

Abstract
Targeted delivery of therapeutics possesses the potential to localize therapeutic agents to a specific tissue as a mechanism to enhance treatment efficacy and abrogate side effects. Antibodies have been used clinically as therapeutic agents and are currently being explored for targeting drug-loaded nanoparticles. Peptides such as RGD peptides are also being developed as an inexpensive and stable alternative to antibodies. In this study, cyclo(1,12)PenITDGEATDSGC (cLABL) peptide was used to target nanoparticles to human umbilical cord vascular endothelial cells (HUVEC) monolayers that have upregulated intercellular cell-adhesion molecule-1 (ICAM-1) expression. The cLABL peptide has been previously demonstrated to possess high avidity for ICAM-1 receptors on the cell surface. Poly(DL-lactic-co-glycolic acid) nanoparticles conjugated with polyethylene glycol and cLABL demonstrated rapid binding to HUVEC with upregulated ICAM-1, which was induced by treating cells with the proinflammatory cytokine, interferon-γ. Binding of the nanoparticles could be efficiently blocked by pre-incubating cells with free peptide suggesting that the binding of the nanoparticles is specifically mediated by surface peptide binding to ICAM-1 on HUVEC. The targeted nanoparticles were rapidly endocytosed and trafficked to lysosomes to a greater extent than the untargeted PLGA-PEG nanoparticles. Verification of peptide-mediated nanoparticle targeting to ICAM-1 may ultimately lead to targeting therapeutic agents to inflammatory sites expressing upregulated ICAM-1.
Keywords: nanoparticle, peptide, targeting, intercellular cell-adhesion molecule-1, poly(DL-lactic-co-glycolic acid)
Introduction
A variety of drug treatments have been investigated in recent decades outlining the potential of targeting specific cytokines or cell adhesion molecules that facilitate leukocyte recruitment in inflammatory diseases (1–4). In particular, monoclonal antibodies (mAbs) have been developed to target and bind molecules to disrupt the inflammatory pathway. However, the application of mAbs may be limited by immunogenicity, production cost, low physicochemical stability, and short in vivo half-life. As an alternative, nanoparticles may be targeted to a specific tissue with up-regulated cell adhesion molecules such as intercellular cell-adhesion molecule-1 (ICAM-1), which is dramatically up-regulated (~10 fold or more) on the vascular endothelium in response to the increased production of proinflammatory cytokines such as interleukin-1-β, interferon-γ, and tumor necrosis factor-α (5). Recent studies have indicated that coupling anti-ICAM-1 mAbs to the surface of nanoparticles facilitated binding and internalization on the vascular endothelium (6, 7). In light of these findings, drugs encapsulated into nanoparticles and targeted to ICAM-1 may have the potential to pool to sites of ICAM-1 upregulation for a localized therapeutic effect (7).
Inhibition of ICAM-1 binding to leukocytes using mAbs has been shown to suppress T-cell activation (8, 9). In addition, therapies combining drugs such as methotrexate with mAbs against proinflammatory cytokines (e.g. TNF-α) have shown great promise in halting the progression of joint deterioration (10, 11). A variety of therapeutic strategies utilizing mAbs against CD4, CD5, CD7, CD25 and CD52 have also been investigated in an effort to interfere with the activation of CD4+T cells, thus, blocking inflammation (1, 12, 13). Previously, Siahaan and others found linear and cyclic peptides derived from the α- and β-subunits of LFA-1 that can inhibit homotypic and heterotypic T-cell adhesion as well as mixed lymphocyte reaction (14). The peptide cyclo(1,12)PenITDGEATDSGC (cLABL) demonstrated the highest avidity for domain-1 (D1) of ICAM-1 and can block ICAM-1-mediated T-cell adhesion. In addition, this peptide was internalized by ICAM-1 into the cytoplasmic domain of activated T cells and endothelial cells (14). cLABL, has also been shown to provide therapeutic benefit by mitigating T-cell adhesion in the pancreatic microvasculature (15). Further enhancement of the demonstrated therapeutic efficacy may be afforded by multivalent peptide/receptor interactions (e.g., on a nanoparticle surface) coupled with sustained, localized release of appropriate therapeutics.
Nanoparticles have garnered attention for use as delivery vehicles for therapeutic drugs since they can be designed to slip between intercellular spaces, enter cells, or transport directly through biological barriers to access disease sites (16–18). Nanoparticles also encapsulate therapeutic agents offering potential protection from enzymatic degradation, metabolism, and filtration. Choosing the proper particle-forming materials allows controlled release of drug over time or in response to a biological cue (19, 20). Delivering drugs in this manner also allows functionalization of the nanoparticle surface without compromising the activity of the drug itself; often a problem when covalently bonding targeting ligands to therapeutics as a prodrug strategy.
Surface modification of nanoparticles is a key requisite for extending circulation half-life and promoting localization. For example, nanoparticles coated with a highly cationic polymer have been used to enhance cellular uptake or open intercellular tight junctions (16, 21). Recently, folate receptors over-expressed on the surface of malignant human cells were targeted by grafting folate on the surface of nanoparticles (22). Studies revealed that the nanoparticles attained a 10-fold higher affinity for the surface folate binding protein than free folate (23). Researchers reasoned that the multivalent form of folate on the nanoparticle surface interacted strongly with folate receptors, which are often present in clusters on the surface of cancer cells, similar to the clustering of ICAM-1 during T-cell adhesion. Finally, research efforts are ongoing to improve nanoparticle performance in vivo by extending nanoparticle circulation and limiting interaction with blood constituents (17, 24–26).
In this report, cLABL was conjugated to the surface of biodegradable PLGA nanoparticles to target ICAM-1 on the surface of human umbilical cord vascular endothelial cells (HUVEC). The amino terminus of modified polyethylene glycol was conjugated to carboxylic acid groups on PLGA nanoparticles via a peptide bond. The density of carboxyl groups on the nanoparticle surface was augmented using poly(ethylene-maleic acid) (PEMA) as a surfactant based on methods recently described by Saltzman and others (27). Particles were further modified by conjugating the N-terminus of cLABL to the carboxylic acid group of PEG on the nanoparticle surface. The cLABL nanoparticles demonstrated rapid binding to HUVEC in comparison to PLGA-PEG particles. Binding of cLABL nanoparticles was enhanced upon upregulation of ICAM-1 receptors by interferon-γ and was interrupted by incubating HUVEC with free cLABL peptide prior to nanoparticle introduction. Labeling lysosomes revealed that cLABL nanoparticles trafficked to lysosomes and accumulated to a much greater extent than PLGA-PEG nanoparticles. This research provides evidence that cLABL can be rapidly and specifically targeted to cells with upregulated ICAM-1, thus, offering the potential for site specific drug delivery.
Experimental Procedures
Materials
Poly(DL-lactic-co-glycolic acid) (50:50) (PLGA, inherent viscosity 0.89, Mw ~150 kDa) was purchased from Absorbable Polymers. Poly(ethylene-maleic anhydride) (PEMA), was purchased from Polysciences, Inc. Fmoc-PEG3400-COOH was purchased from Nektar (San Carlos, CA). Alexa Fluor 488, LysoTracker® Red, and Texas Red® (10,000 kDa, lysine fixable) were purchased from Molecular Probes. Human interferon-γ (hIFN-γ, 100,000 U) was purchased from Roche Applied Science. Six-well glass bottom microwell dishes were purchased from MatTek corporation and 8-well BD Falcon* Culture Slides and 4% paraformaldehyde were purchased from Fisher Scientific. Acrodisc® Syringe Filters with Supor® PES Membrane were acquired from VWR international, Inc. Dulbecco’s modified Eagle’s medium and Triton® X-100 were purchased from Sigma Aldrich. Penicillin-streptomycin, non-essential amino acids, and fetal bovine serum (FBS) were purchased from Invitrogen. Float-A-Lyzers (MWCO 500) were purchased from Spectrum labs. Micro BCA protein assay kit was purchased from Pierce Chemical Company.
Preparation of PLGA nanoparticles
PLGA nanoparticles were prepared by a solvent diffusion method. Briefly, 50 mg PLGA was dissolved in 3.0 mL acetone and the PLGA solution was added into 0.2% PEMA (30 mL) through a syringe pump (20 mL/h) under stirring at 200 rpm in a hood to evaporate acetone. PEMA was utilized as a surfactant to increase the number of carboxyl groups on the particle surface. Saltzman and others have previously reported this method to form stable antibody conjugates on PLGA particles (27). Produced nanoparticles were collected by centrifugation (15,000 rpm, 45 min). The nanoparticles were washed using double-distilled water three times. The sizes and zeta potentials of the PLGA nanoparticles were determined using a ZetaPALS dynamic light scattering system (Brookhaven, ZetaPALS).
Synthesis of PEGylated-PLGA nanoparticles and cLABL conjugation
Deprotection of the amine of Fmoc-PEG3400-COOH was accomplished by stirring Fmoc-PEG3400-COOH in 1.5 mL of 20% piperidine in DMF for 2 h at room temperature. The solution was transferred to polypropylene centrifuge tubes and water was added. Centrifugation and filtration (MWCO 5,000) resulted in essentially complete Fmoc removal. The solution was further subjected to extensive dialysis in 500 MWCO Float-A-Lyzers before being lyophilized to dryness.
Five milligrams of PLGA nanoparticles in suspension (~0.2 mg/mL in double-distilled water) were incubated with 23 mg N-hydroxysuccinimide (NHS, 0.2 mmol), the pH was adjusted to 5.80, and then 153 mg of 1-(3-dimethylaminopropyl)-3-ethylcarbodimide hydrochloride (EDC, 0.8 mmol) was added and incubated for 2 hr at room temperature with gentle stirring. The resulting NHS-activated particles were covalently linked to 10 mg NH2-PEG-COOH. The resulting PLGA-PEG-COOH nanoparticles were again washed, resuspended, and preserved in suspension form in double-distilled water. PLGA-PEG-COOH nanoparticles were activated by NHS as described above. The resulting NHS-activated particles were covalently linked to 5 mg cLABL peptide or 0.2 mg Alexa 488. PLGA-PEG-Alexa 488 particles served as control particles for all studies. The resulting PLGA-PEG-peptide-COOH nanoparticles were again activated by NHS as described above. The resulting NHS-activated particles were covalently linked to 0.2 mg Alexa 488.
Quantification of PEG and cLABL peptide on the surface of nanoparticles
The unreacted PEG or the unreacted cLABL peptide was separated from nanoparticles after the corresponding reaction step by centrifugation at 15,000 rpm for 45 min. The amount of free PEG or cLABL peptide in the supernatant was determined using 2,4,6-trinitro-benzenesulphonic acid (TNBS) as a colorimetric assay (28). Seven hundred microliters of aqueous solution containing free PEG or cLABL peptide was added to 700 μL of 0.1 M sodium borate buffer (pH 9.2). Three hundred and fifty microliters of TNBS aqueous solution (1.65 mg/mL) was added and the solution was rapidly mixed. After incubation at 40°C for 45 min, the reaction was stopped by adding 350 μL of 0.1 M NaH2PO4 containing 1.5 mM Na2SO3 and absorption at 420 nm was determined on a UV/VIS spectrometer (Agilent 8453). The amounts of PEG and cLABL peptide on the surface of nanoparticles were calculated by subtracting the free amount from the total amount added into the reaction system.
The amount of Alexa 488 on the surface of nanoparticles was analyzed by the fluorescence intensity of nanoparticles using a fluorescence plate reader (SpectraMax M5, Ex. 490 nm, Em. 520 nm). Standard curves were obtained by diluting the free Alexa 488 with blank PLGA-PEMA nanoparticles (size = 266 nm) over a concentration range from 0.01–0.1 μg/mL (r = 0.9991).
Binding and uptake of nanoparticles in HUVEC culture
Pooled HUVEC were grown in DMEM medium supplemented with 10% FBS, 0.1% non-essential amino acids, and 0.1% penicillin-streptomycin and were maintained at a density of 1 × 105 to 1 × 106 cells/mL at 37°C in a humidified 5% CO2 atmosphere. HUVEC were plated in 8-well BD Falcon* Culture Slides at a density of 1.0 × 105 cells/mL and used for uptake studies on day 2. Freshly prepared nanoparticles were diluted with serum free medium to give a concentration of 0.3 mg/mL. Two hundred microliters of the nanoparticle suspension was added into each well. The cells were incubated with nanoparticles for different time periods to allow surface binding, washed three times with serum free medium, incubated at 37°C or 4°C for different time periods, washed three times with ice cold PBS, and fixed with 3.7% paraformaldehyde at room temperature.
For studies using free peptide as an inhibitor, HUVEC monolayers were pre-incubated for 30 minutes at 37°C in the presence of different concentrations of free cLABL peptide, washed three times with serum free media, and incubated with nanoparticles as described above. At varying periods of time, the cell monolayers were fixed, lysed, and the fluorescence was determined. For ICAM-1 up-regulation studies, HUVEC were activated with hIFNγ for 16–18 h and then washed three times with serum free media, and incubated with nanoparticles as described above for uptake studies.
Quantification of nanoparticle interaction with HUVEC
Above experiments were terminated by washing the cells three times with ice-cold phosphate-buffered saline (PBS; 8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, and 0.24 g/L KH2PO4 in water at pH 7.4) and solubilizing the cells with 1 mL of 0.5% Triton X-100 in 0.2 M NaOH. Nanoparticle content was quantified by analyzing the cell lysate using a fluorescence plate reader (SpectraMax M5, Ex. 490 nm, Em. 520 nm). Uptake was normalized to the amount of cellular protein. The protein content of the cell lysate was determined using the Micro BCA protein assay kit. The plate reader was calibrated with standard preparations of nanoparticles, which were diluted with a cell lysate solution prepared by solubilizing 2 × 105 HUVEC in 1 mL of 0.5% Triton X-100/0.2 N NaOH solution. Linear calibration curves (r = 0.9991) over the nanoparticle concentration range of 6 to 48 μg/ml were obtained.
Intracellular trafficking of nanoparticles
To follow intracellular trafficking of nanoparticles, HUVEC were incubated for 2 hr at 37°C with Lysotracker to label lysosomes, washed three times with serum free medium, and incubated with nanoparticles as described above for uptake studies. In brief, freshly prepared nanoparticles were diluted to 0.3 mg/mL with serum free medium. Two hundred microliters of the nanoparticle suspension was added into each well. The cells were incubated with nanoparticles for 10 min to allow surface binding, washed three times with serum free medium, incubated at 37°C. At varying periods of time after internalization, the cells were washed three times with ice cold PBS, and fixed with 3.7% paraformaldehyde at room temperature. Then, the co-localization of nanoparticles within Lysotracker-labeled cellular compartments was determined. The results were confirmed by labeling lysosomes with Texas Red dextran (10,000 kDa) following a similar protocol (7, 29).
Results
Preparation of nanoparticles
As previously described by Saltzman and others (27), multiple molecules were conjugated to PLGA nanoparticles in the presence of PEMA surfactant by sequentially activating and conjugating the available terminal carboxyl groups.. Original PLGA particles coated with PEMA were ~175 nm in size with a relatively low polydispersity (Table 1). Dynamic light scattering results were confirmed by scanning electron microscopy, which verified the narrow particle size distribution and revealed a relatively smooth surface morphology (Figure 1). Reacting the terminal amine of PEG to PEMA carboxyl groups resulted in a slight increase in the average particle size and polydispersity. Further addition of the fluorescent dye, Alexa 488, or cLABL produced a further increase in average particle size and polydispersity. This trend continued when linking Alexa 488 to the carboxyl terminus of cLABL. Increasing particle size, presumably due to particle agglomeration, was suspected to result from the addition of NHS when conjugating molecules since NHS presents a relatively hydrophobic intermediate on the particle surface. Sequential conjugations also demonstrated a systematic increase in nanoparticle zeta potential, presumably a result of the occupation of carboxyl sites and increasing particle size.
Table 1.
Nanoparticle properties at specified reaction points.
| PLGA1 | PLGA-PEG | PLGA-PEG-Alexa 488 | PLGA-PEG-cLABL | PLGA-PEG-cLABL-Alexa 488 | |
|---|---|---|---|---|---|
| Size (nm) | 177±11 | 202±11 | 224±14 | 244±15 | 268±19 |
| Polydispersity | 0.052 | 0.080 | 0.116 | 0.215 | 0.181 |
| Zeta potential (mV) | −40.2±3.7 | −31.4±4.0 | −15.4±1.2 | −23.3±1.0 | −8.3±0.9 |
PLGA nanoparticles fabricated using PEMA surfactant.
Figure 1.

Scanning electron micrograph of PEMA-coated PLGA nanoparticles.
The density of molecules on the nanoparticle surface was estimated after each reaction step. The total particle surface area was calculated assuming a normal gaussian particle size distribution. The concentration of molecules was determined indirectly by quantifying the amount of molecules left in solution. Appropriate controls were utilized to assure that this method was accurate (i.e. no loss of molecules through non-specific adsorption to the vial, etc.) The density of PEG on the surface of PLGA particles corresponded well with other reports (Table 2) (30, 31). In addition, the density of peptide was sufficient to facilitate particle interactions in vitro. Alexa 488 surface concentration provided significant signal for detection by fluorescent microscopy and spectroscopy.
Table 2.
Density of conjugated molecules.
| Size (nm) | Total surface area (m2/mg) | Surface PEG (pmol/cm2) | Surface peptide (pmol/cm2) | Surface Alexa 488 (pmol/cm2) | |
|---|---|---|---|---|---|
| PLGA-PEG | 202 | 22.14 | 4.0±0.8 | ||
| PLGA-PEG-Alexa 488 | 224 | 20.05 | 0.011±0.002 | ||
| PLGA-PEG-cLABL | 244 | 18.36 | 2.3±0.9 | ||
| PLGA-PEG-cLABL-Alexa 488 | 269 | 16.7 | 0.012±0.002 |
To effectively determine nanoparticle interactions with cells, colloidal stability is a key requisite. PEGylated nanoparticles with and without peptide possessed good colloidal stability in both water and serum-free cell culture media. A slight increase in particle size, polydispersity, and zeta potential was evident in media. Large precipitates were not obvious in either water or media although larger ~1 μm agglomerates were visible by fluorescence microscopy on rare occasions. Preincubation of nanoparticles with cell culture media in an ultrasound bath prior to dispersing into wells helped to mitigate the agglomeration of nanoparticles.
The cLABL peptide has been previously shown to bind to the D1 domain of ICAM-1 (32). ICAM-1 is expressed at a basal concentration of 5–10 × 104 sites/cell, although the expression may be increased to 3.5 × 106 sites/cell upon treatment of HUVEC with TNF-α (33). To determine the binding avidity to HUVEC expressing basal levels of ICAM-1, nanoparticles were incubated with HUVEC for one hour, washed, and fluorescence was quantified after 2 hours. Increasing the concentration of peptide-labeled nanoparticles resulted in a corresponding increase in the fluorescence intensity (Figure 2). The decreasing effect of increasing particle concentration may be indicative of saturation of ICAM-1 receptors or the removal of ICAM-1 from the surface via endocytosis at high nanoparticle concentrations.
Figure 2.
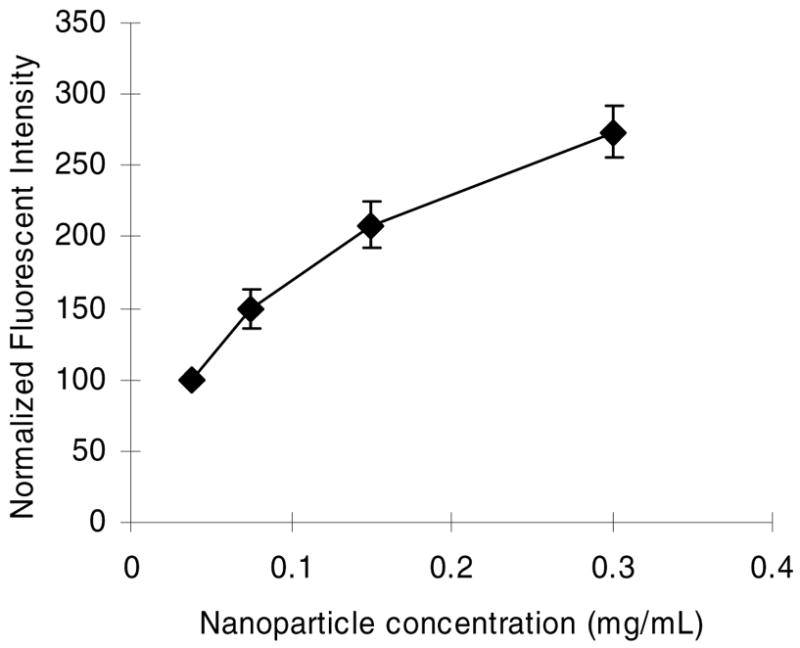
Fluorescent intensity increased in HUVEC as nanoparticle concentration increased. cLABL peptide modified PLGA nanoparticles binding time was one hour, followed by washing and incubation for 2 hours prior to cell lysis.
HUVEC were incubated with PLGA nanoparticles grafted with PEG or PEG-cLABL at 300 μg/mL to assess the specificity of binding mediated by cLABL. The presence of cLABL on nanoparticles resulted in an ~2.5-fold enhancement of the fluorescent intensity in HUVEC (Figure 3). Increasing the binding time decreased the differential between cLABL and non-specific nanoparticle interactions with HUVEC. Fluorescent micrographs were also collected over time to observe the kinetic interaction of nanoparticles with HUVEC. Rapid localization of cLABL nanoparticles to the cell periphery was observed (Figure 4). This observation was consistent with the short nanoparticle binding times determined spectroscopically. Punctate fluorescent patterns developed very quickly (~15 minutes), which suggested rapid internalization of cLABL-nanoparticles as well. Focusing on the cell midline and deconvoluting these images, that is removing of out of focus fluoresent light, provided additional evidence that fluorescent foci were indeed inside the cells. In contrast, fluorescence in PLGA-PEG nanoparticle incubations remained relatively disperse over more than 2 hours and similar localization of fluorescence was not noted until later times, providing circumstantial evidence that these particles did not enter cells to as great an extent.
Figure 3.
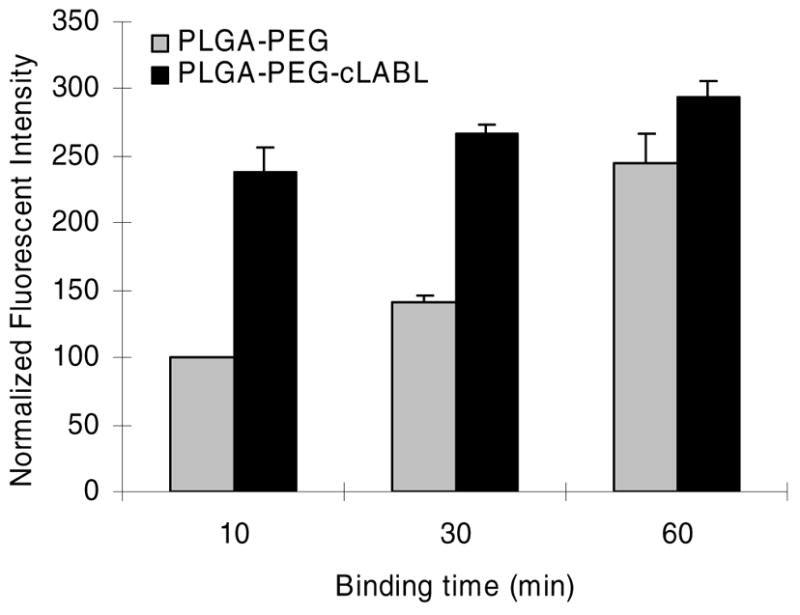
Fluorescent intensity of nanoparticles associated with HUVEC was significantly higher for targeted cLABL nanoparticles for short binding times (p<0.001 for 10 min and 30 min). Nanoparticle binding time was followed by washing and incubation for 2 hours prior to cell lysis.
Figure 4.
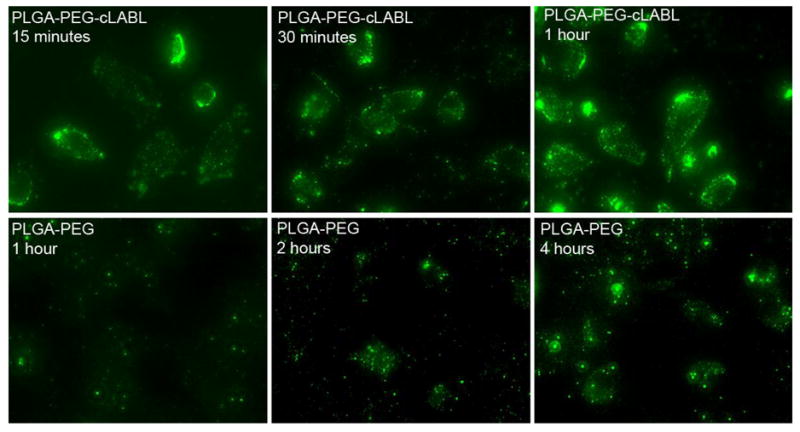
Fluorescent micrographs demonstrate that the binding of PLGA nanoparticles mediated by cLABL occurred rapidly. Significant localized fluorescence was noted at 15 minutes for cLABL nanoparticles; however, PLGA-PEG nanoparticles required >2 hours to demonstrate similar localization to cells. No significant differences in fluorescence were observable after 4 hours. Nanoparticle binding time was 10 minutes, followed by washing and incubation for 2 hours prior to cell lysis or imaging.
Since studies revealed that the binding of cLABL nanoparticles occurred rapidly, experiments were designed to further determine if nanoparticles were indeed entering cells. Reports from Muro and others have verified that ICAM-1 mediates endocytosis along a novel pathway (34) and prior work by Labhasetwar and others demonstrated the endocytosis and exocytosis of PLGA nanoparticles in multiple cell lines (35, 36). Incubating HUVEC at 4°C was utilized to minimize energy dependent endocytic pathways. Uptake of nanoparticles was effectively mitigated at low temperature. For cLABL nanoparticles association with cells was still apparent at 4°C confirming that the binding of these nanoparticles was not substantially reduced at low temperature (Figure 5).
Figure 5.
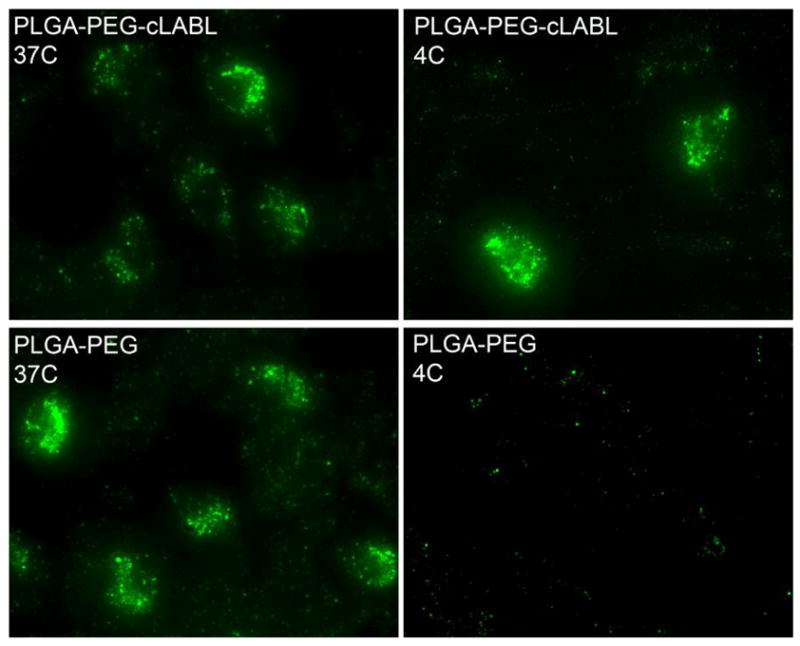
Nanoparticle binding was studied at 4°C to mitigate energy dependant endocytic pathways. Binding of cLABL nanoparticles was still apparent at low temperature Nanoparticle binding time was one hour, followed by washing and incubation for 2 hours prior to cell imaging.
Lysotracker was utilized to determine the rate at which nanoparticles transported to lysosomes in HUVEC. This dye accumulates in lysosomes, staining them with red fluorescence; therefore, nanoparticles (green) accumulating in these organelles produced a yellow color. Some punctate yellow color was noted after only 15 minutes of incubation with cLABL nanoparticles subsequent to the 10 minute binding time (Figure 6). PLGA-PEG nanoparticles remained dispersed for longer periods and did not appear in lysosomes until a minimum of 2 hours had passed. Also, PLGA-PEG nanoparticles were much more disperse in the cell culture. These studies were confirmed using a pulse-chase approach to label lysosomes with texas red dextran as reported by colleagues (7, 29). This method provided additional evidence of the rapid binding and internalization of cLABL nanoparticles. In addition, the green fluorescence of nanoparticles separated from lysosomes at later time points (Figure 7; ~6 hours.) Colocalization of nanoparticles with lysosomes was assessed semi-quantitatively by summing the number of yellow pixels from images and dividing by the number of cells in a particular field of view at selected time points. Kinetic data derived from images showed that cLABL nanoparticles appeared in lysosomes to a much greater extent than PLGA-PEG nanoparticles (Figure 8). In addition, cLABL nanoparticles steadily disappeared from lysosomes after four hours while PLGA-PEG nanoparticle accumulation continued to steadily increase. This result suggested that cLABL nanoparticles, cleaved peptide with dye, or cleaved dye may have entered a recycling endosome, a process that is known to return ICAM-1 to the cell surface (7). Alternatively, cLABL nanoparticles, cleaved peptide with dye, or cleaved dye may have escaped the endo-lysosomal pathway and entered the cytosol; however, this hypothesis was not confirmed.
Figure 6.
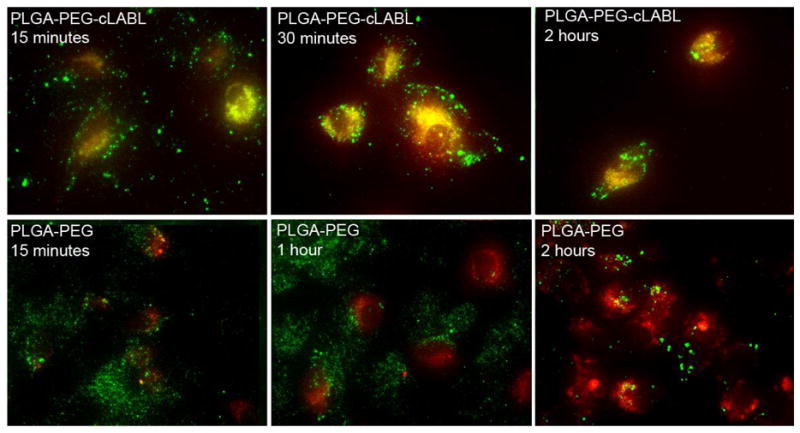
Fluorescent microscopy was utilized to assess the colocalization of nanoparticles (green) with lysosomes using Lysotracker (red). Images confirm the rapid binding of cLABL nanoparticles. In addition, cLABL nanoparticles rapidly trafficked to the lysosome with colocalization visible within 15 minutes. PLGA-PEG nanoparticles began to appear in lysosomes after two hours. Nanoparticle binding time was 10 minutes, followed by washing and incubation for different time prior to cell imaging.
Figure 7.
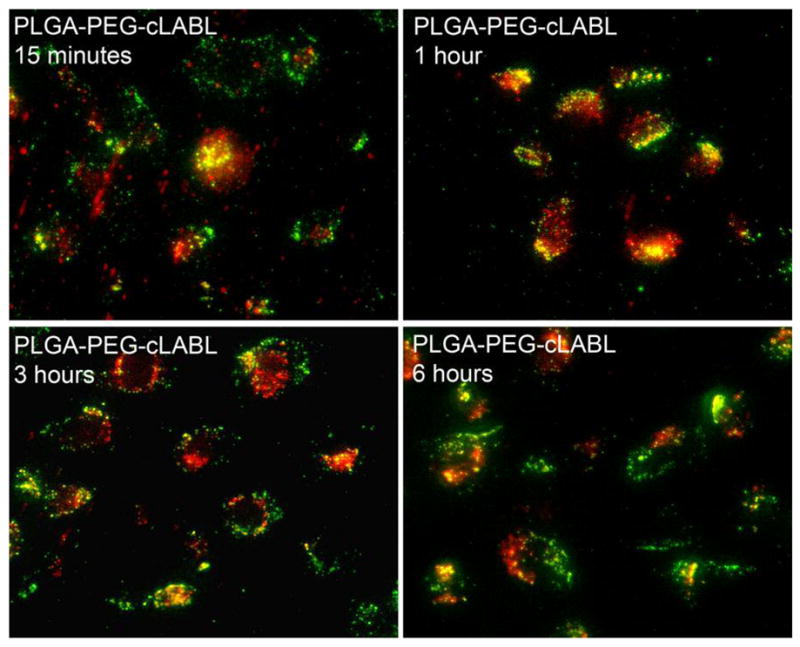
Lysosomes were labeled with fluorescent dextran (red) to confirm results obtained with Lysotracker using cLABL nanoparticles. Again, nanoparticles (green) rapidly colocalized with lysosomes and remained for about 4–6 hours. For times >4–6 hours, colocalization was no longer as apparent. Nanoparticle binding time was 10 minutes, followed by washing and incubation for different time prior to cell imaging.
Figure 8.
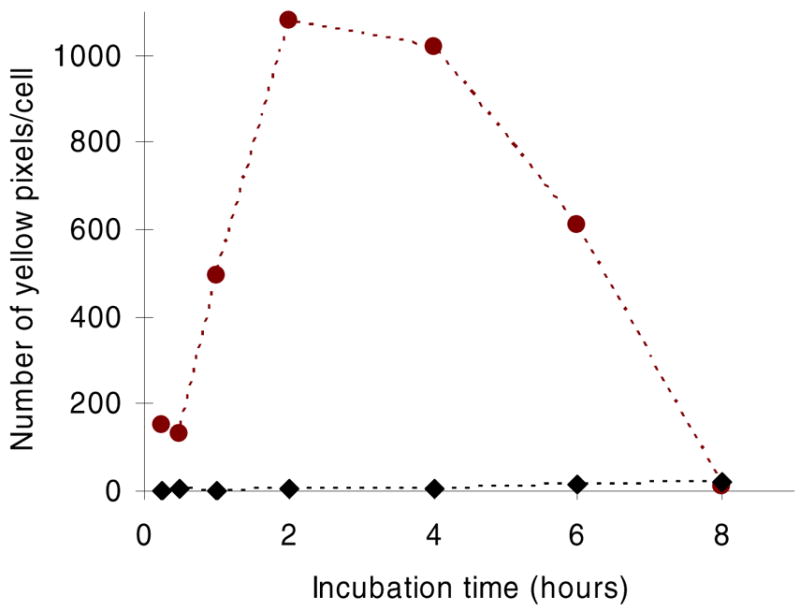
Yellow pixels signifying colocalization of nanoparticles (green) with lysosomes (red) were quantified over time demonstrating the rapid and transient localization of cLABL nanoparticles (circles) with lysosomes. Relatively fewer PLGA-PEG nanoparticles transitioned to lysosomes at later times (>2–4 hours).
HUVEC were grown as monolayers to determine the effect of receptor upregulation on nanoparticle binding. For this study, the surface of HUVEC was incubated for ~16–18 hours with different concentrations of human IFN-γ. The relative ICAM-1 concentration on the surface of HUVEC was determined using fluorescent anti-ICAM-1 mAbs and normalized to basal concentrations of ICAM-1 on control cells (Figure 9A). ICAM-1 concentration on the surface of HUVEC could be enhanced more than 2 fold. Upreg ulated ICAM-1 significantly enhanced the binding of cLABL nanoparticles (Figure 9B). The level of enhancement of nanoparticle interaction with HUVEC corresponded well to the relative enhancement of ICAM-1 expression induced by INF-γ. In contrast, PLGA-PEG nanoparticles showed no sensitivity to ICAM-1 expression levels. Studies were conducted using a long nanoparticle binding time (~1 hour) to achieve similar fluorescence levels of targeted and untargeted nanoparticles at basal ICAM-1 expression levels (see Figure 2). Binding studies using upregulated ICAM-1 receptors were also verified by fluorescence microscopy; in this case, the overall fluorescence for cLABL formulations was enhanced as ICAM-1 expression increased on the cell surface (data not shown).
Figure 9.
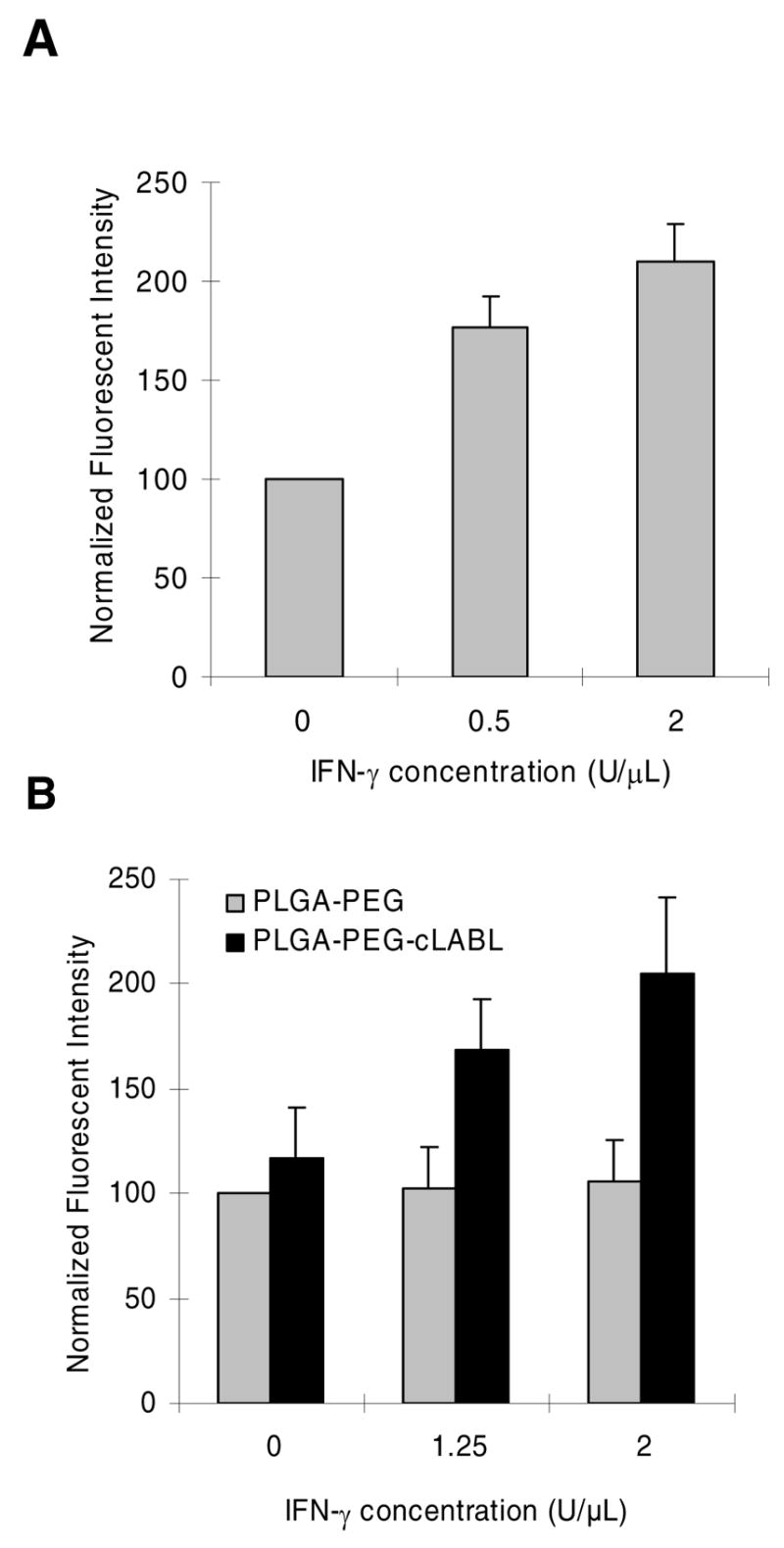
(A) Fluorescence spectrophotometry was used to quantify ICAM-1 expression in response to INF-γ and normalized to basal ICAM-1 expression (blank). (B) Fluorescent spectrophotometry demonstrated that fluorescent intensity increased in HUVEC as IFN-γconcentration increased (p<0.001 for 1.25 U/μL and 2U/μL). Nanoparticle binding time was one hour followed by washing and incubation for 2 hours prior to cell lysis or imaging.
Free cLABL peptide was utilized as a competitive inhibitor of cLABL-modifed nanoparticle binding to HUVEC. Free peptide was incubated at various concentrations with cells prior to introduction of nanoparticles to the media. The extended nanoparticle binding time (1 hour) was again selected to minimize differentials between particle types attributable to binding kinetics (see Figure 3). Increasing free peptide concentration resulted in a corresponding decrease in fluoresence intensity, thus, even for long binding times, inhibition of nanoparticle-cell interactions was evident (Figure 10). Drammatic inhibition of the uptake of cLABL nanoparticles suggests a reduced role for other endocytic pathways in the uptake of these drug delivery vehicles. In a separate experiment, fluorescent micrographs confirmed the trend of free peptide inhibition of cLABL nanoparticle binding (data not shown). No inhibition was evident in PLGA-PEG nanoparticles via spectroscopic measurement of cell lysate fluorescence or in fluorescent micrographs.
Figure 10.

Fluorescent spectrophotometry demonstrates that binding of cLABL nanoparticles was inhibited by pre-incubating HUVEC with free peptide (p<0.001 for 0.5 U/μL and 2 U/μL). PLGA-PEG nanoparticle binding was unaffected. Nanoparticle binding time was 1 hour, followed by washing and incubation for 2 hours prior to cell lysis or imaging.
Discussion
Therapeutic interventions stand to benefit greatly from site specific drug targeting. Substantial research efforts have focused on targeting molecular markers in cancer, infectious disease, and other diseases. The vascular endothelium remains a prime target for localizing drug delivery, and ICAM-1 is an attractive receptor for therapeutic interventions (37) and for targeting therapeutic drug carriers to vascular endothelium because it has shown to be upregulated during a variety of disease states including infections, injuries, ischemia, autoimmune diseases, and certain cancers (38–40). In addition, localized upregulation of ICAM-1 facilitates recruitment of leukocytes suggesting that circulating drug carriers may selectively pool at these regions.
The cLABL peptide was derived from the I-domain of the α-subunit of LFA-1. This peptide has been shown to inhibit ICAM-1/LFA-1-mediated homotypic (41) and heterotypic (42)n T-cell adhesion. Interestingly, the peptide blocked adhesion of T cells but not monocytes to pancreatic islet microvascular endothelium, thus, demonstrating specificity (43). Recently, the cLABL peptide was shown to block the formation of microtubule-organizing center at the cell-cell contact region of human T-lyphotropic virus (HTLV-1)-infected T cells, suggesting that cLABL can be used to inhibit the spreading of HLTV-1 from infected to non-infected human T cells (44).The mechanism of activity of this peptide is partly due to its binding to ICAM-1 at the LFA-1 binding site. Using monoclonal antibodies, it has been shown that cLABL peptide binds to the D1 domain of ICAM-1 (32). The peptide has been specifically designed with a stable conformation including a β-II′ turn at Asp4-Gly5-Glu6-Ala7 and a β-I turn at Pen1-Ile2-Thr3-Asp4 as determined by NMR spectroscopy (45). Molecular docking experiments suggest that this peptide binds to ICAM-1 at F and C strands of D1 domain (45). By itself, fluorescent-labeled cLABL peptide was internalized into activated Molt-3 T cells, presumably by ICAM-1 receptors (32). Recently, we have shown that the Ile2, Asp4, and Glu6 residues in cLABL and the β-turn structure at Asp4-Gly5-Glu6-Ala7 are important for the peptide activity. In addition, a cyclic hexapeptide, cyclo(1,6)ITDGEA, derived from cLABL has been shown to have comparable activity with the parent cLABL; this cyclic peptide is currently being optimized by mutating some of its residues. _In the future, this smaller cyclic peptide may also be used to target drugs or nanoparticles to cells with high expression of ICAM-1 as shown here.
Multiple groups have validated the potential of utilizing ICAM-1 as a molecular marker for targeting therapeutic particles. Eniola and others have reported the importance of multiple ligand-receptor interactions in achieving firm adhesion of particles in flow fields (3, 46). Elegant work by Muro and others has also established ICAM-1 as a valid target for delivering therapeutic nanoparticles. They demonstrated that nanoparticles conjugated with anti-ICAM-1 mAbs were rapidly bound and endocytosed by HUVEC via a novel endocytic pathway (6). Targeting the same receptor, cLABL nanoparticles reported here were also observed to bind rapidly and traffic to lysosomes in ~1–2 hours. Any minor differences in the trafficking kinetics of these two nanoparticle types may be due to the surface concentration of ligand on nanoparticles and/or the relative avidity of targeting molecules (mAbs versus peptides.) Perhaps, the lower density and lower binding strength of cLABL to ICAM-1 may lead to easier disruption of binding events and a slightly more rapid transit to lysosomal compartments. It is also possible that cluster formation of ICAM-1 receptors on the cell surface during binding to cLABL-modified nanoparticles is different than that of anti-ICAM-1-modified particles.
In this work, we have successfully modified the surface of nanoparticles with cLABL peptide to target the vascular endothelium with upregulated ICAM-1. The cLABL peptide effectively mediated rapid nanoparticle binding and internalization. Binding was enhanced upon upregulation of ICAM-1 on HUVEC and this binding could be specifically inhibited by blocking ICAM-1 receptor sites with free cLABL peptide. These results suggest that the cLABL-nanoparticle conjugates may have dual therapeutic applications, namely, inhibition of the infiltration of immune cells via blockage or internalization of ICAM-1 receptors coupled with the ability to localize drug delivery to regions of ICAM-1 upregulation.
Table 3.
Nanoparticle colloidal stability.
| Sizes (nm) | Polydispersity | Zeta potential (mV) | |
|---|---|---|---|
| PLGA-PEG-cLABL- Alexa 488 in water | 269±19 | 0.181 | −8.3±0.9 |
| PLGA-PEG-Alexa 488 in water | 224±14 | 0.116 | −15.4±1.2 |
| PLGA-PEG-cLABL- Alexa 488 in media | 292±8.9 | 0.219 | −13.4±1.7 |
| PLGA-PEG-Alexa 488 in media | 272±13 | 0.141 | −21.3±2.2 |
Acknowledgments
The studies were funded by grants from the Juvenile Diabetes Research Foundation, the American Heart Association, and the NIH (1R03 AR054035-01A1, P20 RR016443 and P20 RR015563). Our sincere thanks is given to the China Scholarship Council in the People’s Republic of China for partial support of Na Zhang as a postdoctoral researcher. Special thanks to Russ Middaugh and to Jeff Krise for the use of laboratory equipment.
References
- 1.Schulze-Koops H, Burkhardt H, Kalden JR. What we have learned from trials of immunomodulatory agents in rheumatoid arthritis: Future directions. Drugs Today (Barc) 1999;35:327–51. doi: 10.1358/dot.1999.35.4-5.552208. [DOI] [PubMed] [Google Scholar]
- 2.Schulze-Koops H, Kalden JR. Where is biological therapy going? Arthritis Research and Therapy. 2000;2:337–341. doi: 10.1186/ar108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eniola AO, Hammer DA. Characterization of biodegradable drug delivery vehicles with the adhesive properties of leukocytes II: effect of degradation on targeting activity. Biomaterials. 2005;26:661–70. doi: 10.1016/j.biomaterials.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Omolola Eniola A, Hammer DA. In vitro characterization of leukocyte mimetic for targeting therapeutics to the endothelium using two receptors. Biomaterials. 2005;26:7136–44. doi: 10.1016/j.biomaterials.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Furuzawa-Carballeda J, Rodriquez-Calderon R, Diaz de Leon L, Alcocer-Varela J. Mediators of inflammation are down-regulated while apoptosis is up-regulated in rheumatoid arthritis synovial tissue by polymerized collagen. Clin Exp Immunol. 2002;130:140–9. doi: 10.1046/j.1365-2249.2002.01955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muro S, Cui X, Gajewski C, Murciano JC, Muzykantov VR, Koval M. Slow intracellular trafficking of catalase nanoparticles targeted to ICAM-1 protects endothelial cells from oxidative stress. Am J Physiol Cell Physiol. 2003;285:C1339–47. doi: 10.1152/ajpcell.00099.2003. [DOI] [PubMed] [Google Scholar]
- 7.Muro S, Gajewski C, Koval M, Muzykantov VR. ICAM-1 recycling in endothelial cells: a novel pathway for sustained intracellular delivery and prolonged effects of drugs. Blood. 2005;105:650–8. doi: 10.1182/blood-2004-05-1714. [DOI] [PubMed] [Google Scholar]
- 8.Jois SDS, Siahaan TJ. A peptide derived from LFA-1 that modulates T-cell adhesion binds to soluble ICAM-1 protein. J Biomol Struc Dyn. 2003;20:635–644. doi: 10.1080/07391102.2003.10506880. [DOI] [PubMed] [Google Scholar]
- 9.Anderson ME, Siahaan TJ. Targeting ICAM-1/LFA-1 interaction for controlling autoimmune diseases: Designing peptide and small molecule inhibitors. Peptides. 2003;24:487–501. doi: 10.1016/s0196-9781(03)00083-4. [DOI] [PubMed] [Google Scholar]
- 10.Breedveld FC, Emery P, Keystone E, Patel K, Furst DE, Kalden JR, St Clair EW, Weisman M, Smolen J, Lipsky PE, Maini RN. Infliximab in active early rheumatoid arthritis. Ann Rheum Dis. 2004;63:149–55. doi: 10.1136/ard.2003.013961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, Harriman GR, Maini RN. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343:1594–602. doi: 10.1056/NEJM200011303432202. [DOI] [PubMed] [Google Scholar]
- 12.Burmester GR, Emmrich F. Anti-CD4 therapy in rheumatoid arthritis. Clin Exp Rheumatol. 1993;11(Suppl 8):S139–45. [PubMed] [Google Scholar]
- 13.Kalden JR, Breedveld FC, Burkhardt H, Burmester GR. Immunological treatment of autoimmune diseases. Adv Immunol. 1998;68:333–418. doi: 10.1016/s0065-2776(08)60564-7. [DOI] [PubMed] [Google Scholar]
- 14.Yusuf-Makagiansar H, Anderson ME, Song X, Murray J, Siahaan TJ. Modulation of adhesion molecules in lymphocyte activation and cell-cell interaction. Curr Top Biochem Res. 2000;2:33–49. [Google Scholar]
- 15.Huang M, Matthews K, Siahaan TJ, Kevil CG. Alpha L-integrin I domain cyclic peptide antagonist selectively inhibits T cell adhesion to pancreatic islet microvascular endothelium. Am J Physiol Gastrointest Liver Physiol. 2005;288:G67–73. doi: 10.1152/ajpgi.00267.2004. [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto H, Kuno Y, Sugimoto S, Takeuchi H, Kawashima Y. Surface-modified PLGA nanosphere with chitosan improved pulmonary delivery of calcitonin by mucoadhesion and opening of the intercellular tight junctions. J Control Release. 2005;102:373–81. doi: 10.1016/j.jconrel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 18.Moghimi SM, Illum L, Davis SS. Physiopathological and physicochemical considerations in targeting of colloids and drug carriers to the bone marrow. Crit Rev Ther Drug Carrier Syst. 1990;7:187–209. [PubMed] [Google Scholar]
- 19.Na K, Lee KH, Bae YH. pH-sensitivity and pH-dependent interior structural change of self-assembled hydrogel nanoparticles of pullulan acetate/oligo-sulfonamide conjugate. J Control Release. 2004;97:513–25. doi: 10.1016/j.jconrel.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Chaw CS, Chooi KW, Liu XM, Tan CW, Wang L, Yang YY. Thermally responsive core-shell nanoparticles self-assembled from cholesteryl end-capped and grafted polyacrylamides:; drug incorporation and in vitro release. Biomaterials. 2004;25:4297–308. doi: 10.1016/j.biomaterials.2003.10.095. [DOI] [PubMed] [Google Scholar]
- 21.Prego C, Garcia M, Torres D, Alonso MJ. Transmucosal macromolecular drug delivery. J Control Release. 2005;101:151–62. doi: 10.1016/j.jconrel.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 22.Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–51. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 23.Stella B, Arpicco S, Peracchia MT, Desmaele D, Hoebeke J, Renoir M, D’Angelo J, Cattel L, Couvreur P. Design of folic acid-conjugated nanoparticles for drug targeting. J Pharm Sci. 2000;89:1452–64. doi: 10.1002/1520-6017(200011)89:11<1452::aid-jps8>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 24.Gref R, Minamitake Y, Peracchia MT, Domb A, Trubetskoy V, Torchilin V, Langer R. Poly(ethylene glycol)-coated nanospheres: potential carriers for intravenous drug administration. Pharm Biotechnol. 1997;10:167–98. doi: 10.1007/0-306-46803-4_6. [DOI] [PubMed] [Google Scholar]
- 25.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–3. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 26.Kim D, El-Shall H, Dennis D, Morey T. Interaction of PLGA nanoparticles with human blood constituents. Colloids Surf B Biointerfaces. 2005;40:83–91. doi: 10.1016/j.colsurfb.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 27.Keegan ME, Falcone JL, Leung TC, Saltzman WM. Biodegradable microspheres with enhanced capacity for covalently bound surface ligands. Macromol. 2004;37:9779–9784. [Google Scholar]
- 28.Zhang N, Ping QN, Huang GH, Xu WF. Investigation of lectin-modified insulin liposomes as carriers for oral administration. Int J Pharm. 2005;294:247–59. doi: 10.1016/j.ijpharm.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 29.Duvvuri M, Krise JP. A novel assay reveals that weakly basic model compounds concentrate in lysosomes to an extent greater than pH-partitioning theory would predict. Mol Pharm. 2005;2:440–8. doi: 10.1021/mp050043s. [DOI] [PubMed] [Google Scholar]
- 30.Vila A, Gill H, McCallion O, Alonso MJ. Transport of PLA-PEG particles across the nasal mucosa: effect of particle size and PEG coating density. J Control Release. 2004;98:231–44. doi: 10.1016/j.jconrel.2004.04.026. [DOI] [PubMed] [Google Scholar]
- 31.Popielarski SR, Pun SH, Davis ME. A nanoparticle-based model delivery system to guide the rational design of gene delivery to the liver. 1 Synthesis and characterization. Bioconjug Chem. 2005;16:1063–70. doi: 10.1021/bc050113d. [DOI] [PubMed] [Google Scholar]
- 32.Yusuf-Makagiansar H, Siahaan TJ. Binding and internalization of an LFA-1-derived cyclic peptide by ICAM receptors on activated lymphocyte: a potential ligand for drug targeting to ICAM-1-expressing cells. Pharm Res. 2001;18:329–335. doi: 10.1023/a:1011007014510. [DOI] [PubMed] [Google Scholar]
- 33.Dustin ML, Springer TA. Lymphocyte function-associated antigen-1 (LFA-1) interaction with intercellular adhesion molecule-1 (ICAM-1) is one of at least three mechanisms for lymphocyte adhesion to cultured endothelial cells. J Cell Biol. 1988;107:321–31. doi: 10.1083/jcb.107.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muro S, Schuchman EH, Muzykantov VR. Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol Ther. 2006;13:135–41. doi: 10.1016/j.ymthe.2005.07.687. [DOI] [PubMed] [Google Scholar]
- 35.Panyam J, Labhasetwar V. Dynamics of endocytosis and exocytosis of poly(D, L-lactide-co-glycolide) nanoparticles in vascular smooth muscle cells. Pharm Res. 2003;20:212–20. doi: 10.1023/a:1022219003551. [DOI] [PubMed] [Google Scholar]
- 36.Qaddoumi MG, Ueda H, Yang J, Davda J, Labhasetwar V, Lee VH. The characteristics and mechanisms of uptake of PLGA nanoparticles in rabbit conjunctival epithelial cell layers. Pharm Res. 2004;21:641–8. doi: 10.1023/b:pham.0000022411.47059.76. [DOI] [PubMed] [Google Scholar]
- 37.Arnold R, Neumann M, Konig W. Peroxisome proliferator-activated receptor-gamma agonists inhibit respiratory syncytial virus-induced expression of intercellular adhesion molecule-1 in human lung epithelial cells. Immunology. 2007;121:71–81. doi: 10.1111/j.1365-2567.2006.02539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weishaupt C, Munoz KN, Buzney E, Kupper TS, Fuhlbrigge RC. T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin Cancer Res. 2007;13:2549–56. doi: 10.1158/1078-0432.CCR-06-2450. [DOI] [PubMed] [Google Scholar]
- 39.Ramudo L, De Dios I, Yubero S, Vicente S, Manso MA. ICAM-1 and CD11b/CD18 Expression During Acute Pancreatitis Induced by Bile-Pancreatic Duct Obstruction: Effect of N-Acetylcysteine. Exp Biol Med (Maywood) 2007;232:737–43. [PubMed] [Google Scholar]
- 40.Eniola AO, Hammer DA. Artificial polymeric cells for targeted drug delivery. J Control Release. 2003;87:15–22. doi: 10.1016/s0168-3659(02)00346-2. [DOI] [PubMed] [Google Scholar]
- 41.Tibbetts SA, Jois SDS, Siahaan TJ, Benedict SH, Chan MA. Linear and cyclic LFA-1 and ICAM-1 peptides inhibit T-cell adhesion and function. Peptides. 2000;21:1161–1167. doi: 10.1016/s0196-9781(00)00255-2. [DOI] [PubMed] [Google Scholar]
- 42.Yusuf-Makagiansar H, Makagiansar IT, Siahaan TJ. Inhibition of the adherence of T-lymphocyte to epithelial cells by a cyclic peptide derived from inserted domain of lymphocyte function-associated antigen-1. Inflammation. 2001;25:203–214. doi: 10.1023/a:1011044616170. [DOI] [PubMed] [Google Scholar]
- 43.Huang M, Matthews K, Siahaan TJ, Kevil CG. Alpha L-integrin I domain cyclic peptide antagonist selectively inhibits T cell adhesion to pancreatic islet microvascular endothelium. Am J Physiol Gastrointest Liver Physiol. 2005;288:G67–73. doi: 10.1152/ajpgi.00267.2004. [DOI] [PubMed] [Google Scholar]
- 44.Barnard AL, Igakura T, Tanaka Y, Taylor GP, Bangham CR. Engagement of specific T-cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood. 2005;106:988–95. doi: 10.1182/blood-2004-07-2850. [DOI] [PubMed] [Google Scholar]
- 45.Xu CR, Yusuf-Makagiansar H, Hu Y, Jois SD, Siahaan TJ. Structural and ICAM-1-docking properties of a cyclic peptide from the I-domain of LFA-1: An inhibitor of ICAM-1/LFA-1-mediated T-cell adhesion. J Biomol Struct Dyn. 2002;19:789–799. doi: 10.1080/07391102.2002.10506785. [DOI] [PubMed] [Google Scholar]
- 46.Eniola AO, Krasik EF, Smith LA, Song G, Hammer DA. I-domain of lymphocyte function-associated antigen-1 mediates rolling of polystyrene particles on ICAM-1 under flow. Biophys J. 2005;89:3577–88. doi: 10.1529/biophysj.104.057729. [DOI] [PMC free article] [PubMed] [Google Scholar]