, DIRY M, BAROUKI R. PXR-dependent induction of human CYP3A4 gene expression by organochlorine pesticides. Biochem Pharmacol. 2002;64(10):1513–1519. doi: 10.1016/s0006-2952(02)01298-4. [DOI] [PubMed] [Google Scholar]
- 18.MASUYAMA H, HIRAMATSU Y, KUNITOMI M, et al. Endocrine disrupting chemicals, phthalic acid and nonylphenol, activate Pregnane X receptor-mediated transcription. Mol Endocrinol. 2000;14(3):421–428. doi: 10.1210/mend.14.3.0424. [DOI] [PubMed] [Google Scholar]
- 19•.GOODWIN B, GAUTHIER KC, UMETANI M, et al. Identification of bile acid precursors as endogenous ligands for the nuclear xenobiotic pregnane X receptor. Proc Natl Acad Sci U S A. 2003;100(1):223–228. doi: 10.1073/pnas.0237082100. This study indicated the existence of a feedforward regulatory loop by which bile acid intermediates activate PXR and accelerate their own metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MNIF W, PASCUSSI JM, PILLON A, et al. Estrogens and antiestrogens activate hPXR. Toxicol Lett. 2007;170(1):19–29. doi: 10.1016/j.toxlet.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 21.GUZELIAN J, BARWICK JL, HUNTER L, et al. Identification of genes controlled by the pregnane X receptor by microarray analysis of mRNAs from pregnenolone 16alpha-carbonitrile-treated rats. Toxicol Sci. 2006;94(2):379–387. doi: 10.1093/toxsci/kfl116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.XIE W, BARWICK JL, SIMON CM, et al. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000;14(23):3014–3023. doi: 10.1101/gad.846800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.GELLNER K, EISELT R, HUSTERT E, et al. Genomic organization of the human CYP3A locus: identification of a new, inducible CYP3A gene. Pharmacogenetics. 2001;11(2):111–121. doi: 10.1097/00008571-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 24•.GUENGERICH FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. An excellent review on human CYP3A4. [DOI] [PubMed] [Google Scholar]
- 25.MACKENZIE PI, GREGORY PA, GARDNER-STEPHEN DA, et al. Regulation of UDP glucuronosyltransferase genes. Curr Drug Metab. 2003;4(3):249–257. doi: 10.2174/1389200033489442. [DOI] [PubMed] [Google Scholar]
- 26.FALKNER KC, PINAIRE JA, XIAO GH, et al. Regulation of the rat glutathione S-transferase A2 gene by glucocorticoids: involvement of both the glucocorticoid and pregnane X receptors. Mol Pharmacol. 2001;60(3):611–619. [PubMed] [Google Scholar]
- 27.GEICK A, EICHELBAUM M, BURK O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276(18):14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 28.TENG S, JEKERLE V, PIQUETTE-MILLER M. Induction of ABCC3 (MRP3) by pregnane X receptor activators. Drug Metab Dispos. 2003;31(11):1296–1299. doi: 10.1124/dmd.31.11.1296. [DOI] [PubMed] [Google Scholar]
- 29.STAUDINGER J, LIU Y, MADAN A, et al. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos. 2001;29(11):1467–1472. [PubMed] [Google Scholar]
- 30.LUO G, CUNNINGHAM M, KIM S, et al. CYP3A4 induction by drugs: correlation between a pregnane X receptor reporter gene assay and CYP3A4 expression in human hepatocytes. Drug Metab Dispos. 2002;30(7):795–804. doi: 10.1124/dmd.30.7.795. [DOI] [PubMed] [Google Scholar]
- 31.ZHU Z, KIM S, CHEN T, et al. Correlation of high-throughput pregnane X receptor (PXR) transactivation and binding assays. J Biomol Screen. 2004;9(6):533–540. doi: 10.1177/1087057104264902. [DOI] [PubMed] [Google Scholar]
- 32.LUO G, GUENTHNER T, GAN LS, et al. CYP3A4 induction by xenobiotics: biochemistry, experimental methods and impact on drug discovery and development. Curr Drug Metab. 2004;5(6):483–505. doi: 10.2174/1389200043335397. [DOI] [PubMed] [Google Scholar]
- 33••.XIE W, BARWICK JL, DOWNES M, et al. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406(6794):435–439. doi: 10.1038/35019116. The first report on Pxr-null and PXR-humanized mouse models. [DOI] [PubMed] [Google Scholar]
- 34•.STAUDINGER JL, GOODWIN B, JONES SA, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98(6):3369–3374. doi: 10.1073/pnas.051551698. One of the first reports showing that activation of PXR regulating the homeostasis of bile acids. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35•.BERTILSSON G, HEIDRICH J, SVENSSON K, et al. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A. 1998;95(21):12208–12213. doi: 10.1073/pnas.95.21.12208. One of the first reports showing the regulation of CYP3A by PXR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.IYER M, RESCHLY EJ, KRASOWSKI MD. Functional evolution of the pregnane X receptor. Expert Opin Drug Metab Toxicol. 2006;2(3):381–397. doi: 10.1517/17425255.2.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.WATKINS RE, NOBLE SM, REDINBO MR. Structural insights into the promiscuity and function of the human pregnane X receptor. Curr Opin Drug Discov Devel. 2002;5(1):150–158. [PubMed] [Google Scholar]
- 38.XUE Y, MOORE LB, ORANS J, et al. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol Endocrinol. 2007;21(5):1028–1038. doi: 10.1210/me.2006-0323. [DOI] [PubMed] [Google Scholar]
- 39••.MA X, SHAH Y, CHEUNG C, et al. The Pregnane X receptor gene-humanized mouse: a model for investigating drug-drug interactions mediated by cytochromes P450 3A. Drug Metab Dispos. 2007;35(2):194–200. doi: 10.1124/dmd.106.012831. The first report on PXR-humanized mouse model generated by bacterial artificial chromosome (BAC) transgenesis. [DOI] [PubMed] [Google Scholar]
- 40.ZHANG H, LECULYSE E, LIU L, et al. Rat pregnane X receptor: molecular cloning, tissue distribution, and xenobiotic regulation. Arch Biochem Biophys. 1999;368(1):14–22. doi: 10.1006/abbi.1999.1307. [DOI] [PubMed] [Google Scholar]
- 41.GONG H, SINGH SV, SINGH SP, et al. Orphan nuclear receptor pregnane X receptor sensitizes oxidative stress responses in transgenic mice and cancerous cells. Mol Endocrinol. 2006;20(2):279–290. doi: 10.1210/me.2005-0205. [DOI] [PubMed] [Google Scholar]
- 42.LAZAROU J, POMERANZ BH, COREY PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279(15):1200–1205. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 43••.NIEMI M, BACKMAN JT, FROMM MF, et al. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet. 2003;42(9):819–850. doi: 10.2165/00003088-200342090-00003. An excellent review on rifampicin mediated drug-drug interactions. [DOI] [PubMed] [Google Scholar]
- 44•.REIMERS D, JEZEK A. [The simultaneous use of rifampicin and other antitubercular agents with oral contraceptives] Prax Pneumol. 1971;25(5):255–262. One of the first reports showing the adverse interactions between rifampicin and oral contraceptives. [PubMed] [Google Scholar]
- 45.BACKMAN JT, OLKKOLA KT, NEUVONEN PJ. Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin Pharmacol Ther. 1996;59(1):7–13. doi: 10.1016/S0009-9236(96)90018-1. [DOI] [PubMed] [Google Scholar]
- 46.POLK RE, BROPHY DF, ISRAEL DS, et al. Pharmacokinetic Interaction between amprenavir and rifabutin or rifampin in healthy males. Antimicrob Agents Chemother. 2001;45(2):502–508. doi: 10.1128/AAC.45.2.502-508.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.GRUB S, BRYSON H, GOGGIN T, et al. The interaction of saquinavir (soft gelatin capsule) with ketoconazole, erythromycin and rifampicin: comparison of the effect in healthy volunteers and in HIV-infected patients. Eur J Clin Pharmacol. 2001;57(2):115–121. doi: 10.1007/s002280100277. [DOI] [PubMed] [Google Scholar]
- 48.Website Herbs at a glance: St. John’s Wort. [Access 29 February 2008]; Available at: http://nccam.nih.gov/health/stjohnswort/
- 49•.RUSCHITZKA F, MEIER PJ, TURINA M, et al. Acute heart transplant rejection due to Saint John’s wort. Lancet. 2000;355(9203):548–549. doi: 10.1016/S0140-6736(99)05467-7. One of the first clinical reports showing the life-threatening adverse drug-drug interactions mediated by St John’s wort. [DOI] [PubMed] [Google Scholar]
- 50••.MOORE LB, GOODWIN B, JONES SA, et al. St. John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci U S A. 2000;97(13):7500–7502. doi: 10.1073/pnas.130155097. This is the first study which provided a molecular mechanism for the interaction of St. John’s wort with drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MURAKAMI Y, TANAKA T, MURAKAMI H, et al. Pharmacokinetic modelling of the interaction between St John’s wort and ciclosporin A. Br J Clin Pharmacol. 2006;61(6):671–676. doi: 10.1111/j.1365-2125.2006.02606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MAI I, BAUER S, PERLOFF ES, et al. Hyperforin content determines the magnitude of the St John’s wort-cyclosporine drug interaction. Clin Pharmacol Ther. 2004;76(4):330–340. doi: 10.1016/j.clpt.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 53.LEE WM. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology. 2004;40(1):6–9. doi: 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- 54.CRIPPIN JS. Acetaminophen hepatotoxicity: potentiation by isoniazid. Am J Gastroenterol. 1993;88(4):590–592. [PubMed] [Google Scholar]
- 55.SZYMANSKA JA, SWIETLICKA EA, PIOTROWSKI JK, et al. Effects of 3-methylcholanthrene or diethyl maleate on the hepatotoxicity of acetaminophen. J Appl Toxicol. 1992;12(6):415–419. doi: 10.1002/jat.2550120609. [DOI] [PubMed] [Google Scholar]
- 56•.GUO GL, MOFFIT JS, NICOL CJ, et al. Enhanced acetaminophen toxicity by activation of the pregnane X receptor. Toxicol Sci. 2004;82(2):374–380. doi: 10.1093/toxsci/kfh286. This is the first study revealing the potential role of PXR and CYP3A in acetaminophen hepatotoxicity. [DOI] [PubMed] [Google Scholar]
- 57.ARMAS LA, HOLLIS BW, HEANEY RP. Vitamin D2 is much less effective than vitamin D3 in humans. J Clin Endocrinol Metab. 2004;89(11):5387–5391. doi: 10.1210/jc.2004-0360. [DOI] [PubMed] [Google Scholar]
- 58.COATES ME. Requirements of different species for vitamins. Proc Nutr Soc. 1968;27(2):143–148. doi: 10.1079/pns19680039. [DOI] [PubMed] [Google Scholar]
- 59.HOLICK MF. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am J Clin Nutr. 2004;80(6 Suppl):1678S–1688S. doi: 10.1093/ajcn/80.6.1678S. [DOI] [PubMed] [Google Scholar]
- 60•.PASCUSSI JM, ROBERT A, NGUYEN M, et al. Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest. 2005;115(1):177–186. doi: 10.1172/JCI21867. The first report indicating the role of PXR in vitamin D homeostasis via the regulation of CYP24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.XU Y, HASHIZUME T, SHUHART MC, et al. Intestinal and hepatic CYP3A4 catalyze hydroxylation of 1alpha,25-dihydroxyvitamin D(3): implications for drug-induced osteomalacia. Mol Pharmacol. 2006;69(1):56–65. doi: 10.1124/mol.105.017392. [DOI] [PubMed] [Google Scholar]
- 62.D’ERASMO E, RAGNO A, RAEJNTROPH N, et al. [Drug-induced osteomalacia] Recenti Prog Med. 1998;89(10):529–533. [PubMed] [Google Scholar]
- 63.WILLIAMS SE, WARDMAN AG, TAYLOR GA, et al. Long term study of the effect of rifampicin and isoniazid on vitamin D metabolism. Tubercle. 1985;66(1):49–54. doi: 10.1016/0041-3879(85)90053-4. [DOI] [PubMed] [Google Scholar]
- 64.BRODIE MJ, BOOBIS AR, DOLLERY CT, et al. Rifampicin and vitamin D metabolism. Clin Pharmacol Ther. 1980;27(6):810–814. doi: 10.1038/clpt.1980.115. [DOI] [PubMed] [Google Scholar]
- 65.IGARASHI M, YOGIASHI Y, MIHARA M, et al. Vitamin K induces osteoblast differentiation through PXR-mediated transcriptional control of the Msx2 gene. Mol Cell Biol. 2007;27(22):7947–7954. doi: 10.1128/MCB.00813-07. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.SINAL CJ, TOHKIN M, MIYATA M, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 67.INOUE Y, YU AM, INOUE J, et al. Hepatocyte nuclear factor 4alpha is a central regulator of bile acid conjugation. J Biol Chem. 2004;279(4):2480–2489. doi: 10.1074/jbc.M311015200. [DOI] [PubMed] [Google Scholar]
- 68.KALAANY NY, MANGELSDORF DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–191. doi: 10.1146/annurev.physiol.68.033104.152158. [DOI] [PubMed] [Google Scholar]
- 69.ZHANG J, HUANG W, QATANANI M, et al. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J Biol Chem. 2004;279(47):49517–49522. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- 70.LI T, CHIANG JY. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha-hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2005;288(1):G74–84. doi: 10.1152/ajpgi.00258.2004. [DOI] [PubMed] [Google Scholar]
- 71•.HOENSCH HP, BALZER K, DYLEWIZC P, et al. Effect of rifampicin treatment on hepatic drug metabolism and serum bile acids in patients with primary biliary cirrhosis. Eur J Clin Pharmacol. 1985;28(4):475–477. doi: 10.1007/BF00544371. One of the first clinical studies on the mechanism of rifampicin in the therapeutics of primary biliary cirrhosis. [DOI] [PubMed] [Google Scholar]
- 72.GALEAZZI R, LORENZINI I, ORLANDI F. Rifampicin-induced elevation of serum bile acids in man. Dig Dis Sci. 1980;25(2):108–112. doi: 10.1007/BF01308307. [DOI] [PubMed] [Google Scholar]
- 73.TAILLAN B, CHICHMANIAN RM, FUZIBET JG, et al. [Jaundice caused by rifampicin: 3 cases] Rev Med Interne. 1989;10(5):409–411. doi: 10.1016/s0248-8663(89)80045-1. [DOI] [PubMed] [Google Scholar]
- 74.BACHS L, PARES A, ELENA M, et al. Effects of long-term rifampicin administration in primary biliary cirrhosis. Gastroenterology. 1992;102(6):2077–2080. doi: 10.1016/0016-5085(92)90335-v. [DOI] [PubMed] [Google Scholar]
- 75.PRINCE MI, BURT AD, JONES DE. Hepatitis and liver dysfunction with rifampicin therapy for pruritus in primary biliary cirrhosis. Gut. 2002;50(3):436–439. doi: 10.1136/gut.50.3.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76•.ZHOU J, ZHAI Y, MU Y, et al. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem. 2006;281(21):15013–15020. doi: 10.1074/jbc.M511116200. This study revealed the role of PXR in lipid homeostasis in mouse liver. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.ZHOU J, FEBBRAIO M, WADA T, et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology. 2008;134(2):556–567. doi: 10.1053/j.gastro.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 78.DAI G, HE L, BU P, et al. Pregnane X receptor is essential for normal progression of liver regeneration. Hepatology. 2008;47(4):1277–1287. doi: 10.1002/hep.22129. [DOI] [PubMed] [Google Scholar]
- 79.MITRO N, VARGAS L, ROMEO R, et al. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett. 2007;581(9):1721–1726. doi: 10.1016/j.febslet.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 80.MORERE P, NOUVET G, STAIN JP, et al. [Information obtained by liver biopsy in 100 tuberculous patients] Sem Hop. 1975;51(31–34):2095–2102. [PubMed] [Google Scholar]
- 81.TRUHAUT R, CLAUDE JR, WARNET JM, et al. [Liver steatogenic power of high doses of rifampicin in rats] C R Acad Sci Hebd Seances Acad Sci D. 1978;286(6):493–497. [PubMed] [Google Scholar]
- 82.PIRIOU A, MAISSIAT R, JACQUESON A, et al. Ultrastructural changes in the parenchymal liver cells of rats treated with high doses of rifampicin. Br J Exp Pathol. 1987;68(2):201–207. [PMC free article] [PubMed] [Google Scholar]
- 83•.ZHAI Y, PAI HV, ZHOU J, et al. Activation of pregnane X receptor disrupts glucocorticoid and mineralocorticoid homeostasis. Mol Endocrinol. 2007;21(1):138–147. doi: 10.1210/me.2006-0291. This study revealed the role of PXR in glucocorticoid and mineralocorticoid homeostasis in mouse. [DOI] [PubMed] [Google Scholar]
- 84.BUTERS JT, KORZEKWA KR, KUNZE KL, et al. cDNA-directed expression of human cytochrome P450 CYP3A4 using baculovirus. Drug Metab Dispos. 1994;22(5):688–692. [PubMed] [Google Scholar]
- 85.FAYER JL, PETULLO DM, RING BJ, et al. A novel testosterone 6 beta-hydroxylase activity assay for the study of CYP3A-mediated metabolism, inhibition, and induction in vitro. J Pharmacol Toxicol Methods. 2001;46(2):117–123. doi: 10.1016/s1056-8719(02)00168-5. [DOI] [PubMed] [Google Scholar]
- 86.KONISHI H, TANAKA K, MINOUCHI T, et al. Urinary 6beta-hydroxycortisol/17-hydroxycorticosteroids ratio as a measure of hepatic CYP3A4 capacity after enzyme induction. Ann Clin Biochem. 2004;41(Pt 4):335–337. doi: 10.1258/0004563041201527. [DOI] [PubMed] [Google Scholar]
- 87.NIWA T, YABUSAKI Y, HONMA K, et al. Contribution of human hepatic cytochrome P450 isoforms to regioselective hydroxylation of steroid hormones. Xenobiotica. 1998;28(6):539–547. doi: 10.1080/004982598239290. [DOI] [PubMed] [Google Scholar]
- 88.LEE AJ, KOSH JW, CONNEY AH, et al. Characterization of the NADPH-dependent metabolism of 17beta-estradiol to multiple metabolites by human liver microsomes and selectively expressed human cytochrome P450 3A4 and 3A5. J Pharmacol Exp Ther. 2001;298(2):420–432. [PubMed] [Google Scholar]
- 89.BADAWI AF, CAVALIERI EL, ROGAN EG. Role of human cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16alpha-hydroxylation of 17beta-estradiol. Metabolism. 2001;50(9):1001–1003. doi: 10.1053/meta.2001.25592. [DOI] [PubMed] [Google Scholar]
- 90•.YU AM, FUKAMACHI K, KRAUSZ KW, et al. Potential role for human cytochrome P450 3A4 in estradiol homeostasis. Endocrinology. 2005;146(7):2911–2919. doi: 10.1210/en.2004-1248. This study revealed the role of CYP3A in estradiol homeostasis homeostasis in mouse. [DOI] [PubMed] [Google Scholar]
- 91.Website Safety information of rifampicin from intergovernmental organizations, International Program on Chemical Safety. [Access 29 February 2008]; Available at: http://www.inchem.org/documents/pims/pharm/rifam.htm.
- 92.DRING MM, GOULDING CA, TRIMBLE VI, et al. The pregnane X receptor locus is associated with susceptibility to inflammatory bowel disease. Gastroenterology. 2006;130(2):341–348. doi: 10.1053/j.gastro.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 93•.LANGMANN T, MOEHLE C, MAUERER R, et al. Loss of detoxification in inflammatory bowel disease: dysregulation of pregnane X receptor target genes. Gastroenterology. 2004;127(1):26–40. doi: 10.1053/j.gastro.2004.04.019. The first study indicating the association between PXR and inflammatory bowel diseases. [DOI] [PubMed] [Google Scholar]
- 94.MARTINEZ A, MARQUEZ A, MENDOZA J, et al. Role of the PXR gene locus in inflammatory bowel diseases. Inflamm Bowel Dis. 2007;13(12):1484–1487. doi: 10.1002/ibd.20252. [DOI] [PubMed] [Google Scholar]
- 95•.SHAH YM, MA X, MORIMURA K, et al. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol. 2007;292(4):G1114–1122. doi: 10.1152/ajpgi.00528.2006. The first study showing the benefit of PXR activation in DSS-induced colitis mouse model. [DOI] [PubMed] [Google Scholar]
- 96.CHEN C, SHAH YM, MORIMURA K, et al. Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. 2008;7(2):135–147. doi: 10.1016/j.cmet.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.MAIER A, ZIMMERMANN C, BEGLINGER C, et al. Effects of budesonide on P-glycoprotein expression in intestinal cell lines. Br J Pharmacol. 2007;150(3):361–368. doi: 10.1038/sj.bjp.0706992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.LAUSTSEN G, WIMMETT L. 2004 drug approval highlights: FDA update. Nurse Pract. 2005;30(2):14–29. doi: 10.1097/00006205-200502000-00004. [DOI] [PubMed] [Google Scholar]
- 99.SHAFRAN I, JOHNSON LK. An open-label evaluation of rifaximin in the treatment of active Crohn’s disease. Curr Med Res Opin. 2005;21(8):1165–1169. doi: 10.1185/030079905x53252. [DOI] [PubMed] [Google Scholar]
- 100.GIONCHETTI P, RIZZELLO F, MORSELLI C, et al. Management of inflammatory bowel disease: does rifaximin offer any promise? Chemotherapy. 2005;51(Suppl 1):96–102. doi: 10.1159/000081995. [DOI] [PubMed] [Google Scholar]
- 101.GUSLANDI M, PETRONE MC, TESTONI PA. Rifaximin for active ulcerative colitis. Inflamm Bowel Dis. 2006;12(4):335. doi: 10.1097/01.MIB.0000215092.85116.6c. [DOI] [PubMed] [Google Scholar]
- 102•.MA X, SHAH YM, GUO GL, et al. Rifaximin is a gut-specific human pregnane X receptor activator. J Pharmacol Exp Ther. 2007;322(1):391–398. doi: 10.1124/jpet.107.121913. This study indicated that rifaximin is a gut-specific human PXR activator. [DOI] [PubMed] [Google Scholar]
- 103.HARMSEN S, MEIJERMAN I, BEIJNEN JH, et al. The role of nuclear receptors in pharmacokinetic drug-drug interactions in oncology. Cancer Treat Rev. 2007;33(4):369–380. doi: 10.1016/j.ctrv.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 104.CHEN Y, TANG Y, WANG MT, et al. Human pregnane X receptor and resistance to chemotherapy in prostate cancer. Cancer Res. 2007;67(21):10361–10367. doi: 10.1158/0008-5472.CAN-06-4758. [DOI] [PubMed] [Google Scholar]
- 105.DOTZLAW H, LEYGUE E, WATSON P, et al. The human orphan receptor PXR messenger RNA is expressed in both normal and neoplastic breast tissue. Clin Cancer Res. 1999;5(8):2103–2107. [PubMed] [Google Scholar]
- 106.MASUYAMA H, HIRAMATSU Y, KODAMA J, et al. Expression and potential roles of pregnane X receptor in endometrial cancer. J Clin Endocrinol Metab. 2003;88(9):4446–4454. doi: 10.1210/jc.2003-030203. [DOI] [PubMed] [Google Scholar]
- 107.PFRUNDER A, GUTMANN H, BEGLINGER C, et al. Gene expression of CYP3A4, ABC-transporters (MDR1 and MRP1-MRP5) and hPXR in three different human colon carcinoma cell lines. J Pharm Pharmacol. 2003;55(1):59–66. doi: 10.1111/j.2042-7158.2003.tb02434.x. [DOI] [PubMed] [Google Scholar]
- 108.MIKI Y, SUZUKI T, KITADA K, et al. Expression of the steroid and xenobiotic receptor and its possible target gene, organic anion transporting polypeptide-A, in human breast carcinoma. Cancer Res. 2006;66(1):535–542. doi: 10.1158/0008-5472.CAN-05-1070. [DOI] [PubMed] [Google Scholar]
- 109.ZHOU J, LIU M, ZHAI Y, et al. The antiapoptotic role of pregnane x receptor in human colon cancer cells. Mol Endocrinol. 2008;22(4):868–880. doi: 10.1210/me.2007-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.MAREK CJ, TUCKER SJ, KONSTANTINOU DK, et al. Pregnenolone-16alpha-carbonitrile inhibits rodent liver fibrogenesis via PXR (pregnane X receptor)-dependent and PXR-independent mechanisms. Biochem J. 2005;387(Pt 3):601–608. doi: 10.1042/BJ20041598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.HAUGHTON EL, TUCKER SJ, MAREK CJ, et al. Pregnane X receptor activators inhibit human hepatic stellate cell transdifferentiation in vitro. Gastroenterology. 2006;131(1):194–209. doi: 10.1053/j.gastro.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 112.Gong H, Singh SV, Singh SP, et al. Orphan nuclear receptor pregnane X receptor sensitizes oxidative stress responses in transgenic mice and cancerous cells. Mol Endocrinol. 2006;20(2):279–290. doi: 10.1210/me.2005-0205. [DOI] [PubMed] [Google Scholar]