

Abstract
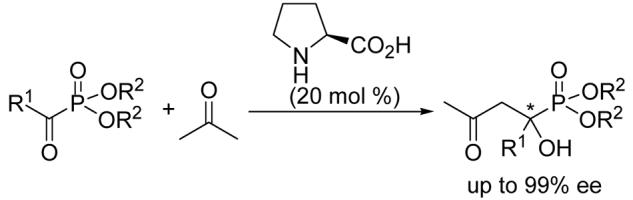 Tertiary α-hydroxy phosphonates have been synthesized in good yields and high enantiomeric purity (up to 99% ee) through a novel L-proline-catalyzed cross aldol reaction of α-keto phosphonates and ketones.
Tertiary α-hydroxy phosphonates have been synthesized in good yields and high enantiomeric purity (up to 99% ee) through a novel L-proline-catalyzed cross aldol reaction of α-keto phosphonates and ketones.
Although their biological activities have not been extensively explored,1 α-hydroxy phosphonic acid derivatives have been shown to be very important enzyme inhibitors. For example, they are inhibitors of such important medicinal enzymes as renin2 or human immunodeficiency virus (HIV) protease and polymerase.3 They also show anti-virus4 and anti-cancer activities.5 It is well-known that, in a racemic mixture, normally only one enantiomer shows the observed activities and, therefore, an enantioselective method for the synthesis of these compounds is highly desirable. Optically active α-hydroxy phosphonates are very useful precursors to other optically active α-hydroxy phosphonic acid derivatives. However, to achieve high enantioselectivity in the synthesis of α-hydroxy phosphonates is a still a challenging task for organic chemists. The optically enriched forms of these compounds are mainly obtained through enzymatic methods,6 such as kinetic resolution of racemic mixture by bacteria, fungi or lapses7 or through asymmetric reduction of α-keto phosphonate with baker's yeast or fungi.6,8 A few available chemical methods9 include asymmetric reduction of α-keto phosphonates,10 asymmetric oxidation of benzyl phosphonates11 and diastereoselective addition of dialkyl phosphites to aldehydes (phosphoaldol reaction).12a,b These methods are either not catalytic, using special reagents that are difficult to handle, or having very limited substrate scope. A catalytic method based on the phosphoaldol reaction was also reported,12c,d but the enantioselectivities obtained were dependent on the substrates. Furthermore, all the reported method are only suitable for the synthesis of secondary α-hydroxy phosphonates. To the best of our knowledge, there is no general method for the synthesis of optically active tertiary α-hydroxy phosphonates.13
Recent progresses14 in the proline-catalyzed asymmetric cross aldol reaction of activated ketones have revealed that compound such as glyoxylate is a good substrate for asymmetric aldol reaction.14d,e These results prompt us to study the possibility of using such a cross aldol reaction for the asymmetric synthesis of optically active α-hydroxy phosphonates. Due to the susceptibility of α-keto phosphonate toward nucleophilic attack and the leaving-group ability of the phosphonate group, it can be considered as a synthetic equivalent of acid chloride.15 It is, therefore, not surprising that the cross aldol reaction of α-keto phosphonate and ketone has never been studied in the literature, even with non-chiral reagents. However, when we carefully examined the proposed aldol reaction of enamine with α-keto phosphonate, it appeared the reaction intermediate can either lead to the desired α-hydroxy phosphonate or a 1,3-diketone product through the elimination of the phosphonate group.14c,15 Wiemer and co-workers have shown that tertiary α-hydroxy phosphonates are reasonably stable.13 Based on their results, we hypothesized that formation of α-hydroxy phosphonate should be feasible if the reaction conditions are appropriate. Herein we wish to report our preliminary results of the first highly enantioselective synthesis of tertiary α-hydroxy phosphonates based on a novel cross aldol reaction of α-keto phosphonates and ketones.
By using diethyl benzoylphosphonate and acetone as the model compounds, we first screened some readily available proline derivatives (Figure 1) as the catalyst. The cross aldol reaction went smoothly at room temperature in acetone16 with all these catalysts and excellent yields of the aldol product were obtained. Although L-prolinamide is a more reactive catalyst, only a moderate enantioselectivity was obtained (54% ee).17 L-Proline tetrazole induced a poor 34% ee for the same reaction.17 Nevertheless, the results of L-proline were promising, which are collected in Table 1.
Figure 1.
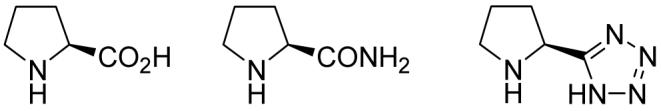
Catalysts Screened for the Cross Aldol Reaction
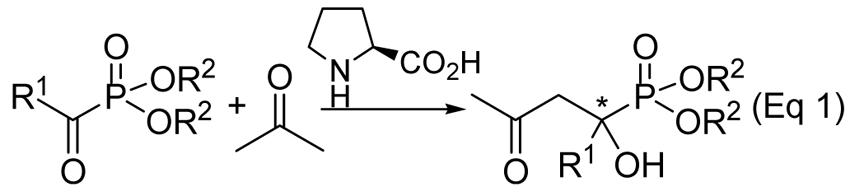
Table 1.
Enantioselective Synthesis of α-Hydroxy Phosphonatesa
 | ||||||
|---|---|---|---|---|---|---|
| entry | R1 | R2 | T (°C) |
time (h) |
yield (%)b |
ee (%)c |
| 1 | Ph | Et | rt | 24 | 85 | 71 |
| 2 | Ph | Et | −30 | 96 | 65 | 87 |
| 3 | Ph | Me | −30 | 96 | 66 | 95 |
| 4 | Ph | i-Pr | −30 | 120 | 60 | 96 |
| 5 | p-Cl-C6H4 | Et | −30 | 96 | 68 | 91 |
| 6 | p-Cl-C6H4 | i-Pr | −30 | 96 | 63 | 95 |
| 7 | p-F-C6H4 | Et | −30 | 120 | 47 | 80 |
| 8 | p-F-C6H4 | i-Pr | −30 | 120 | 68d | 96 |
| 9 | p-Br-C6H4 | Et | −30 | 96 | 66 | >99 |
| 10 | p-I-C6H4 | Et | −30 | 96 | 67 | 94 |
| 11 | p-Me-C6H4 | Et | −30 | 96 | 63d | 85 |
| 12 | p-MeO-C6H4 | Et | −30 | 96 | 32d | 86 |
| 13 | Me | Et | rt | 24 | 94 | 92 |
| 14 | Me | Et | 0 | 72 | 91 | 97 |
| 15 | PhCH2 | Et | rt | 24 | 86d | 92 |
| 16 | PhCH2CH2 | Et | rt | 48 | 76 | 81 |
| 17 | trans-CH3CH=CH | Et | −30 | 108 | 67 | 98 |
All reactions were carried out with the ketophosphonate (0.50 mmol) in dry acetone (2.0 mL), with L-proline (0.10 mmol, 20 mol %) as the catalyst for the specified reaction time and temperature, unless otherwise specified.
Yield of isolated product after column chromatography.
Enantioselectivity was determined by HPLC analyses, the absolute configuration of the major enantiomer not determined.
With L-proline (0.25 mmol, 50 mol %) as the catalyst.
With 20 mol % L-proline as the catalyst, a good ee value of 71% of the desired aldol product was obtained in a yield of 85% at room temperature (entry 1). When the reaction was carried out at −30 °C, a much better ee value of 87% was obtained (entry 2). The corresponding methyl ester is even more temperature sensitive: While at room temperature a poor ee value of 35% was obtained,17 an excellent enantioselectivity of 95% was obtained at −30°C (entry 3). The iso-propyl ester also yields an excellent enantioselectivity of 96% at this subambient temperature (entry 4). Some 4-substituted benzoyl substrates were also studied for this reaction (entries 5-12). It appears that the enantioselectivity is dependent on both the R1 and R2 group. When the R2 group maintains the same (e.g., Et), the electronic nature of the para substituents plays some role on the enantioselectivity: With the exception of the fluoro substituent, electron-withdrawing groups generally perform better in enantioselectivity than electron-donating groups. Also they are more reactive. The effects of the size of the R2 group are also evident. For example, with benzoylphosphonate, the methyl and iso-propyl esters perform the best (95% and 96% ee, respectively); while for 4-fluorobenzoyl phosphonate, the iso-propyl ester produces the best enantioselectivity (96% ee). Catalyst loading was found to be critical to high enantioselectivity: With a larger 50 mol % loading, although the reaction is slightly faster, worse enantioselectivities of the products were obtained in most cases. For example, with a 20 mol % loading, an ee value of 99% was obtained for the para-bromo derivative (entry 9); similar reaction with 50 mol % loading produced a product of only 74% ee.17 Control experiments showed this was due to slow racemization of the product caused by the excessive catalyst.
Alkyl-substituted ketophosphonates are also good substrates for this reaction. Diethyl acetylphosphonate yields the aldol product in excellent yield with a high ee value of 92% (entry 13) at room temperature. The ee improved to 97% when the temperature was lowered to 0 °C (entry 14). Similarly, phenylacetylphosphonate also produces excellent ee value of the product (92% ee, entry 15). However, when the chain length was further extended to benzylacetyl, a significant drop of the enantioselectivity (to 81%) was observed (entry 16).
α-Hydroxy phosphonates with unsaturated side-chain are very useful,13 as the side chain can be readily elaborated to introduce other functional groups. Thus, we also briefly studied the aldol reaction of diethyl trans-2-butenoylphosphonate. The reaction proceeded smoothly at −30 °C, and the desired aldol product was obtained in 67% yield and 98% ee (entry 17). Although for this specific substrate Michael addition is possible, no such product was observed in the crude reaction mixture.
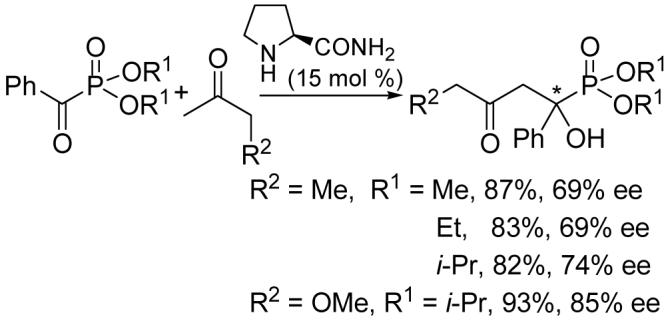
Ketones like 2-butanone and methoxyacetone also participate in this reaction if L-prolinamide is used as the catalyst. Such a cross aldol reaction with benzoylphosphonates produced only one regioselective product in good enantioselectivities (Scheme 1).
Scheme 1.
In summary, we have developed an organocatalytic highly enantioselective cross aldol reaction of α-ketophosphonates and ketones for the highly enantioselective synthesis of optically active tertiary α-hydroxyphosphonates. This method can be applied to α-ketophosphonates with alkyl, aryl, and alkenyl substituents.
Supplementary Material
Acknowledgement
The authors thank the Welch Foundation (Grant No. AX-1593) and the NIH-MBRS program (Grant No. S06 GM 008194) for the generous financial support of this project.
Footnotes
Supporting Information Available: Detailed experimental procedures, NMR spectra for all new compounds and HPLC analysis data. This material is available free of charge via Internet at http://pubs.acs.org.
References
- 1.For review, see: Kolodiazhnyi OI. Tetrahedron: Asymm. 2005;16:3295–3340.
- 2.(a) Dellaria JF, Jr., Maki RG, Stein HH, Cohen J, Whittern D, Marsh K, Hoffman DJ, Plattner JJ, Perun TJ. J. Med. Chem. 1990;33:534–542. doi: 10.1021/jm00164a011. [DOI] [PubMed] [Google Scholar]; (b) Tao M, Bihovsky R, Wells GJ, Mallamo JP. J. Med. Chem. 1998;41:3912–3916. doi: 10.1021/jm980325e. [DOI] [PubMed] [Google Scholar]
- 3.Stowasser B, Budt K-H, Li J-Q, Peyman A, Ruppert D. Tetrahedron Lett. 1992;33:6625–6628. [Google Scholar]
- 4.Snoeck R, Holy A, Dewolf-Peeters C, Van Den Oord J, De Clercq E, Andrei G. Antimicrob. Agents Chemother. 2002;46:3356–3361. doi: 10.1128/AAC.46.11.3356-3361.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Peters ML, Leonard M, Licata AA. Clev. Clin. J. Med. 2001;68:945–951. doi: 10.3949/ccjm.68.11.945. [DOI] [PubMed] [Google Scholar]; (b) Leder BZ, Kronenberg HM. Gastroenterology. 2000;119:866–869. doi: 10.1053/gast.2000.17841. [DOI] [PubMed] [Google Scholar]
- 6.For review, see: Kafarski P, Lejczak B. J. Mol. Cat. B: Enzym. 2004;29:99–104.
- 7.(a) Li Y-F. Tetrahedron: Asymm. 1993;4:109–120. [Google Scholar]; (b) Drescher M, Li Y-F, Hammerschmidt F. Tetrahedron. 1995;51:4933–4946. [Google Scholar]; (c) Drescher M, Hammerschmidt F, Kahling H. Synthesis. 1995:1267–1272. [Google Scholar]; (d) Wuggenig F, Hammerschmidt F. Monatsh. Chem. 1998;129:423–436. [Google Scholar]; (e) Khushi T, O'Toole KJ, Sime JT. Tetrahedron Lett. 1993;34:2375–2378. [Google Scholar]
- 8.(a) Brzezinska-Rodak M, Zymanczyk-Duda E, Kafarski P, Lejczak B. Biotechnol. Prog. 2002;18:1287–1291. doi: 10.1021/bp0200090. [DOI] [PubMed] [Google Scholar]; (b) Maly A, Lejczak B, Kafarski P. Tetrahedron: Asymm. 2003;19:1019–1024. [Google Scholar]
- 9.For reviews, see: Wiemer DF. Tetrahedron. 1997;53:16609–16644.Gröger H, Hammer B. Chem. Eur. J. 2000;6:943–948.
- 10.(a) Meier C, Laux WHG. Tetrahedron: Asymm. 1996;7:89–94. [Google Scholar]; (b) Meier C, Laux WHG. Tetrahedron: Asymm. 1995;6:1089–1092. [Google Scholar]; (c) Meier C, Laux WHG. Tetrahedron. 1996;52:589–598. [Google Scholar]; (d) Gajda T. Tetrahedron:Asymm. 1994;5:1965–1972. [Google Scholar]; (e) Nesterov V, Kolodyazhnyi OI. Russ. J. Gen. Chem. 2005;75:1161–1162. [Google Scholar]
- 11.(a) Pogatchnik DM, Wiemer DF. Tetrahedron Lett. 1997;38:3495–3498. [Google Scholar]; (b) Cermak DM, Du Y, Wiemer DF. J. Org. Chem. 1999;64:388–393. [Google Scholar]; (c) Skropeta D, Schmidt RR. Tetrahedron: Asymm. 2003;14:265–273. [Google Scholar]
- 12.For examples, see: Wroblewski AE, Balcerzak KB. Tetrahedron:Asymm. 2001;12:427–431.Yokomatsu T, Yamagishi T, Shibuya S. Tetrahedron: Asymm. 1993;4:1401–1404.Rowe BJ, Spilling CD. Tetrahedron:Asymm. 2001;12:1701–1708.Arai T, Bougauchi M, Sasai H, Shibasaki M. J. Org. Chem. 1996;61:2926–2927. doi: 10.1021/jo960180o.
- 13.For an example of the synthesis of racemic tertiary α-hydroxy phosphonates via allylation, see: Wiemer DF, Kim DY. Tetrahedron Lett. 2003;44:2803–2805.
- 14.(a) Enders D, Grondal C. Angew. Chem., Int. Ed. 2005;44:1210–1212. doi: 10.1002/anie.200462428. [DOI] [PubMed] [Google Scholar]; (b) Luppi G, Cozzi PG, Monari M, Kaptein B, Broxterman QB, Tomasini C. J. Org. Chem. 2005;70:7418–7421. doi: 10.1021/jo050257l. [DOI] [PubMed] [Google Scholar]; (c) Shen Z, Li B, Wang L, Zhang Y. Tetrahedron Lett. 2005;46:8785–8788. [Google Scholar]; (d) Tokuda O, Kano T, Gao W-G, Ikemoto T, Maruoka K. Org. Lett. 2005;7:5103. doi: 10.1021/ol052164w. [DOI] [PubMed] [Google Scholar]; (e) Tang Z, Cun L-F, Cui X, Mi A-Q, Jiang Y-Z, Gong L-Z. Org. Lett. 2006;8:1263. doi: 10.1021/ol0529391. [DOI] [PubMed] [Google Scholar]; (f) Samanta S, Zhao C-G. Tetrahedron Lett. 2006;47:3383. [Google Scholar]
- 15.For examples, see: Maeda H, Takahashi K, Ohmori H. Tetrahedron. 1998;54:12233–12242.Afarinkia K, Twist AJ, Yu H.-w. J. Organomet. Chem. 2005;690:2688–2691.Afarinkia K, Twist AJ, Yu H.-w. J. Org. Chem. 2004;69:6500–6503. doi: 10.1021/jo049133j.
- 16.DMF, DMSO, CH2Cl2 are worse solvents for this reaction.
- 17.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.