

Carbocations have fascinated organic chemists for almost a century and the properties of this reactive intermediate have been thoroughly studied1. Carbocations have been proposed as intermediates in several enzyme-catalyzed reactions, such as the prenyl transfer and cyclization reactions involved in terpene biosynthesis,2,3 the pyrrole tetramerization involved in porphyrin biosynthesis,4 the glycosyl transfer reactions involved in a wide variety of glycosylations5 and, the topic of this communication, the thiazole/pyrimidine coupling reaction involved in thiamin phosphate biosynthesis6. This reaction, catalyzed by thiamin phosphate synthase (TP-synthase) is the penultimate step in the biosynthesis of thiamin pyrophosphate, the active form of vitamin B1. All organisms that synthesize thiamin pyrophosphate have some form of this coupling enzyme. Previous structural and biochemical data suggest a dissociative mechanism for TP-synthase (Scheme 1)6-8. Here we describe a study which allowed the direct measurement of the rate constant for pyrimidine carbocation 3 formation using transient state kinetic methods.
Scheme 1.

Prior to this work the rate limiting steps for catalysis by TP-synthase were unknown; therefore, we sought to inspect the formation of thiamin phosphate (TP) on the millisecond time scale under “burst” conditions in order to more clearly understand the kinetic pathway. As shown in Figure 1c (blue dots) a pre-steady state burst of product formation was observed upon rapidly mixing TP-synthase with an excess of HMP-PP and Thz-P in a chemical quench-flow apparatus. The reaction mixture for each time point was quenched by the rapid addition of 0.25 M NaOH and the amount of product was quantified by the oxidation of TP using K3Fe(CN)6 to yield the intensely fluorescent thiochrome phosphate 7 (Figure 1b)7. The data were fit by non-linear regression to the following burst equation:
Figure 1. Pre-steady state product formation catalyzed by TP-synthase using HMP-PP and two HMP-PP analogs (5 and 6).
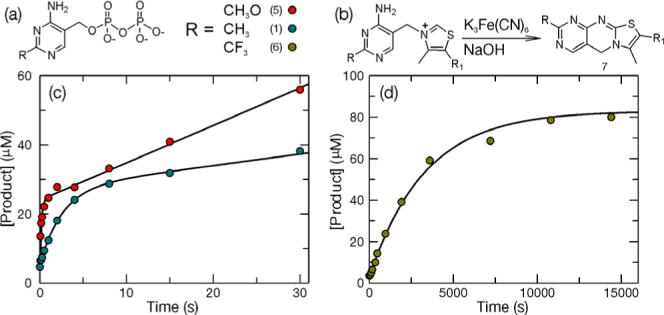
(a) The structures of the three substrates used to probe the rate of catalysis by TP-synthase are shown. (b) The concentration of product was quantified by taking advantage of the oxidation of TP by K3Fe(CN)6 under basic conditions to yield the highly fluorescent compound thiochrome 7 (or thiochrome derivatives). (c) A pre-steady state burst of product formation was observed upon rapidly mixing TP-synthase with an excess (500 μM) of either HMP-PP or MeOHMP-PP 5 and Thz-P (500 μM). (d) Shown is a single-turnover experiment using CF3HMP-PP 6 as a substrate. The reaction was performed under the following conditions: [TP-synthase] = 100 μM, [CF3HMP-PP] = 300 μM and [Thz-P] = 80 μM.
A burst phase rate (λ) of 0.39 ± 0.02 s−1 and a steady state phase rate (kss/[E]) of 0.0125 ± 0.0005 s−1 were obtained. The non-zero value for [TP] in the t = 0 time point results from a fraction of TP-synthase that co-purifies with TP3. If the reaction to form TP is irreversible at the active site, the amplitude (A = 21.9 ± 0.5 μM) of the burst phase should be nearly equal to the active enzyme concentration. However, in this case an additional level of complexity exists because the enzyme co-purifies with a fraction of product bound (both TP and PPi). Therefore, the sum of the amplitude and the Y-intercept (C = 5.3 ± 0.3 μM) is ∼equal to the enzyme concentration used in the burst experiment. To investigate this matter further, an active site titration was performed using the burst amplitude as a measure of active enzyme concentration. This was accomplished by taking advantage of a change in the intrinsic protein fluorescence upon addition of PPi to the TP-synthase/TP complex (see supporting information). The burst amplitude and the active site titration agreed well and indicated that the enzyme co-purifies with slightly less PPi than TP. It is also important to note that both the burst rate and the burst amplitude were independent of HMP-PP and Thz-P concentration under all pseudo first order conditions examined.
Observation of a pre-steady state burst of product formation can often indicate that the overall rate constant for catalysis (kcat) is limited by product dissociation. TP-synthase is no exception as the rate constant for the dissociation of TP nearly fully limits kcat (results not shown). Therefore, probing the mechanism of catalysis by TP-synthase under steady state conditions is not informative. The evidence at hand suggests that the observed rate of the burst phase is largely a measure of a single irreversible step. Several possibilities exist regarding the identity of this step and may likely be narrowed to: pyrimidine carbocation formation 3, pyrimidine carbocation trapping by the Thz-P or a conformational change of the protein. To differentiate between these, the sensitivity of the burst phase rate to carbocation stabilizing (methoxy) and dest abilizing ( trifluoromethyl) groups was determined. If the rate of the burst phase is limited by carbocation formation, we anticipate a large substituent effect (i.e. methoxy >> trifluoromethyl). In contrast, if the rate of the burst phase is limited by carbocation trapping by Thz-P, we predict a small and opposite substituent effect (i.e. methoxy < trifluoromethyl). A conformational change is likely to be relatively insensitive to the pyrimidine substituent (i.e. methoxy = trifluoromethyl).
Data from a burst experiment performed with MeO-HMP-PP 59 are shown in Figure 1c (red dots). The observed burst phase rate (6.2 ± 1.0 s−1) was 16-times greater than that observed for HMP-PP. The steady state rate also increased, although only by a factor of about 3 and is likely due to an increase in the rate of dissociation of methoxythiamin-phosphate from the active site. The reaction using CF3-HMP-PP 6 was extremely slow and it was necessary to carry out this reaction under single-turnover conditions ([TP-synthase] > [Thz-P]) to accurately measure catalysis (Figure 1d). The reaction proceeded to completion in a monophasic fashion and was analyzed by fitting the data to a single exponential equation ([Product] = A·(1–e−λ·t) + C) to obtain an observed rate of (3.01 ± 0.23) × 10−4 s−1. The relative rates of reaction for CH3O-HMP-PP, CH3-HMP-PP, and CF3-HMP-PP were 16, 1 and 7.7 × 10−4 respectively.
A Hammett plot for these substituent effects is shown in Figure 2b. Also shown is a plot for the solvolysis of tert-cumyl chloride derivatives 8 - a well studied reaction that proceeds via a carbocation intermediate, and a plot for the SN2 displacement reaction of benzyl chloride derivatives 9 (Figure 2a)10−12. These plots demonstrate that the enzymatic reaction shows similar sensitivity to carbocation stabilizing/destabilizing substituents as the tert-cumyl chloride system (ρenz = −3.14 ± 0.20, ρmodel = −4.02) and the opposite sensitivity to the benzyl chloride substitution (ρ = +0.73). This supports the previous exclusion of the SN2 mechanism and suggests that the observed burst rate is largely a measure of the rate constant for pyrimidine carbocation formation. This analysis also indicates that subsequent to carbocation formation Thz-P reacts relatively rapidly to produce TP. The rate constant for this reaction is most likely greater than ∼20 s−1 given that no lag in TP formation was observed, even in the case of the methoxy substituted HMP-PP analog. Furthermore, information gained from this analysis suggests that protein isomerization does not play a significant role in limiting the rate of the burst phase.
Figure 2. Analysis of the substituent effect on the TP-synthase catalyzed reaction.
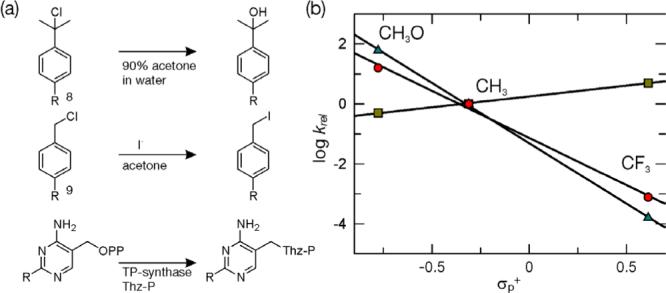
(a) Comparison of the SN1 solvolysis of p-substituted tert-cumyl chloride derivatives 8 to the SN2 displacement of benzyl chloride derivatives 9 and to the reaction catalyzed by TP-synthase. (b) Hammett plot (logarithm (kobs/kMe) versus σp+) of the model SN2 displacement reactions (green squares), the model SN1 solvolysis reactions (blue triangles) and the enzymatic reactions (red dots). A linear fit to the enzymatic reaction data gives a ρ value (slope) of −3.14 ± 0.20.
The crystal structure of the S130A mutant of thiamin phosphate synthase, with a trapped intermediate in the active site, has been previously reported6. The C7 carbon of the pyrimidine is 2.93Å from the closest pyrophosphate oxygen and 2.97Å from the thiazole nitrogen (Figure 3). As the corresponding C-O (in substrate, HMP-PP) and C-N (in product, TP) bond lengths are 1.44Å and 1.57Å respectively, the structure clearly shows that there is no bonding between the pyrimidine and the thiazole or the pyrophosphate and suggests that the observed intermediate is a pyrimidine carbocation 3. One of the pyrophosphate oxygen atoms is located 3.03Å from the nitrogen of the pyrimidine amino group. This suggests that proton transfer or hydrogen bonding of the pyrimidine amino group to the pyrophosphate oxygen plays an important role in stabilizing the bound carbocation.
Figure 3. Structure of TP-synthase with the bound pyrimidine carbocation intermediate (PDB ID 1G67).

(a) Stereo view of the active site (cross-eye viewing). (b) Schematic representation of the key active site interactions.
The TP synthase catalyzed reaction proceeds via a carbocation intermediate. The high stability of this intermediate, relative to other enzyme bound carbocations, makes this enzyme a uniquely tractable experimental system. This stability has enabled us to determine the structure of the enzyme intermediate complex and the rate constant for carbocation formation at the active site.
Supplementary Material
Acknowledgment
This research was supported by NIH grants to DK44083 to TPB and DK67081 to SEE.
Footnotes
Supporting Information Available: Detailed experimental procedures for kinetic experiments are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Smith MB, March J. March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure. 5th Edition 2001. [Google Scholar]
- 2.Gibbs RA. In: Comprehensive Biological Catalysis. Sinnott M, editor. Vol. 1. Academic Press; San Diego: 1998. pp. 31–118. [Google Scholar]
- 3.Poulter CD, Wiggins PL, Le AT. J. Am. Chem. Soc. 1981;103:3926–3927. [Google Scholar]
- 4.Battersby AR, Leeper FJ. Chem. Rev. 1990;90:1261–1274. [Google Scholar]
- 5.Davies G, Sinnott ML, Witters SG. In: Comprehensive Biological Catalysis. Sinnott M, editor. Vol. 1. Academic Press; San Diego: 1998. pp. 119–207. [Google Scholar]
- 6.Peapus DH, Chiu HJ, Campobasso N, Reddick JJ, Begley TP, Ealick SE. Biochemistry. 2001;40:10103–10114. doi: 10.1021/bi0104726. [DOI] [PubMed] [Google Scholar]
- 7.Reddick JJ, Nicewonger R, Begley TP. Biochemistry. 2001;40:10095–10102. doi: 10.1021/bi010267q. [DOI] [PubMed] [Google Scholar]
- 8.Chiu HJ, Reddick JJ, Begley TP, Ealick SE. Biochemistry. 1999;38:6460–6470. doi: 10.1021/bi982903z. [DOI] [PubMed] [Google Scholar]
- 9.Reddick JJ, Saha S, Lee JM, Melnick JS, Perkins J, Begley TP. Bioorg. Med. Chem. Lett. 2001;11:2245–2248. doi: 10.1016/s0960-894x(01)00373-0. [DOI] [PubMed] [Google Scholar]
- 10.Carroll FA. Perspectives on structure and mechanism in organic chemistry. Brooks Cole Publishing Co.; Pacific Grove, CA: 1998. [Google Scholar]
- 11.Brown HC, Okamoto Y. J. Am. Chem. Soc. 1958;80:4979–87. [Google Scholar]
- 12.Jaffe HH. Chem. Rev. 1953;53:191–261. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.