

Abstract
Thiazole synthase catalyzes the formation of the thiazole moiety of thiamin pyrophosphate. The enzyme from Saccharomyces cerevisiae (THI4) copurifies with a set of strongly bound adenylated metabolites. One of them has been characterized as the ADP adduct of 5-(2-hydroxyethyl)-4-methylthiazole-2-carboxylic acid. Attempts towards yielding active wild type Thi4 by releasing protein-bound metabolites have failed so far. Here we describe the identification and characterization of two partially active mutants (C204A and H200N) of THI4. Both mutants catalyzed the release of the nicotinamide moiety from NAD to produce ADP-ribose, which was further converted to ADP-ribulose. In the presence of glycine, both the mutants catalyzed the formation of an advanced intermediate. The intermediate was trapped with ortho-phenylenediamine yielding a stable quinoxaline derivative, which was characterized by NMR spectroscopy and ESI-MS. These observations confirm NAD as the substrate for THI4 and elucidate the early steps of this unique biosynthesis of the thiazole moiety of thiamin in eukaryotes.
Introduction
Thiamin pyrophosphate is an essential cofactor in all living systems where it plays an important role in amino acid and carbohydrate metabolism1, 2. Bacteria, fungi and plants can biosynthesize this cofactor whereas most other eukaryotes cannot. They need to obtain it as an essential vitamin from the diet (vitamin B1). Thiamin was the first vitamin discovered and consists of a thiazole linked to a pyrimidine. The biosynthesis of thiamin involves the separate biosyntheses of the thiazole and pyrimidine moieties, which are then coupled3-5. The biosyntheses of the thiazole moiety in prokaryotes is well characterized and involves a complex oxidative condensation of 1-deoxy-D-xylulose-5-phosphate, glycine (or tyrosine) and cysteine, which is catalyzed by five different enzymes3. In eukaryotes, the biosynthesis of the thiazole occurs by a different route, the details of which are only beginning to emerge6-8.
Extensive mutagenesis in Saccharomyces cerevisiae has resulted in the identification of only one gene required for thiazole formation (THI4)9. We recently reported the characterization of an adenylated thiazole (ADT, 15, Figure 1) tightly bound to the active site of THI4 (Figure 2, peak D)7, 8. This identification suggested NAD 1 as the probable precursor to ADT 15 and provided key insights into the mechanism of thiazole biosynthesis in eukaryotes (Figure 1). Two other major adenylated metabolites were also found tightly bound to the active site (Peak A and B, Figure 2). These could not be characterized due to their instability. Peak C was always a minor component and its abundance varied between different protein preparations. All attempts to reconstitute active THI4 by releasing the bound metabolites failed, making it impossible to experimentally confirm the novel proposal that NAD was the thiamin-thiazole precursor in S. cerevisiae.
Figure 1.

Mechanistic proposal for the THI4 catalyzed formation of ADT, 15 (Peak D, Figure 2)
Figure 2.
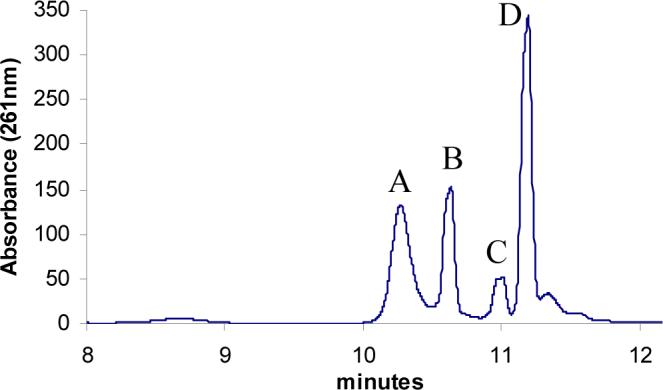
Metabolites associated with THI4 that are released upon denaturing the protein. Peak D is ADT (15). Peaks A and B were unstable and degraded during the isolation process.
In an attempt to generate product free enzyme able to catalyze the early steps of thiazole formation, several THI4 mutants were constructed based on the recently solved crystal structure8. Here we report the identification and characterization of two partially active mutants (C204A and H200N), which show informative activities. In the presence of glycine 5, these mutants catalyzed the conversion NAD 1 to one of the previously observed unstable THI4-bound metabolites (peak A, Figure 2) via the intermediacy of ADP-ribose 3 and ADP-ribulose 4 confirming NAD as the thiazole precursor. The unstable metabolite, corresponding to peak A, could be efficiently trapped with ortho-phenylenediamine (oPDA, 16) suggesting that it contains a 1, 2-diketone or equivalent functionality. A mechanistic proposal consistent with these observations is described (Figure 1).
RESULTS
Mutagenesis studies on THI4 to identify product-free partially-active mutants
Extensive mutagenesis on wild type THI4 was performed in an attempt to identify partially active mutants. Each mutant was analyzed for tightly bound metabolites by HPLC. The mutants were also incubated with NAD 1 and the reaction mixture was analyzed by HPLC. Mutants D238A, C205S, C204A and H200N were shown to degrade NAD 1 (peak G) to produce ADPr 3 (peak E), and nicotinamide 2 (peak H) as products (Figure 3 and Table 1). All the other mutants (H237N, H237A, R301Q, R301A, P304A, E97A, E97Q, and D207A) did not show any activity towards NAD and did not contain bound metabolites. These observations confirm that NAD 1 is one of the substrates for THI4 and demonstrate that the hydrolysis of NAD 1 to form ADPr 3 and nicotinamide 2 is the first step in the THI4 catalyzed biosynthesis of ADT 15.
Figure 3.
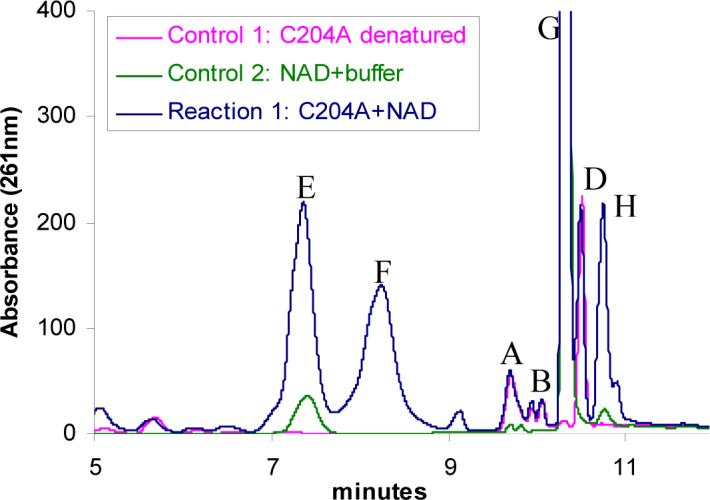
Conversion of NAD (1, peak G) to ADPr (3, peak E), nicotinamide (2, peak H) and an unknown species (peak F) catalyzed by the partially active mutant C204A. Reaction 1 represents the HPLC analysis of the reaction mixture containing the mutant C204A and NAD. Control 1 represents an experiment where the mutant C204A, with no NAD, was treated identically as reaction 1. It represents the protein bound small molecules (A, B and D). Control 2 represents an enzyme free reaction mixture where NAD was incubated in the same reaction buffer and was treated identically as for reaction 1. The condition (heat) applied to denature the protein causes minor hydrolysis of NAD to produce ADPr and nicotinamide. The ADPr and the nicotinamide peaks were identified by co-migration with ADPr and nicotinamide standards.
Table 1.
Catalytic properties of THI4 mutants.
| Mutant | Bound metabolites | Products (# turnovers after overnight incubation) |
|---|---|---|
| D238A | Wild type (A, B and D) | ADPr 3, ADPrl 4 and nicotinamide 2 (<0.1) |
| C205S | None | ADPr 3, ADPrl 4 and nicotinamide 2 (<0.1) |
| C204A | <Wild type (A, B and D) | ADPr 3, ADPrl 4 and nicotinamide 2 (∼0.8) |
| H200N | Only peak A | ADPr 3, ADPrl 4 and nicotinamide 2 (∼0.8) |
| Other Mutants* | None | None |
H237N, H237A, R301Q, R301A, P304A, E97A, E97Q, and D207A
Enzyme catalyzed generation of ADP-ribulose (ADPrl 4) from ADP-ribose (ADPr 3)
Incubation of the partially active mutants (D238A, C205S, C204A and H200N) with NAD also resulted in the formation of a new adenylated species (Figure 3, peak F) along with ADPr 3. The same adenylated species could be generated directly from ADPr 3 (peak E), by incubating ADPr with these mutants (Figure 4). A standard ADP-ribulose sample (ADPrl, 4) was shown to co-migrate with this new adenylated metabolite (Figure 4). Attempts at the direct isolation and characterization of the peak F compound failed, as the compound decomposed during the isolation process. However it wsas possible to reduce the peak F compound with NaBD4 and the resulting stable deuterated reduction product was characterized by NMR. These observations establish the isomerization of ADPr 3 to ADPrl 4 as the second step in the THI4 catalyzed biosynthesis of ADT 15 from NAD 1.
Figure 4.
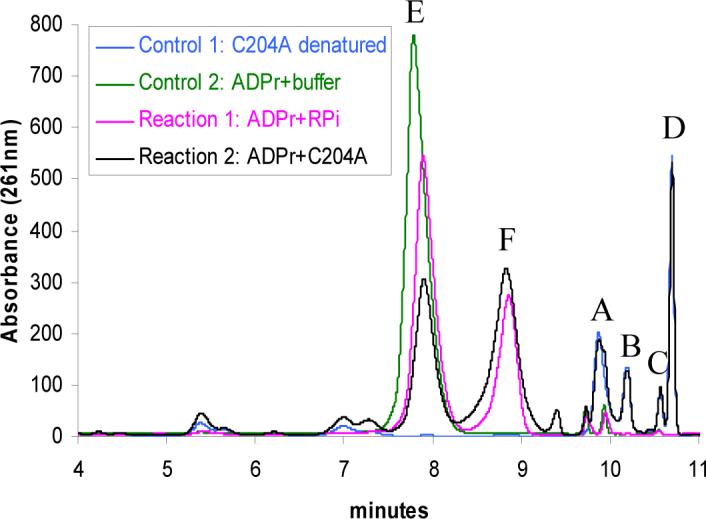
Conversion of ADP-ribose (3, peak E) to ADP-ribulose (4, peak F) catalyzed by THI4 C204A mutant and ribose-5-phosphate isomerase (RPi). Reaction 1 and 2 represent the reactions of ADP-ribose catalyzed by the RPi and C204A respectively. Control 1 represents a sample of the C204A THI4 protein which was denatured and analyzed for bound metabolites. It shows the enzyme bound metabolites A, B, C and D. Control 2 shows the ADP-ribose control where ADP-ribose was incubated with the same reaction buffer and treated identically as reaction 1 and 2.
Glycine dependent conversion of ADPr 3 and ADPrl 4 to a new species
Prolonged incubation of NAD 1 or ADPr 3 with the C204A or H200N mutants resulted in a small increase in the intensity of one of the three Thi4-bound adenylated metabolites (peak A, Figure 2). This increase varied with different protein preparations. However, in the presence of glycine 5, incubation of these mutants with NAD 1 or ADPr 3 always resulted in a significant increase in the intensity of peak A, and a concomitant decrease in the intensities of the peak E (ADPr 3) and the peak F (ADPrl 4) when compared to a similar reaction mixture without glycine (Figure 5). This observation indicates that glycine is important for the further conversion of ADPrl 4 to the advanced intermediate represented by peak A in Figure 2.
Figure 5.

The C204A mediated conversion of NAD (1, peak G) to the protein bound metabolite A, in the presence of glycine 5. HPLC chromatograms: Reaction 1 represents a reaction mixture containing C204A and NAD (no glycine). Reaction 2 represents a reaction mixture containing C204A, NAD and glycine. Control 1 represents the experiment where a sample of C204A was incubated in absence of both NAD and glycine (to reveal protein bound metabolites A, B and D). Control 2 represents the experiment where NAD and glycine were incubated in the same reaction buffer, in the absence of protein, under the same conditions as for reactions 1 and 2. Peaks for ADPr (3, peak E), ADPrl (4, peak F), nicotinamide (2, peak H), NAD (1, peak G) and protein bound molecules (peaks A, B and D) are labeled. The glycine-dependent decrease in the intensity of E and F and a concomitant increase in the intensity of A are illustrated.
Characterization of the peak A compound by trapping with ortho-phenylenediamine (oPDA)
The attempts at direct isolation and characterization of the compound eluting as peak A (Figure 2) failed due to its instability. However, incubation with ortho-phenylenediamine (oPDA, 16,) resulted in its clean conversion to a stable new species, which could be purified by HPLC (peak J, Figure 6). The other protein bound metabolites were stable under the mild trapping conditions used. This new species had absorption maxima at 260nm and 321nm, suggesting the presence of an adenine moiety as well as a putative quinoxaline moiety (Figure 7A). Treatment of this derivative with nucleotide pyrophosphatase (pptase) followed by HPLC analysis revealed the formation of AMP and another species (peak K) that had an absorption maximum only at 321nm (Figure 7B). This suggests a general structure where a quinoxaline moiety is attached to an ADP unit (structure 18, Figure 7C).
Figure 6.

Trapping of the THI4-bound metabolite A with oPDA (16, peak I) as a new species J. The blue trace represents the reaction mixture where the metabolites released from THI4 (by heat denaturing) were incubated with an excess of oPDA at room temperature. The purple trace represents a control reaction where oPDA was not added (only THI4 bound metabolites).
Figure 7.

A: UV-Vis spectrum of the quinoxaline adduct, J, formed as a result of the trapping of the THI4 bound metabolite A by oPDA, 16 (Peak I, Figure 6). B: The purified peak J compound can be cleaved with nucleotide pyrophosphatase (pptase) to produce AMP and another species K (19) that has only one absorption maximum at 321 nm. The blue and the purple traces represent the same reaction mixture containing the purified quinoxaline adduct J and pptase, observed at 261 nm and 321 nm respectively. The green trace represents a control reaction, where pptase was not added, observed at 261 nm. C: Proposed diketone (or equivalent) trapping with oPDA 16 and pyrophosphate hydrolysis with pptase.
Analysis of 1H, (1H,1H)-dqfCOSY, HMQC, and HMBC NMR spectra obtained for the Peak J compound confirmed the presence of a 5′-phosphorylated adenosine (Figure 9, 1H NMR of J). Additionally, a three proton spin system representing a -CHOH-CH2-OP fragment (CH2 at 4.24 and 4.35 ppm, m; CH at 5.40 ppm, dd), a methyl singlet (2.73 ppm) and a 1,2-disubstituted benzene (7.77, m, 1H; 7.65, m, 1H; 7.62−7.58, m, 2H) were identified. In HMBC spectra, these four aromatic protons showed correlations to two quaternary carbons (139.4 ppm and 139.8 ppm), one of which also showed a weak correlation to the methyl singlet. Furthermore, the HMBC spectra showed strong correlations of the methyl protons and the CHOH methine proton to two additional quaternary aromatic carbons (y, 153.06 ppm, and w, 152.91 ppm in structure 20, Figure 8). As the cross-peaks representing these two carbons in the HMBC spectrum overlapped considerably, the exact chemical shift values for these two carbons were determined from a 13C-NMR spectrum. Spatial proximity of the methyl group to the CHOH-CH2-OP spin system was confirmed by a ROESY spectrum that revealed a strong NOE between the methyl and the methine protons (on v) and a weaker NOE between the methyl and the methylene protons. In summary, the analyses of the NMR spectroscopic data, coupled with the observations implying the presence of a quinoxaline moiety, suggested the partial structure 20 (Figure 8). The combined results of the mass spectrometric analysis (described below), the required presence of an ADP unit and partial structure 20, unambiguously identify 21 as the structure of the oPDA derivative (peak J) of the compound corresponding to Peak A. In negative mode ESI-MS spectra (Figure 10) both the monoanionic (m/z = 612) and the dianionic (m/z = 305.6) species were visible and extensive fragmentation analyses performed for both the monoanionic and the dianionic species further corroborated structure 21. Isolation of the nucleotide pyrophosphatase cleavage product of 21 (peak K) by HPLC, followed by 1H NMR (Figure 11) analysis revealed the expected quinoxaline 23, serving as a further confirmation for the structure 21.
Figure 9.

1H NMR spectrum of the purified quinoxaline adduct. The peaks have been assigned as shown in the structure.
Figure 8.

Structure 20 represents the partial structure deduced from the NMR experiments and the observations suggesting the presence of a quinoxaline moiety. The five carbon atoms attached to ADP, derived from ADP-ribose, are labeled as u, v, w, y and z. 21 represents the actual structure deduced from additional negative mode ESI-MS experiments and other observations. The structure 21 must be generated from the 1, 2-diketone containing species 22 (or equivalent) and oPDA (16).
Figure 10.

Negative mode ESI-MS analysis of the quinoxaline derivative 21. The m/z for the monoanionic species is 612 and for the di-anionic species is 305.6. Peaks with m/z = 477, 426.1, 346.1 and 185 result from the fragmentation of 21 under spraying conditions.
Figure 11.

1H NMR spectrum of the HPLC-purified quinoxaline phosphate unit (23) obtained from 21 by its hydrolysis catalyzed by nucleotide pyrophosphatase (pptase).
DISCUSSION
Recombinant wild type THI4 copurifies with a set of tightly bound adenylated metabolites (Peaks A-D, Figure 2). One of these was shown to be an adenylated thiazole derivative (ADT 15, Peak D)7. This discovery provided the first clues to the enzymology of thiamin-thiazole formation in eukaryotes and suggested that the precursor to ADT 15 might be an adenylated pentose/pentulose such as ADP-ribose 3. It was reasonable to propose NAD 1 as the precursor to ADT 15, because NAD could be converted to ADPr 3 by a well-characterized reaction similar to that involved in protein ADP-ribosylation10 and the conversion of ADPr 3 to ADT 15 could be accomplished using reasonable chemistry (Figure 1). In addition, sequence analysis of THI4 predicted a nucleotide binding motif. However, this proposal could not be tested using recombinant THI4 overexpressed in Escherichia coli or in S. cerevisiae because the tightly bound metabolites in the active site of this protein inhibited catalysis. All attempts to prepare active enzyme by releasing the bound metabolites, under a variety of denaturing conditions, failed.
We therefore carried out extensive mutagenesis on THI4 to identify a partially active mutant with empty active sites. This could either arise from a weaker affinity of a mutant towards its reaction products, or from a catalytically impaired mutant that is unable to make the late intermediates or the product. The early steps of the complex reaction sequence leading to ADT 15 could be characterized if such a mutant retained some activity. The recently published crystal structure of THI4 enabled us to select a set of conserved active site residues for mutagenesis (Table 2 and Figure 12)
Table 2.
Summary of all the THI4 mutants constructed.
| Amino acid | Possible role(s) | Mutants |
|---|---|---|
| E97 | Binding adenylated intermediates/product via H-bonding to the ribose part of the ADP moiety | E97A, E97Q |
| H200a | Catalysis of thiazole formation | H200N |
| C204a, b | Catalysis of thiazole formation | C204A |
| C205a | Only conserved cysteine. Catalysis of thiazole formation. Possibly important in the sulfur transfer. | C205S |
| D207a | Catalysis of thiazole formation | D207A |
| H237 | Catalysis of thiazole formation | H237N, H237A |
| D238 | Catalysis of thiazole formation | D238A |
| R301 | Binding advanced intermediates via H-bonding to the carboxylate unit coming from glycine. Other catalytic role. | R301Q, R301A |
| P304 | Providing structural rigidity to a loop spanning the active site. | P304A |
These conserved residues are part of a flexible loop that spans the active site of a different monomer in the octameric structure (see figure 12).
This cysteine residue is replaced by a conserved serine in most THI4 orthologs.
Figure 12.

Stereoview of the active site of THI4 with the ADT (15) carbon atoms labeled in cyan. Two separate monomers contribute to the ADT binding site. Carbon atoms from one monomer are labeled with green and those from the other are labeled in yellow. His200 is on a disordered region and is not shown.
The D238A and C205S mutants showed very weak activities towards NAD 1 (peak G), the proposed substrate, to produce ADPr 3 (peak E) and nicotinamide 2 (peak H) as products (unpublished data). This same activity was much stronger for the C204A and H200N mutants (Figure 3). FAD and NADP were also tested as substrates for these two active mutants but no reaction was detected. These observations confirm the hypothesis that NAD 1 is one of the substrates for THI4 and identify the hydrolysis of NAD 1 to form ADPr 3 as the first step of thiazole biosynthesis in eukaryotes.
In addition to the formation of ADPr 3 and nicotinamide 2 from NAD 1, the partially active mutants also catalyzed the formation of a new adenylated species (peak F, Figure 3). The production of the same unknown species was observed when the C204A and H200N mutants were treated with ADPr (Figure 4). To determine if this intermediate was ADP-ribulose (ADPrl, 4), the next intermediate on our proposed pathway (Figure 1), a reference sample of this compound was prepared by the ribose-5-phosphate isomerase (RPi) catalyzed isomerization of ADPr to ADPrl11. This reference compound co-migrated with the compound corresponding to Peak F generated by treating ADPr with either of the C204A or H200N mutants of THI4 (Figure 4). To further confirm the identity of the peak F compound as ADPrl 4, it was reduced with NaBD4 to produce a stable deuterated reduction product, which was isolated by HPLC and characterized by NMR. These observations establish the isomerization of ADPr to ADPrl as the second step in the THI4-catalyzed biosynthesis of ADT from NAD.
In the absence of glycine, the C204A and H200N mutants catalyzed low levels of conversion of ADPrl 4 to the compound corresponding to peak A after prolonged incubation. The amount of the peak A compound formed over the background was always low and varied between experiments using different protein preparations. In the presence of glycine, however, the same conversion occurred at a much higher level (Figure 5). This reaction, for the first time, led to the reconstitution of the biosynthesis of one of the previously observed unstable THI4-bound adenylated metabolites (peak A, Figure 2).
Direct characterization of the compound corresponding to peak A could not be achieved due to its instability. The mechanistic proposal outlined in Figure 1 suggested a possible presence of a 1,2-diketone or a 1,2-ketoimine functionality in this intermediate compound (e.g. structures 9 and 10). Facile decomposition of the isolated peak A compound to produce ADP as the major product is consistent with this prediction (unpublished results). Treatment of the reaction mixture with ortho-phenylenediamine (oPDA, 16), which traps reactive 1,2-diketones as stable quinoxalines12, 13, resulted in the selective and efficient conversion of the peak A compound to a stable adduct, which was isolated and its structure determined as the quinoxaline 21 (discussed in the results section). While this assignment does not unambiguously determine the structure of the compound generating peak A, it is consistent with structure 9. The observation, that the conversion of ADPrl 4 to the peak A compound is greatly facilitated by glycine, is consistent with the mechanism outlined in Figure 1, in which, imine formation facilitates deprotonation reactions at the C3 carbon of the ribose. In this mechanism, imine formation between ADPrl 4 and glycine 5 gives 6, imine deprotonation at C3 gives 7, which eliminates water and tautomerizes to give 9, the proposed intermediate giving rise to peak A (Figure 1). Model studies have demonstrated that imine deprotonation is about 108 times faster than the corresponding ketone deprotonation14, 15. This pKa enhancement strategy is used by many enzymes including the prokaryotic thiazole synthase16.
Much still remains to be understood about this protein and its function in the eukaryotic cell. None of the mutant THI4 enzymes were able to catalyze multiple turnovers presumably because the products in each case bind tightly to the enzyme. In addition, it was not possible to advance the thiazole biosynthesis beyond the compound corresponding to peak A (9). This could be due to the fact that the correct sulfur donor has not yet been identified. The following observations reported on this enzyme also require further investigation: THI4 appears to be involved in mitochondrial DNA damage prevention and other stress related pathways17-21. THI4 in Neurospora crassa is highly expressed (1.5% of the entire proteome) in the exponential growth phase and seems to be post-translationally modified and forms a stable complex with a cyclophilin (cis/trans proline isomerase)22, 23.
The identification of reaction intermediates is a valuable tool in the investigation of an enzyme mechanism. In most cases, this involves extensive kinetic characterization and insightful design of substrate analogs to block the reaction at intermediate stages. THI4 allows a remarkable departure from this traditional strategy for investigating enzyme mechanism. This protein copurifies with a set of tightly bound metabolites that identify its function as well as provide a series of molecular “snapshots” that reveal the mechanism of the complex reaction sequence that it catalyzes.
Experimental Methods
All the chemicals and snake venom nucleotide pyrophosphatase were purchased from Sigma-Aldrich Corporation (USA) unless otherwise mentioned. For all the HPLC analysis, a Supelco LC-18-T (150×4.6 mm, 3 μm ID) reverse phase column was used. The ribose-5-phosphate isomerase plasmid was a kind gift from Prof. Sherry L. Mowbray (Swedish University of Agricultural Sciences).
Construction of the THI4 mutants
All the THI4 mutants were constructed by the Cornell Protein Purification Facility. Standard methods were used for DNA manipulations24, 25. Plasmid DNA was purified with the Qiagen Miniprep kit. E. coli strain MachI (Invitrogen) was used as a recipient for transformations during plasmid construction and for plasmid propagation and storage.
Site-directed mutagenesis was performed on pThi4.28 by a standard PCR protocol using PfuTurbo DNA polymerase per the manufacturer's instructions (Invitrogen) and DpnI (New England Biolabs) to digest the methylated parental DNA prior to transformation.
For the C205S, R301Q, R301A, H237N, H237A, D238A, C304A, E97A, E97Q, and C204A mutants, primers were designed to introduce or remove a diagnostic restriction enzyme site to facilitate screening for a mutated clone. Only clones producing the anticipated restriction pattern were sequenced. For the H200N and D207A mutants, a third primer was designed to screen for the presence of the mutant by colony PCR with an appropriate vector-specific primer. Only clones that produced a PCR product were sequenced. In every case, the mutagenesis primer pair consisted of the primer whose sequence is in the table below and its reverse complement.
Overexpression and purification of THI4 and its mutants
E. coli BL21(DE3) containing the overexpression plasmid (containing a full length clone of THI4, or its mutant, in the pET28 vector) was grown in LB medium containing kanamycin (40 μg/mL) with shaking at 37 °C until the OD600 reached 0.6. Isopropyl-β-D-thiogalactopyranoside (IPTG) was then added (final concentration = 2 mM) and cell growth was continued at 15 °C for 16 h. The cells were then harvested by centrifugation and the resulting cell pellets were stored at −20 °C until needed. To purify the protein, the cell pellets from 1L of culture were resuspended in 20 mL lysis buffer (10 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, pH 8) and lysed by sonication (Heat systems Ultrasonics model W-385 sonicator, 2 s cycle, 50% duty). The resulting cell lysate was clarified by centrifugation and the THI4 protein was purified on Ni-NTA resin following the manufacturer's (Qiagen) instructions. After elution, the protein was desalted using a PD-10 column (Amersham) pre-equilibrated with 50 mM potassium phosphate buffer, pH 8.0. All the THI4 mutant proteins expressed well and were well behaved except for the E97A and E97Q mutants. These two mutants mostly precipitated immediately after purification.
HPLC analysis of THI4 and its mutants for bound metabolites
The purified enzyme was denatured by heat (100 °C, 40 s) and then rapidly cooled on dry ice. The precipitated protein was removed by centrifugation. The supernatant was filtered through a 10 kDa MW cut off filter (Microcon YM-10, Millipore) and 100 μL of the filtrate was analyzed by reverse phase HPLC. The following linear gradient (method 1, in some experiments minor modifications to the method were made to get better resolution at certain parts of the chromatogram) was used at 1 mL/min flow rate: solvent A is water, solvent B is 100 mM KPi, pH 6.6, solvent C is Methanol; 0 min 100% B, 5 min 10% A 90% B, 8 min 25% A 60% B 15% C, 14 min 25% A 60% B 15% C, 19 min 30% A 40% B 30% C, 21 min 100% B, 30 min 100% B. The absorbance at 220 nm, 261 nm and 320 nm was monitored. The UV-Vis spectra of the resolved components were analyzed by an inline diode-array UV detector.
Activity assays with the mutants
The purified mutants (100 μM) were incubated with 300 μM NAD or ADP-ribose (in the presence or absence of 300 μM glycine) for 10 hours at room temperature (25 °C). Control reactions were set up with the enzyme (without the substrate) or with the substrate(s) in the reaction buffer. Subsequently the enzyme in the reaction mixture was heat denatured (100 °C, 40 s) and the precipitated enzyme was removed by centrifugation. The supernatant was filtered through a 10 kDa MWCO filter (Microcon YM-10, Millipore). The control reactions were treated identically. The filtrate was analyzed by HPLC as described above.
Preparation of ADP-ribulose
E. coli ribose-5-phosphate isomerase was overexpressed in E. coli strain BL21(DE3) containing the overexpression plasmid (containing a full length clone of the E. coli ribose-5-phosphate isomerase gene in the pET15b vector) in LB medium as described above. The protein was purified on Ni-NTA resin following the manufacturer's (Qiagen) instructions and desalted into 50 mM Tris-HCl, pH 7.5 containing 100 mM NaCl. 50 μM of the protein was incubated with 250 μM of ADP-ribose for 5 h. The reaction mixture was heat denatured (100 °C, 40 s) and the precipitated protein was removed by centrifugation. The supernatant was filtered through a 10 kDa MWCO filter (Microcon YM-10, Millipore) and the filtrate was analyzed by HPLC as described above.
Characterization of NaBD4 reduced peak F compound
1 mM ADP-ribose was incubated with the C204A THI4 mutant (100 μM) overnight at room temperature. The reaction mixture was heat denatured (100 °C, 40 s) and the precipitated protein was removed by centrifugation. The supernatant was filtered through a 10 kDa MWCO filter (Microcon YM-10, Millipore). From the filtrate thus obtained, the peak F compound (putative ADPrl) was purified by HPLC. The pulled fractions were combined (∼20ml) and directly reduced with NaBD4 (2 mM, 20 min, room temperature), neutralized with 1 M HCl and the entire sample was subjected to HPLC purification and the collected fractions were lyophilized. The lyophilized product was dissolved in 0.5 mL of “100%” D2O (Sigma) and was re-lyophilized. The residue thus obtained was dissolved in about 0.25 mL of “100%” D2O and the solution was placed in a Shigemi NMR tube (standardized for D2O). NMR experiments were acquired on a Varian INOVA 600 MHz instrument equipped with a 5 mm triple-gradient inverse-detection HCN probe. The NaBD4 reduction yielded two species observed by HPLC with approximately 1:1 ratio, both of which were isolated. One of these was easily identified by NMR as the NaBD4 reduction product of ADPrl. The NMR analysis of the other product revealed very similar characteristics and most likely is the borate adduct of the reduction product.
Trapping of the peak A compound with ortho-phenylenediamine (analytical sample)
500 μL of a concentrated wild type THI4 sample was heat-denatured to release all the bound metabolites and the precipitated protein was removed by centrifugation. The supernatant was filtered through a 10 kDa MWCO filter (Microcon YM-10, Millipore). To 150 μL of the filtrate was added a freshly prepared aqueous solution of ortho-phenylenediamine to a final concentration of 4 mM. The mixture was incubated on ice for 15 min and analyzed by HPLC.
Purification of the trapped species
All the THI4 protein obtained from a 3 L culture (∼150mg) was concentrated to 6 mL (Amicon Utra-4, Millipore, 10 kDa cut off) and divided into ten 600 μL aliquots. Each aliquot was heat denatured (100 °C) and the precipitated protein was removed by centrifugation. The supernatants were combined and filtered through a 10 kDa MW cut off filter (Amicon Ultra-4, Millipore). Ortho-phenylenediamine (oPDA) was added to the filtrate to a final concentration of 4 mM and the mixture was incubated for 15 min on ice. A different HPLC method was developed for the purification of the adduct, because the buffer system had to be compatible with the ESI-MS analysis. The following linear gradient was used at 1mL/min flow rate (method 2): solvent A is ammonium acetate buffer (20 mM, pH 7) and 1% methanol, solvent B is 10% ammonium acetate buffer (20 mM, pH 7) and 90% methanol; 0 min 100% A, 3 min 100% A, 12 min 70% A 30% B, 14 min 70% A 30% B, 16 min 100% A, 25 min 100% A. The peak corresponding to the adduct was collected, frozen with dry ice and lyophilized. The product residue was redissolved in water and checked for purity by HPLC. It was found to be stable when kept cold (on ice) or frozen.
NMR spectroscopy and negative mode ESI-MS analysis of the purified adduct
The lyophilized product was dissolved in 0.5 mL of “100%” D2O (Sigma) and was re-lyophilized. The residue thus obtained was dissolved in about 0.25 mL of “100%” D2O and the solution was placed in a Shigemi NMR tube (standardized for D2O). The 13C-NMR experiment was acquired on a Varian INOVA 500 MHz instrument. All other NMR experiments were acquired on a Varian INOVA 600 MHz instrument equipped with a 5 mm triple-gradient inverse-detection HCN probe.
For ESI-MS analysis the lyophilized product was dissolved in 100 μL of water. Right before spraying into the instrument (An Esquire ion trap instrument, Bruker) this aqueous solution was mixed with 100 μL methanol containing 0.1% triethyl amine. The sample was analyzed in the negative ion mode and the peaks thus obtained were isolated and fragmented.
Cleavage of the quinoxaline containing adduct 21 by nucleotide pyrophosphatase and the isolation of the quinoxaline fragment
To a 100 μL solution of the purified quinoxaline-ADP adduct (21) was added 200 μg of lyophilized snake venom nucleotide pyrophosphatase (EC 3.6.1.9, Sigma) and MgCl2 to a final concentration of 1 mM (20 mM Tris-HCl, pH 8.0). A control reaction was set up with only MgCl2 and no enzyme. Both the samples were incubated at 37 °C for two hours, filtered (10 kDa MW cut off filter) and the filtrates were analyzed by HPLC (method 1 described above). The same reaction was repeated on a large scale and the quinoxaline fragment (23) peak was collected (method 2 described above) and lyophilized. The product thus obtained was dissolved in 0.25 mL of “100%” D2O and the solution was placed in a Shigemi NMR tube (standardized for D2O) and analyzed by 1H-NMR. 1H-NMR (600 MHz, D2O): δ 8.12−8.06 (m, 1H, aromatic), δ 8.01−7.96 (m, 1H, aromatic), δ 7.87−7.79 (m, 2H, aromatic), δ 5.46 (dd, 1H, J = 3.66, 7.57 Hz), δ 4.16−4.03 (m, 2H), δ 2.83 (s, 3H).
Supplementary Material
Table 3.
Primers used in the mutagenesis process.
| Mutant | Mutagenesis Primer Sequence, top strand | Restriction Site |
|---|---|---|
| C205S | CCCAAGCTCACGGTACTCAATGTTCCATGGACCCTAACG | StyI |
| R301Q | GGATGGATTAAACCAAATGGGACCAACTTTTGGAGCTATGGC | BsmFI |
| R301A | CTGGATGGATTAAACGCCATGGGTCCAACTTTTGGAGCTATGGC | NcoI |
| H237N | GGTGTCATTTTATCCACTACCGGCAATGATGGTCCATTTGGTGC | BsrDI |
| H237A | GGTGTCATTTTATCCACTACCGGAGCGGATGGTCCATTTGGTGC | BsrBI |
| D238A | GGTGTCATTTTATCCACTACCGGTCATGCGGGACCATTTGGTGC | BsmFI |
| C304A | GGATTAAACCGTATGGGCGCCACTTTTGGAGCTATGGC | KasI |
| E97A | CTTGAAGGTTTGTATTATCGCGAGTTCAGTTGCACCAGGTGG | NruI |
| E97Q | CTTGAAGGTTTGTATTATCCAGAGCTCAGTTGCACCAGGTGG | HgiAI |
| C204A | CCAAGCTCACGGTACTCAAGCTTGCATGGACCCTAACG | HindIII |
| H200N | GTTAGTTACCCAAGCTAACGGCACTCAATGTTGCATGG | N/A |
| D207A | GGTACTCAATGTTGCATGGCCCCTAACGTAATTGAATTGG | N/A |
Table 4.
Screening primers used only for H200N and D207A mutants.
| Mutant | Screening Primer Sequence |
|---|---|
| H200N | TAGTTACCCAAGCTAACGGC |
| D207A | GGTACTCAATGTTGCATGGC |
ACKNOWLEDGMENT
We thank Dr. Cynthia Kinsland (Cornell Protein Overexpression and Characterization Facility) for the preparation of the THI4 mutants and Prof. Sherry L. Mowbray for sending us the RPi plasmid. This research was funded by NIH grants DK44083 (to T.P.B.) and DK67081 (to S.E.E.)
Footnotes
Supporting Information Available: NMR data for the characterization of ADPrl 4, ESI-MS fragmentation analysis, 2-D NMR data, summary of chemical shifts and NMR correlations for compound 21.
REFERENCES
- 1.Butterworth RF. Thiamin deficiency and brain disorders. Nutr. Res. Rev. 2003;16(2):277–283. doi: 10.1079/NRR200367. [DOI] [PubMed] [Google Scholar]
- 2.Jordan F. Current mechanistic understanding of thiamin diphosphate-dependent enzymatic reactions. Nat. Prod. Rep. 2003;20(2):184–201. doi: 10.1039/b111348h. [DOI] [PubMed] [Google Scholar]
- 3.Settembre E, Begley TP, Ealick SE. Structural biology of enzymes of the thiamin biosynthesis pathway. Curr. Opin. Struct. Biol. 2003;13(6):739–747. doi: 10.1016/j.sbi.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Begley TP, Downs DM, Ealick SE, McLafferty FW, Van Loon APGM, Taylor S, Campobasso N, Chiu H-J, Kinsland C, Reddick JJ, Xi J. Thiamin biosynthesis in prokaryotes. Arch. Microbiol. 1999;171(5):293–300. doi: 10.1007/s002030050713. [DOI] [PubMed] [Google Scholar]
- 5.Spenser ID, White RL. Biosynthesis of vitamin B1 (thiamin): an instance of biochemical diversity. Angew. Chem. Int. Ed. Engl. 1997;36(10):1032–1046. [Google Scholar]
- 6.Godoi PHC, Galhardo RS, Luche DD, Van Sluys M-A, Menck CFM, Oliva G. Structure of the Thiazole Biosynthetic Enzyme THI1 from Arabidopsis thaliana. J. Biol. Chem. 2006;281(41):30957–30966. doi: 10.1074/jbc.M604469200. [DOI] [PubMed] [Google Scholar]
- 7.Chatterjee A, Jurgenson CT, Schroeder FC, Ealick SE, Begley TP. Thiamin Biosynthesis in Eukaryotes: Characterization of the Enzyme-Bound Product of Thiazole Synthase from Saccharomyces cerevisiae and Its Implications in Thiazole Biosynthesis. J. Am. Chem. Soc. 2006;128(22):7158–7159. doi: 10.1021/ja061413o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jurgenson CT, Chatterjee A, Begley TP, Ealick SE. Structural Insights into the Function of the Thiamin Biosynthetic Enzyme Thi4 from Saccharomyces cerevisiae. Biochem. 2006;45(37):11061–11070. doi: 10.1021/bi061025z. [DOI] [PubMed] [Google Scholar]
- 9.Praekelt UM, Byrne KL, Meacock PA. Regulation of THI4 (MOL1), a thiamine-biosynthetic gene of Saccharomyces cerevisiae. Yeast. 1994;10(4):481–90. doi: 10.1002/yea.320100407. [DOI] [PubMed] [Google Scholar]
- 10.Ueda K, Hayaishi O. ADP-ribosylation. Annu. Rev. Biochem. 1985;54:73–100. doi: 10.1146/annurev.bi.54.070185.000445. [DOI] [PubMed] [Google Scholar]
- 11.Franco L, Guida L, Zocchi E, Silvestro L, Benatti U, De Flora A. Adenosine diphosphate ribulose in human erythrocytes: A new metabolite with membrane binding properties. Biochem. Biophys. Res. Comm. 1993;190(3):1143–8. doi: 10.1006/bbrc.1993.1169. [DOI] [PubMed] [Google Scholar]
- 12.Hauck T, Bruehlmann F, Schwab W. Formation of 4-hydroxy-2,5-dimethyl-3[2H]-furanone by Zygosaccharomyces rouxii: Identification of an intermediate. Appl. Environ. Microbiol. 2003;69(7):3911–3918. doi: 10.1128/AEM.69.7.3911-3918.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu J, Patel R, Pei D. Catalytic Mechanism of S-Ribosylhomocysteinase (LuxS): Stereochemical Course and Kinetic Isotope Effect of Proton Transfer Reactions. Biochem. 2004;43(31):10166–10172. doi: 10.1021/bi0491088. [DOI] [PubMed] [Google Scholar]
- 14.Bender ML, Williams A. Ketimine intermediates in amine-catalyzed enolization of acetone. J. Am. Chem. Soc. 1966;88(11):2502–8. [Google Scholar]
- 15.Roberts RD, Ferran HE, Jr., Gula MJ, Spencer TA. Superiority of very weakly basic amines as catalysts for alpha-proton abstraction via iminium ion formation. J. Am. Chem. Soc. 1980;102(23):7054–8. [Google Scholar]
- 16.Dorrestein PC, Zhai H, Taylor SV, McLafferty FW, Begley TP. The biosynthesis of the thiazole phosphate moiety of thiamin (vitamin B1): the early steps catalyzed by thiazole synthase. J. Am. Chem. Soc. 2004;126(10):3091–3096. doi: 10.1021/ja039616p. [DOI] [PubMed] [Google Scholar]
- 17.Machado CR, Costa de Oliveira RL, Boiteux S, Praekelt UM, Meacock PA, Menck CFM. Thi1, a thiamine biosynthetic gene in Arabidopsis thaliana, complements bacterial defects in DNA repair. Plant Mol. Biol. 1996;31(3):585–593. doi: 10.1007/BF00042231. [DOI] [PubMed] [Google Scholar]
- 18.Machado CR, Praekelt UM, De Oliveira RC, Barbosa ACC, Byrne KL, Meacock PA, Menck CFM. Dual role for the yeast THI4 gene in thiamine biosynthesis and DNA damage tolerance. J. Mol. Biol. 1997;273(1):114–121. doi: 10.1006/jmbi.1997.1302. [DOI] [PubMed] [Google Scholar]
- 19.Choi GH, Marek ET, Schardl CL, Richey MG, Chang S, Smith DA. sti35, A stress-responsive gene in Fusarium spp. J. Bact. 1990;172(8):4522–8. doi: 10.1128/jb.172.8.4522-4528.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medina-Silva R, Barros MP, Galhardo RS, Netto LES, Colepicolo P, Menck CFM. Heat stress promotes mitochondrial instability and oxidative responses in yeast deficient in thiazole biosynthesis. Res. Microbiol. 2006;157(3):275–281. doi: 10.1016/j.resmic.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Wang G, Ding X, Yuan M, Qiu D, Li X, Xu C, Wang S. Dual Function of Rice OsDR8 Gene in Disease Resistance and Thiamine Accumulation. Plant Mol. Biol. 2006;60(3):437–449. doi: 10.1007/s11103-005-4770-x. [DOI] [PubMed] [Google Scholar]
- 22.Faou P, Tropschug M. A Novel Binding Protein for a Member of CyP40-type Cyclophilins: N. crassa CyPBP37, a Growth and Thiamine Regulated Protein Homolog to Yeast Thi4p. J. Mol. Biol. 2003;333(4):831–844. doi: 10.1016/j.jmb.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Faou P, Tropschug M. Neurospora crassa CyPBP37: A Cytosolic Stress Protein that is able to Replace Yeast Thi4p Function in the Synthesis of Vitamin B1. J. Mol. Biol. 2004;344(4):1147–1157. doi: 10.1016/j.jmb.2004.09.097. [DOI] [PubMed] [Google Scholar]
- 24.Ausubel FM, Brent R, et al., editors. Current Protocols in Molecular Biology. John Wiley & Sons; New York: 1987. [Google Scholar]
- 25.Sambrook J, Fritsch EF, et al. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Plainview, NY: 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.