

Abstract
A nine-octapeptide insertional mutation in the prion protein (PrP) causes a fatal neurodegenerative disorder in both humans and transgenic mice. To determine the precise cellular localization of this mutant PrP (designated PG14), we have generated transgenic mice expressing PG14-EGFP, a fluorescent fusion protein that can be directly visualized in vivo. Tg(PG14-EGFP) mice develop an ataxic neurological illness characterized by astrogliosis, PrP aggregation, and accumulation of a partially protease-resistant form of the mutant PrP. Strikingly, PG14-EGFP forms numerous fluorescent aggregates in the neuropil and white matter of multiple brain regions. These aggregates are particularly prominent along axonal tracts in both brain and peripheral nerve, and similar intracellular deposits are visible along the processes of cultured neurons. Our results reveal intra-axonal aggregates of a mutant PrP, which could contribute to the pathogenesis of familial prion disease by disrupting axonal transport.
Keywords: prion, transgenic, protein aggregation, axon, mutation, green fluorescent protein
INTRODUCTION
Prions are infectious proteins associated with several fatal neurodegenerative diseases in mammals (Prusiner, 2004). Prion diseases result from conversion of the cellular prion protein (PrPC) into a conformationally altered isoform (PrPSc) that is aggregated and protease-resistant. Dominantly inherited mutations in the gene encoding PrP are responsible for familial forms of prion disease (Kong et al., 2004). One mutant, designated PG14, harbors a nine-octapeptide repeat insertion in the N-terminal region of PrP that is associated with ataxia, dementia, and cerebellar PrP plaques in several families (Duchen et al., 1993; Krasemann et al., 1995; Owen et al., 1992). Tg(PG14) mice expressing the mouse homolog of the PG14 mutant develop an ataxic neurological illness characterized by non-amyloid PrP deposits, apoptosis of cerebellar granule neurons, and loss of synaptophysin-positive nerve terminals (Chiesa et al., 2000; Chiesa et al., 2005; Chiesa et al., 1998).
Elucidating the mechanisms by which PG14 and other mutant PrPs induce neuropathology requires information about the localization of these molecules at the anatomical and subcellular levels. However, immunolocalization of PG14 PrP deposits in brain tissue has proven to be technically challenging due to the poor antibody reactivity of the mutant protein as a result of conformational changes and/or aggregation. Like PrPSc, PG14 PrP possesses hidden epitopes that prevent antibody recognition without the use of harsh antigen retrieval techniques, such as hydrolytic autoclaving or treatment with guanidine thiocyanate (Kitamoto et al., 1987; Kitamoto et al., 1992; Van Everbroeck et al., 1999). However, these techniques, which denature or partially hydrolyze proteins, necessarily introduce a number of potential artifacts.
Our previous immunohistochemical studies identified punctate, most likely extracellular deposits of PG14 PrP in the cerebellum and other brain regions of Tg(PG14) mice (Chiesa et al., 2000; Chiesa et al., 1998). However, intracellular aggregates of the protein in neuronal cell bodies or axons were never observed. This result is surprising, given the known cellular trafficking patterns of PrP along secretory, endocytic and axonal transport pathways (Harris, 2003), as well as our own previous observation that mutant PrP molecules are partially retained in the endoplasmic reticulum of cultured cells (Ivanova et al., 2001). These considerations suggest that conventional immunohistochemical methods may be providing an incomplete picture of the localization of PG14 in brain tissue.
To overcome the limitations of immunocytochemical detection, we have developed lines of transgenic mice expressing PrP-EGFP, a fusion protein incorporating enhanced green fluorescent protein (EGFP) inserted near the C-terminal, glycolipid attachment site of PrP. In mice that express PrP-EGFP incorporating wild-type (WT) PrP, the fluorescent protein is correctly synthesized and posttranslationally modified, and is distributed in an anatomic and subcellular pattern similar to that of untagged PrP (Barmada et al., 2004). In addition, the fusion protein retains functional activity, as assayed by a genetic test (Barmada et al., 2004). We have used Tg(WT-EGFP) mice to monitor the distribution of PrPSc after scrapie inoculation (Barmada and Harris, 2005).
In the present study, we report the construction and characterization of transgenic mice expressing the PG14 version of WT-EGFP. Our results provide, for the first time, evidence for an intra-axonal localization of a mutant PrP, and they suggest that disruption of axonal transport may play a role in the phenotype of familial prion disorders.
MATERIALS AND METHODS
Transgenic mice
The PG14-EGFP construct was generated as described by Ivanova et al. (2001) by inserting the EGFP open reading frame into the StuI site (within codon 223, wild-type numbering) of a plasmid encoding murine PG14 PrP tagged with the 3F4 epitope. The PG14-EGFP open reading frame was then amplified by PCR with SalI ends, and cloned initially into pGEM-T and then into the XhoI site of the MoPrP.XhoI transgenic vector (Borchelt et al., 1996) as described by Barmada et al. (2004). The transgene was excised with NotI and microinjected into the pronuclei of fertilized eggs from an F2 cross of C57BL/6J × CBA/J F1 parental mice. Founder animals were identified by PCR screening of tail DNA using primers P1 (AACCGAGCTGAAGCATT) and P4 (CACGAGAAATGCGAAGGAACAAGC). Tg(PG14-EGFP) lines were established by breeding transgene-positive founders to a recombinant inbred strain of C57BL/6J × CBA/J mice. All mice used in this study were bred onto a Prn-p+/+ (C57BL/6J × CBA/J) background, with the exception of one (Fig. 3D), which had been bred onto a Prn-p0/0 (C57BL/6J × 129) background (Büeler et al., 1992). All mice were housed in a pathogen-free environment and were cared for following the guidelines set forth by the Washington University Policy on Animal Care.
The following mouse lines have been described previously: Tg(WT-EGFP) (line A) (Barmada et al., 2004), Tg(PG14) (line A2) (Chiesa et al., 1998), Tg(WT) (line E1) (Chiesa et al., 1998), and Prn-p0/0 (Büeler et al., 1992).
Mice were checked weekly for symptoms of neurological dysfunction. Kyphosis, seizure, foot clasp, and hyperexcitability were determined by visual observation, while ataxia was tested by placing mice in the center of a horizontally oriented grill (45 × 45 cm) consisting of 3 mm diameter steel rods spaced 7 mm apart. Mice unable to maneuver around the grid were scored as ataxic. Animals that exhibited at least two symptoms were scored as ill.
Paraffin sections
Mice were injected intraperitoneally with heparin anticoagulant (1,000 U/ml), then anesthetized by injection of ketamine (Fort Dodge Animal Health, Fort Dodge, IA) and xylazine (Butler Animal Health Supply, Dublin, OH). Animals were perfused intracardially with 50 ml of saline solution, followed by 40 ml of 4% paraformaldehyde in 0.1M sodium phosphate buffer (pH 7.2). Brains were removed and then post-fixed in the same solution for 48 hrs. After bisecting the brain along the mid-sagittal plane, each hemisphere was dehydrated in graded ethanol solutions, cleared in xylene, and then embedded in paraffin. Six μm sagittal sections were cut and mounted on polylysine coated glass slides.
Hematoxylin and eosin staining was performed after dewaxing sections in xylene and rehydrating them in graded ethanol solutions. For assessing astrocytosis, dewaxed and rehydrated sections were stained with anti-GFAP monoclonal antibody (DAKO, Carpinteria, CA), followed by incubation with Alexa 594-coupled goat anti-mouse IgG (Molecular Probes, Eugene, OR). Sections were imaged using a Nikon TE2000-E inverted fluorescence microscope.
Fluorescence microscopy
Animals were fixed by intracardiac perfusion as above. Brains were then removed and then post-fixed in the same solution for 2 hrs before transfer to 0.1M sodium phosphate buffer (pH 7.2) containing 0.02% sodium azide for storage at 4°C. A vibratome (The Vibratome Company, St. Louis, MO) was used to cut the tissue into 60 μm thick sagittal sections. Sections were mounted on glass slides using Gel/Mount (Biomeda, Foster City, CA). Intrinsic EGFP fluorescence was imaged using a Zeiss LSM 510 inverted confocal microscope with an Axiovert 200 laser scanning system.
To visualize PrP, vibratome sections were stained with antibodies P45-66 (Lehmann and Harris, 1995), 3F4 (Bolton et al., 1991), or 8H4 (Zanusso et al., 1998). Some sections were stained with antibodies directed against giantin (Covance, Berkeley, CA), TRAP (Upstate, Charlottesville, VA), LAMP1 (1D4B, Developmental Studies Hybridoma Bank, Univ. of Iowa, Iowa City, IA), MAP2 (Sigma, St. Louis, MO), or GFP (gift of Maurine Linder, Washington University). Primary antibodies were visualized using Alexa 488-coupled goat anti-rabbit IgG (P45-66, GFP), Alexa 488-coupled anti-mouse IgG (3F4, 8H4), or Alexa 594-coupled goat anti-mouse IgG (giantin, TRAP, LAMP1, MAP2). Sections were then imaged as described above.
Biochemical analyses
Brain homogenates (10% w/v) were prepared in ice-cold PBS using a Teflon-glass apparatus with pestle revolving at 3,500 rpm (Wheaton Science Products, Millville, NJ). A postnuclear supernatant was obtained by centrifuging homogenates at 1,000 × g for 5 min. Protein concentration was determined using a BCA assay (Pierce, Rockford, IL). Homogenates were analyzed by SDS-PAGE followed by Western blot using anti-PrP antibodies 8H4 (Zanusso et al., 1998) or 3F4 (Bolton et al., 1991).
To quantitate protein expression levels, serial dilutions of Tg(PG14-EGFP)+/o mouse brain homogenate were analyzed by Western blot using Image J software (National Institutes of Health, USA). The amount of PG14-EGFP was calibrated by comparison to the level of endogenous PrP.
To assay protease resistance, frozen brain hemispheres were homogenized in detergent buffer (DB: 10 mM Tris-HCl, pH 7.4, 0.5% sodium deoxycholate, 0.5% NP-40, 150 mM NaCl), then assayed for protein concentration as described above. Two hundred μg of total protein were diluted in DB to a final concentration of 1μg/μl. The solution was mixed for 10 min at 4°C, then centrifuged at 1,500 × g for 5 min at 4°C. 0.5–2μg/ml of proteinase K was added to the supernatant and the mixture was incubated at 37°C for 30 min. Phenylmethylsulfonyl fluoride (PMSF; 10 mg/ml) was added to terminate digestion. Proteins were isolated using methanol precipitation, then analyzed by SDS-PAGE and Western blotting.
To assay detergent insolubility, brain homogenates prepared in DB were diluted to 0.4μg/μl in the presence of protease inhibitors (1μg/ml pepstatin and leupeptin, 0.5 mM PMSF, and 2 mM EDTA), then incubated for 20 min at 4°C. The sample was centrifuged at 1,500×g for 5 min at 4°C. The supernatant was then recovered and centrifuged for 75 min at 135,000×g at 4°C to separate soluble and insoluble fractions. Proteins from the supernatant of this subsequent centrifugation were recovered by methanol precipitation, and then both the supernatant and pellet fractions were analyzed by SDS-PAGE.
To test sensitivity to phosphatidylinositol-specific phospholipase C (PIPLC), 200μg of postnuclear supernatant was centrifuged at 16,000×g at 4°C for 5 min to collect membranes. Membrane pellets were resuspended in PBS with B. thuringiensis PIPLC (prepared as described in Shyng et al. (1995)) at a final concentration of 1 unit/ml, then incubated on ice for 2.5 hrs. Membranes were then collected again by centrifugation at 16,000×g, and proteins released into the supernatant were precipitated with methanol. Membrane pellets and proteins precipitated from the supernatant were resuspended in gel loading buffer and analyzed by SDS-PAGE and Western blotting.
To immunoprecipitate aggregated PrP with antibody 15B3, we followed the procedure recommended by Prionics (Zurich, Switzerland), utilizing the buffers supplied by them. First, a 100 μl aliquot of mouse anti-IgM Dynabeads (Dynal, Carlsbad, CA) was coated with 20 μg of mAb 15B3. Ten μl of 15B3-coated Dynabeads were then added to 200 μg of total protein from brain homogenates. Samples were incubated on a rotating wheel for 2 hr at 25°C, after which beads were washed three times with 1 ml of 15B3 Wash Buffer (Prionics, Zurich, Switzerland). Washed beads were suspended in 40μl of 2X 15B3 Loading Buffer (Prionics) and heated for 5 min at 96°C. Immunoprecipitated proteins were resolved by SDS-PAGE followed by Western blotting with 6D11 antibody (Pankiewicz et al., 2006).
Neuronal cell culture and transfection
Cerebellar granule neurons (CGNs) were isolated from 4 day old mouse pups according to methods described previously (Miller and Johnson, 1996). Neurons were plated in CGN medium (basal medium Eagle’s with Earle’s salts, 10% fetal calf serum, 2 mM glutamine, 25 mM KCl, 0.1 mg/ml gentamycin). Cells were plated at a density of 375,000–450,000/cm2 onto 35 mm glass-bottom dishes pre-coated with poly-D-lysine. Cultures were stained with FM 4-64 (Invitrogen) according to the manufacturer’s directions.
EGFP-WT and EGFP-PG14 constructs (in which EGFP is inserted near the N-terminus of PrP) were generated by first cleaving the EGFP open reading frame from the pEGFP-C1 plasmid (Clontech, Mountain View, CA) using restriction enzymes NcoI and EcoRI. Both ends were blunted, then ligated into the AgeI site (within codon 33) of a pcDNA3 plasmid encoding murine WT or PG14 PrP tagged with the 3F4 epitope. The resulting plasmids were introduced into CGNs cultured from non-transgenic mice by transfection using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Cells were analyzed 24 hrs after transfection.
Granule neurons were imaged in the living state using a Zeiss LSM 510 inverted confocal microscope with an Axiovert 200 laser scanning system.
RESULTS
Weak immunostaining of native PG14 PrP in brain sections
In all of our previous studies using immunostaining to localize PG14 PrP in paraffin-embedded brain sections from Tg(PG14) mice, antigen retrieval treatments (guanidine thiocyanate plus hydrolytic autoclaving) were applied to partially denature the mutant PrP and so enhance its immunoreactivity (Chiesa et al., 2000; Chiesa et al., 2005; Chiesa et al., 1998; Chiesa et al., 2003). To test the reactivity of PG14 PrP in the native state, we immunostained vibratome sections without pre-treatment. We observed that, while brain sections from Tg(WT) mice (expressing wild-type PrP) stained strongly with antibodies directed against three different regions of the PrP molecule (supplemental Fig. 1A, D, G), sections from Tg(PG14) mice produced only a low level of fluorescence (supplemental Fig. 1B, E, H). Western blots confirmed that the expression levels of PG14 and WT PrP are similar (Chiesa et al., 1998). We noted that, although the fluorescence signal observed in Tg(PG14) brains was low with all three antibodies, it was detectably above the background level seen in Prn-p0/0 mice that do not express any PrP (supplemental Fig. 1C, F, I). This residual signal is most likely due to the presence of a sub-population of PG14 PrP molecules (designated PG14Sol) that are soluble and that possess all of the biochemical properties of PrPC, including reactivity with antibodies that recognize PrPC-accessible epitopes (Biasini et al., manuscript submitted).
Construction of Tg(PG14-EGFP) mice
To allow antibody-independent localization of PG14 PrP, we generated transgenic mice expressing PG14-EGFP, a fusion protein in which the EGFP moiety is inserted near the C-terminal glycolipid attachment site of PrP harboring the PG14 mutation (Fig. 1A). For comparison, we used Tg(WT-EGFP) mice expressing the wild-type version of PrP-EGFP, which have been described previously (Barmada et al., 2004) (Fig. 1A). Both WT-EGFP and PG14-EGFP were constructed with an epitope tag for the 3F4 antibody (Bolton et al., 1991), which allows discrimination of transgenically encoded PrP from endogenous PrP. The PG14-EGFP fusion construct was cloned into the MoPrP.XhoI vector (Borchelt et al., 1996), which contains a partial promoter sequence from the endogenous PrP gene. This promoter drives protein expression in a developmental and tissue-specific pattern comparable to that of endogenous PrP, with the exception that the transgene is not expressed in Purkinje cells (Barmada et al., 2004; Fischer et al., 1996).
FIGURE 1. Structure and expression of WT-EGFP and PG14-EGFP in transgenic mice.

(A) Schematic of the structures of WT-EGFP and PG14-EGFP. WT-EGFP contains an N-terminal signal sequence (SS), five octapeptide repeats (OR, blue), and a C-terminal GPI addition signal (GPI). The EGFP tag is inserted at codon 223. PG14-EGFP is similar to WT-EGFP, but contains nine additional octapeptide repeats (OR, red), resulting in a total of 14 repeats. (B, C) Expression of WT-EGFP and PG14-EGFP. Brain homogenates from non-transgenic (lane 1), Tg(PG14) (lane 2), Tg(WT-EGFP) (lane 3), and Tg(PG14-EGFP) (lane 4) mice were analyzed by Western blotting using anti-PrP antibodies 3F4 (B) and 8H4 (C). All mice were on the Prn-p+/+ background. ▲, PG14-EGFP; △, WT-EGFP; solid bracket, PG14 PrP; dotted bracket, endogenous WT PrP. Molecular size markers are given in kilodaltons.
Four separate lines of Tg(PG14-EGFP) mice were established (D, X1, X3, and X4). Anatomical localization of PG14-EGFP within the brain was unusual in line D, which showed preferential expression of the fluorescent protein in the mossy fibers, alveus, and stratum lacunosum moleculare, but low expression elsewhere (supplemental Fig. 2B). This unusual expression pattern is likely due to the site of transgene integration. In contrast, the gross neuroanatomical distribution of fluorescent protein in the X1, X3 and X4 lines was similar to that of WT-EGFP (Barmada et al., 2004) (supplemental Fig. 2D–O), and to the known distribution of endogenous PrPC (Moya et al., 2000; Salès et al., 1998). However, only the X4 line expressed PG14-EGFP at a sufficiently high level to be detected by fluorescence microscopy without the aid of anti-GFP labeling (supplemental Fig. 2). Although the X4 line served as the primary source of data for the experiments described below, the subcellular distribution of PG14-EGFP (including the presence of axonal aggregates, see results) was confirmed in the D line using the intrinsic fluorescence of EGFP and in the X1 and X3 lines by staining with anti-GFP antibody (supplemental Fig. 2, and data not shown).
Western blot analysis of brain homogenates confirmed expression of PG14-EGFP protein in transgenic animals. PG14-EGFP migrates at approximately 85 kDa, larger than WT-EGFP which migrates at 60–70 kDa (Fig. 1B,C). Antibody 8H4 recognizes both endogenous and transgenic PrPs (Fig. 1C), whereas 3F4 identifies only transgenically encoded PrP molecules (PG14, WT-EGFP and PG14-EGFP), each of which carries the 3F4 epitope tag (Fig 1B). Based on quantitative Western blotting, we determined that expression of PG14-EGFP in animals of the X4 line was ~0.15X that of endogenous PrP in mice carrying a hemizygous transgene array, and ~0.3X in those carrying a homozygous transgene array (data not shown).
Tg(PG14-EGFP) mice develop a spontaneous neurological illness
Tg(PG14-EGFP) mice that were homozygous for the transgene array developed spontaneous neurological disease at 391 ± 54 days (Table 1). Symptoms in these animals included kyphosis, ataxia, foot clasp, poor grooming, hyperexcitability, and seizures. The same clinical features were also present in approximately 10% of Tg(PG14-EGFP) hemizygotes, but they appeared only at much later ages (~630 days). All Tg(WT-EGFP) mice remained healthy (Table 1), as reported previously (Barmada et al., 2004).
TABLE 1.
Disease onset in Tg(PG14-EGFP) animals.
| Genotype | Age of Onset |
|---|---|
| WT-EGFP+/o | >600 (0/7) |
| PG14-EGFP+/+ | 391 ± 54 (6/7) |
| PG14-EGFP+/o | 630d ± 43 (2/27) |
| PG14+/o | 235 ± 10 (61/61)a |
Age of onset is recorded in days. Numbers in parentheses indicate the number of ill mice over the total number of animals observed.
Data taken from Chiesa et. al. (2000).
Histological analysis revealed prominent astrogliosis in the cerebella and hippocampi of homozygous Tg(PG14-EGFP) mice compared with Tg(WT-EGFP) controls (Fig. 2A, C, D, F). Healthy Tg(PG14-EGFP) heterozygotes exhibited some astrogliosis, although of a lesser severity than their homozygote counterparts (Fig. 2B, E). Cerebellar sections from either homozygous or heterozygous Tg(PG14-EGFP) mice stained with hematoxylin and eosin did not show significant granule cell loss or other obvious histological abnormalities (Fig. 2G–I). Consistent with these observations, we did not detect positive staining in the cerebellum by the TUNEL method, which reveals dying cells undergoing DNA fragmentation (data not shown).
FIGURE 2. Tg(PG14-EGFP) mice exhibit astrogliosis but not loss of cerebellar granule cells.

Paraffin sections from cerebellum (A–C, G–I) and dentate gyrus (D–F) were stained with an antibody against GFAP (A–F), or with hematoxylin and eosin (G–I). Sections were obtained from age-matched healthy Tg(WT-EGFP+/0) mice (A, D, G), healthy Tg(PG14-EGFP+/0) mice (B, E, H), and clinically ill Tg(PG14-EGFP+/+) mice (C, F, I). The abbreviations are: ML, molecular layer; CGL, cerebellar granule cell layer; DGL, dentate granule cell layer; MF, mossy fibers. Scale bars are 20μm for A–F and 200μm for G–I.
PG14-EGFP possesses PrPSc-like biochemical properties
To investigate whether PG14-EGFP displays abnormal biochemical properties like untagged PG14 PrP, we performed assays for protease resistance, detergent insolubility, phospholipase sensitivity, and immunoprecipitation by the PrPSc-reactive antibody 15B3.
Like untagged PG14 PrP, PG14-EGFP is weakly protease resistant, producing a PrP 27–30 core fragment when subjected to digestion with proteinase K (PK) concentrations of 0.5–2μg/ml (Fig. 3A, lanes 1–4, 9). In contrast, WT-EGFP was completely digested by PK under the same conditions (Fig 3A, lanes 5–8).
FIGURE 3. PG14-EGFP displays abnormal biochemical properties like untagged PG14 PrP.

(A) Assay for protease resistance. Brain homogenates from Tg(PG14-EGFP) mice (lanes 1–4), Tg(WT-EGFP) mice (lanes 5–8) and Tg(PG14) mice (lane 9) were treated with the indicated concentrations of PK, and then subjected to Western blotting with 3F4 antibody. PG14-EGFP and PG14, but not WT-EGFP, give rise to PrP 27–30 fragments. (B) Assay for detergent insolubility. Brain homogenates from Tg(PG14-EGFP) mice (lanes 1, 2), Tg(PG14) mice (lanes 3, 4) and Tg(WT-EGFP) mice (lanes 5, 6) were subjected to ultracentrifugation, followed by Western blotting of supernatant (S lanes) and pellet fractions (P lanes) using 3F4 antibody. PG14-EGFP and PG14 PrP, but not WT-EGFP, are partially detergent insoluble. (C) Assay for PIPLC release. Brain membranes from Tg(WT-EGFP) mice (lanes 1, 2, 5, 6) and Tg(PG14-EGFP) mice (lanes 3, 4, 7, 8) were incubated without (lanes 1–4) or with (lanes 5–8) PIPLC. Membranes were then collected by centrifugation, and PrP in pellets (P lanes) and supernatants (S lanes) was analyzed by Western blotting with 8H4 antibody. WT-EGFP, but not PG14-EGFP, is partially released by PIPLC. (D) Test of reactivity with antibody 15B3. Brain homogenates from the following mice were subjected to immunoprecipitation with 15B3, followed by Western blotting with 6D11 antibody: Tg(PG14)/Prn-p0/0 (lane 1); Tg(PG14-EGFP)/Prn-p+/+ (lane 2); Tg(PG14-EGFP)/Prn-p0/0 (lane 3); non-Tg (lane 4); and Tg(WT-EGFP) (lane 5). One-fifth as much brain homogenate was used as starting material in lane 1 as in the other lanes. All Tg(PG14-PrP) mice were hemizygous for the transgene array, and all had a Prn-p+/+ genetic background unless otherwise stated.
To test detergent insolubility, detergent lysates of brain were subjected to ultracentrifugation to separate soluble from insoluble protein fractions. As expected, WT-EGFP is recovered almost entirely in the supernatant (S, soluble) fraction in this assay (Fig 3B, lanes 5, 6). In contrast, PG14-EGFP, like untagged PG14 PrP, was found in both supernatant and pellet (P, insoluble) fractions (Fig 3B, lanes 1–4). In multiple experiments, the proportion of insoluble PG14-EGFP varied from 25–50% (data not shown).
PIPLC is a bacterial enzyme that cleaves the glycosyl-phosphatidylinositol (GPI) anchor that attaches PrP to cellular membranes, thereby releasing the protein into the extracellular medium. PG14 PrP is partially resistant to the action of PIPLC, probably due to aggregation and/or conformational changes at the C-terminus of the protein, as opposed to aberrant GPI anchor incorporation (Chiesa et al., 1998; Lehmann and Harris, 1995; Narwa and Harris, 1999). After PIPLC treatment of brain membranes, approximately half of the total amount of WT-EGFP shifts into the supernatant (S) fraction, indicating partial release of the protein (Fig 3C, lanes 1, 2, 5, 6). Incomplete release of WT PrP from brain membranes has been observed previously (Chiesa et al., 1998; Ivanova et al., 2001), and is probably attributable to the mixed topology of the membranous vesicles produced by homogenization. In contrast, the majority of PG14-EGFP remains associated with the membrane (P) fraction, demonstrating that the protein is partially resistant to PIPLC cleavage, like untagged PG14 (Fig 3C, lanes 3, 4, 7, 8). Also similar to untagged PG14, a small fraction of PG14-EGFP is found to be PIPLC-sensitive (Fig. 3C, lane 7), demonstrating that the protein contains a functional GPI anchor. Thus, the presence of the EGFP tag does not interfere with GPI anchoring of either WT or PG14 PrP.
15B3 is a monoclonal antibody that was originally reported to react specifically with PrPSc and not PrPC (Korth et al., 1997). Recently, we have shown that this antibody recognizes multiple forms of aggregated PrP, both infectious and non-infectious, including PG14 PrP from both transfected cells and transgenic mouse brain (Biasini et al., manuscript submitted). The antibody shows no reactivity with monomeric PrPC, even when present in vast excess. We found that 15B3 immunoprecipitated both PG14-EGFP and untagged PG14 PrP, but did not recognize wild-type PrP from both Tg(WT) and non-transgenic mice (Fig. 3D). Thus, PG14-EGFP and PG14 PrP share aggregation-specific, 15B3-reactive epitopes.
Collectively, these results show that PG14-EGFP behaves like untagged PG14 in four different assays that measure PrPSc-like biochemical properties.
PG14-EGFP forms aggregates in multiple brain regions
To compare the distributions of PG14-EGFP and WT-EGFP in brain tissue, vibratome sections from transgenic mice were examined using fluorescence microscopy. Consistent with our previous analysis (Barmada et al., 2004), we found that WT-EGFP was concentrated primarily in neuropil areas that are rich in synapses as well as along axon tracts, and was present only at low levels in dendrites and neuronal somata. In the hippocampus, for example, fluorescence was distributed in the stratum oriens and stratum radiatum of the CA1 region (Fig. 4A). Mossy fibers in the dentate gyrus were also fluorescent (Fig. 4C). In the cerebellum, WT-EGFP was present in the molecular layer, as well as in neuropil of the granule cell layer (Fig. 4E). In these three brain regions, the fluorescence signal had a relatively uniform distribution, with the exception of fluorescent puncta in the cell bodies of pyramidal and granule neurons, corresponding to the location of the Golgi apparatus in these cells (Barmada et al., 2004) (arrows in Fig. 4A, C, E).
FIGURE 4. PG14-EGFP forms aggregates in multiple brain regions.

(A–F) Comparison of the distributions of WT-EGFP and PG14-EGFP in brain sections. Vibratome sections from healthy Tg(WT-EGFP+/0) mice (A, C, E) and ill, age-matched Tg(PG14-EGFP+/+) mice (B, D, F) were prepared from the CA1 region of the hippocampus (A, B), the dentate gyrus (C, D), and the cerebellar cortex (E, F), and were imaged by fluorescence microscopy. The inset in panel A shows a brain section from a non-transgenic, Prn-p+/+ mouse, to illustrate the background level of fluorescence. PG14-EGFP forms numerous fluorescent aggregates, whereas WT-EGFP has a much more uniform distribution. (G-L) The number of PG14-EGFP aggregates is higher in mice with a homozygous transgene array. Sections from healthy Tg(PG14-EGFP+/0) mice (G, I, K) and ill, age-matched Tg(PG14-EGFP+/+) mice (H, J, L) were prepared from the neocortex (G, H), the striatum (I, J), and the CA3 region of the hippocampus (K, L). Aggregate concentration is increased in animals expressing twice the amount of the transgenic mutant protein. The arrowheads in D and J indicate linear aggregates of PG14-EGFP that probably lie within individual axons. The arrows in A, C and E indicate accumulations of WT-EGFP in the Golgi apparatus of neuronal cell bodies. The abbreviations are: OR, stratum oriens; PYR, pyramidal cell layer; RA, stratum radiatum; DGL, dentate granule cell layer; MF, mossy fibers; CGL, cerebellar granule cell layer; ML, molecular layer, PC, Purkinje cell layer. All scale bars represent 20 μm.
The distribution PG14-EGFP was markedly different from that of WT-EGFP. In Tg(PG14-EGFP) mice, bright, intensely fluorescent aggregates were visible in multiple brain areas. In the CA1 region of the hippocampus, PG14-PrP aggregates were found in the stratum oriens, and to a lesser extent in the stratum radiatum and the pyramidal cell layer (Fig. 4B). Intensely fluorescent aggregates of PG14-EGFP were also evident along mossy fibers of the hippocampus (Fig. 4D, K, L), as well as in the molecular layer of the cerebellum (Fig. 4F), the neocortex (Fig. 4G. H), and the striatum (Fig. 4I, J). In general, PG14-EGFP aggregates were concentrated in the same brain regions that displayed high levels of WT-EGFP in Tg(WT-EGFP) mice (Barmada et al., 2004). The aggregates were often distributed in a linear pattern that seemed to correspond to the course of individual neuronal processes (arrowheads in Fig. 4D, J). Although much of the PG14-EGFP signal was present in the form of discrete fluorescent aggregates, these aggregates were superimposed on a more uniform, but less intense background of fluorescence that was similar in appearance to the pattern observed in Tg(WT-EGFP) mice (Fig. 4A-F). This latter signal was specific, since it was absent in non-transgenic mice (inset, Fig. 4A), and is likely attributable to non-aggregated forms of PG14-EGFP (see Discussion).
Although fluorescent aggregates were observed in all Tg(PG14-EGFP) animals, we found that aggregate concentration was directly correlated with the level of transgene expression. Thus, Tg (PG14-EGFP) animals that were homozygous for the transgene array accumulated more numerous fluorescent aggregates than animals that were hemizygous for the transgene array (Fig. 4G–L).
PG14-EGFP aggregates are present in axons but not dendrites
Aggregates of PG14-EGFP were found at highest density in myelinated and unmyelinated axon bundles, and could often be observed arrayed along the course of individual axons. In the striatum, axonal fibers cut in cross-section were intensely fluorescent, and fibers cut longitudinally displayed bright fluorescent puncta along their length (Figs. 4J, 5F). Aggregates were also visible in the alveus (Fig. 5G) and corpus callosum (Fig. 5H) which contain myelinated axons, as well as along unmyelinated mossy fibers in the hippocampus (Fig. 4D). Aggregates were prominent in peripheral as well as central axons, for example, along fibers of the sciatic nerve (Fig. 5J). PG14-EGFP deposition did not occur along all axonal tracts, however. For example, aggregates were sparse along white matter tracts of the cerebellum (Fig 5I). In contrast to PG14-EGFP, WT-EGFP displayed a relatively homogeneous, non-aggregated distribution in each of these areas (Fig. 5A–E). While WT-EGFP appeared to uniformly coat the surface of axonal fibers, PG14-EGFP fluorescence was restricted to punctate deposits that seemed to be intra-axonal. This conclusion was borne out by analysis of neurons in culture (see below).
FIGURE 5. PG14-EGFP aggregates are present in axons but not dendrites.

(A–J) Comparison of the distributions of WT-EGFP and PG14-EGFP in several axon-rich regions of the brain, and in peripheral nerve. Vibratome sections from healthy Tg(WT-EGFP+/0) mice (A–E) and ill, age-matched Tg(PG14-EGFP+/+) mice (F–J) were prepared from the striatum (A, F), the alveus (B, G), the corpus callosum (C, H), and the cerebellar white matter (D, I). Sciatic nerves were examined as whole mounts (E, J). (K–M) PG14-EGFP does not co-localize with a somatodendritic marker. A section from the CA3 area of the hippocampus from an ill Tg(PG14-EGFP+/+) mouse was immunostained for MAP2. The section was then viewed for EGFP fluorescence (K), MAP2 staining (L), and as a merged image of the two signals (M). The abbreviations are: PYR, pyramidal cell layer; LUC, stratum lucidum. The scale bars in A (applicable to A, C–F, H–J), B (applicable to B, G), and K (applicable to K–M) represent 20 μm.
To determine whether PG14-EGFP aggregates were present in dendrites as well as axons, we stained brain sections with an antibody to MAP2, a somatodendritic marker protein. We found that fluorescent deposits of PG14-EGFP did not co-localize with MAP2, for example in the apical dendrites of pyramidal neurons in the stratum lucidum of the CA3 region of the hippocampus (Fig. 5K–M). We conclude that PG14-EGFP, like WT-EGFP (Barmada et al., 2004), is present primarily in axons, and is largely absent from dendrites.
PG14-EGFP aggregates do not co-localize with markers for the ER, Golgi, or lysosomes
We reported previously that mutant PrP molecules, including those harboring the PG14 mutation, are partially retained in the ER of non-neuronal cells (Ivanova et al., 2001). In addition, PrPSc has been localized to the Golgi apparatus (Barmada and Harris, 2005) as well as to lysosomes (Laszlo et al., 1992) in brain tissue. To determine if PG14-EGFP is found in these intracellular organelles in neurons, we analyzed the distribution of the fluorescent protein in brain sections that were stained for markers representing the ER, Golgi, and lysosomes. We found that most PG14-EGFP aggregates did not co-localize in neuronal cell bodies with markers for the ER (TRAP), Golgi (giantin), or lysosomes (LAMP1) (Fig. 6). As is the case for WT-EGFP (Barmada et al., 2004), a few PG14-EGFP puncta in the perinuclear region of large neurons appeared to co-localize with the Golgi marker, giantin, presumably representing protein in transit through the secretory pathway (not shown).
FIGURE 6. PG14-EGFP aggregates do not co-localize with markers for the ER, Golgi, or lysosomes.

Vibratome sections from the brains of ill Tg(PG14-EGFP+/+) mice were stained with antibodies to TRAP (an ER marker) (A–C), giantin (a Golgi marker) (D–F), or LAMP1 (a lysosomal marker) (G–I). Sections were derived from the CA3 area of the hippocampus (A–F) or the dentate gyrus (G–I). Sections were viewed for EGFP fluorescence (A, D, G), for marker protein staining (B, E, H), or as a merged image of the two signals (C, F, I). The abbreviations are: PYR, pyramidal cell layer; LUC, stratum lucidum; DGL, dentate granule cell layer; MF, mossy fibers. The scale bar is 20 μm (applicable to all panels).
PG14-EGFP forms aggregates along neurites of cultured neurons and is decreased at the cell surface
To determine the subcellular localization of WT-EGFP and PG14-EGFP more precisely, we analyzed cultures of cerebellar granule neurons (CGNs) prepared from neonatal transgenic mice. WT-EGFP was distributed in a rim around neuronal cell bodies, as well as along neuritic processes (Fig. 7A, B). The protein showed a relatively uniform distribution, with only a few, small, perinuclear puncta corresponding to the location of the Golgi apparatus (not shown). In contrast, PG14-EGFP was distributed in numerous, large, intensely fluorescent aggregates along neuritic processes (Fig. 7C, D).
FIGURE 7. PG14-EGFP forms aggregates along neurites of cultured neurons and is decreased at the cell surface.
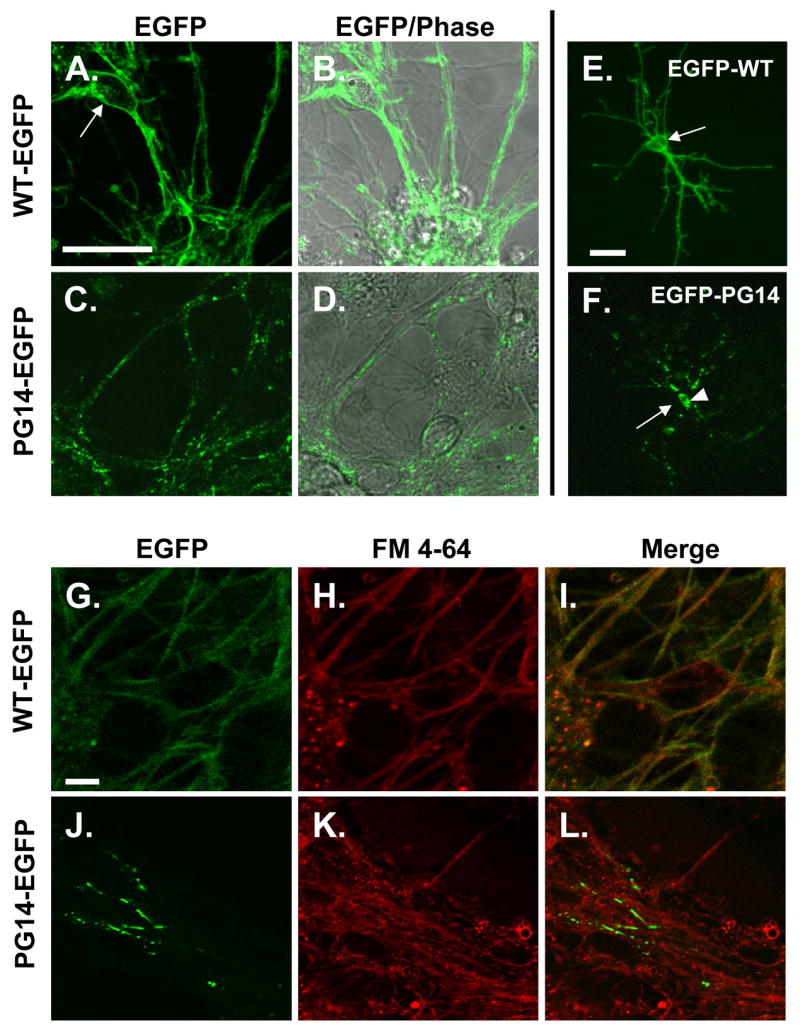
(A–D) Comparison of the distribution of WT-EGFP and PG14-EGFP. Cerebellar granule neurons were cultured from neonatal Tg(WT-EGFP) mice (A, B) or Tg(PG14-EGFP) mice (C, D). After 4 days in culture, cells were viewed by fluorescence (A, C), or by fluorescence superimposed on phase contrast (B, D). The arrow in A points to a neuronal cell body, which is rimmed by fluorescence. (E, F) Distribution of N-terminal EGFP fusion proteins in individual neurons. Cerebellar granule neurons cultured from non-Tg mice were transfected with plasmids encoding EGFP-WT (E) or EGFP-PG14 (F), and were viewed 24 hrs later by fluorescence microscopy. The arrow in E points to a neuronal cell body, which is rimmed by fluorescence. The arrow in F points to the position of the surface membrane of the soma (visible in phase contrast; not shown), which is devoid of fluorescence. The arrowhead in F indicates intracellular accumulations of EGFP-PG14. (G–L) Localization of EGFP fusion proteins with respect to the plasma membrane. Cerebellar granule neurons cultured from Tg(WT-EGFP) mice (G–I) or Tg(PG14-EGFP) mice (J–L) were stained with FM 4-64 dye at 4°C, and then imaged to reveal EGFP fluorescence (G, J), FM 4-64 staining (H, K), or a merge of the two signals (I, L). WT-EGFP co-localizes extensively with FM 4-64, while PG14-EGFP shows little overlap. All scale bars represent 20 μm.
In order to visualize the fluorescence signal in individual neurons more clearly, we transiently transfected cultures of non-transgenic CGNs with EGFP expression constructs. Because of the low efficiency of transfection (~1%), isolated, fluorescent neurons could then be observed against a background of non-fluorescent neurons. For these experiments, we employed plasmids encoding C-terminal PrP-EGFP fusions analogous to those used to construct the Tg(WT-EGFP) and Tg(PG14-EGFP) mice (data not shown), as well as N-terminal fusions in which EGFP was fused at codon 33, ten amino acids beyond the signal peptide cleavage site (Fig. 7E, F). N- and C-terminal fusion proteins displayed similar distributions, arguing that the location of the EGFP moiety has no effect on protein localization. We observed that EGFP-WT uniformly filled the entire neuritic tree out to the smallest, terminal branches, and also formed a rim around the cell soma (Fig. 7E). In contrast, EGFP-PG14 was distributed in numerous, fluorescent puncta along the length of individual neurites, and was also visible in the cell soma in the form of cytoplasmic aggregates that were located at a distance from the surface membrane (Fig. 7F).
To determine whether the fluorescent proteins were localized on the plasma membrane, we stained cultures with FM 4-64, a red fluorescent dye that selectively integrates into the lipid bilayer at the cell surface (Betz et al., 1992). We found that while WT-EGFP almost completely co-localized with FM 4-64 (Fig. 7G–I), aggregates of PG14-EGFP showed little co-localization (Fig. 7J–L).
Taken together, our studies of cultured CGNs demonstrate that neuronal PG14-EGFP aggregates are primarily intracellular (not on the cell surface), and are concentrated in neuritic processes. Since the culture conditions we used do not induce axo-dendritic polarization of CGNs (Powell et al., 1997), it is not possible to specify whether the neuritic PG14-EGFP aggregates we observe are in axons or dendrites.
DISCUSSION
In this study, we have characterized Tg(PG14-EGFP) mice that express the EGFP-tagged version of a mutant PrP molecule carrying a nine-octapeptide insertion. This PG14 mutant is associated with an inherited dementia in humans (Duchen et al., 1993; Krasemann et al., 1995; Owen et al., 1992), and we have reported previously that it causes a strong neurodegenerative phenotype when expressed as a non-EGFP-tagged molecule in Tg(PG14) transgenic mice (Chiesa et al., 2000; Chiesa et al., 2005; Chiesa et al., 1998). We show here that Tg(PG14-EGFP) mice recapitulate key clinical, neuropathological and biochemical features of our original Tg(PG14) mice. However, the presence of the EGFP moiety has allowed us to visualize the anatomical and subcellular localization of the mutant protein without the need for antigen retrieval techniques typically required for immunohistochemical detection of aggregated, misfolded forms of PrP. Using Tg(PG14-EGFP) mice, we describe for the first time intracellular aggregates of mutant PrP in central and peripheral axons. Our results provide an entirely new picture of the localization of mutant PrP molecules in a familial prion disease, and they suggest a novel mechanism by which these proteins might induce neuropathology via interference with axonal transport.
Tg(PG14-EGFP) mice model a familial prion disease
Like Tg(PG14) mice, Tg(PG14-EGFP) animals spontaneously develop a progressive neurological disease characterized clinically by ataxia, kyphosis, and seizure. In addition, both kinds of mice exhibit astrogliosis and PrP deposition. Finally, PG14-EGFP displays abnormal biochemical properties like PG14 PrP. In contrast, Tg(WT-EGFP) mice do not develop neurological illness or neuropathology, and they do not accumulate biochemically abnormal PrP. Taken together, these results argue that the C-terminal addition of EGFP does not significantly alter the molecular properties or pathogenic effects of PG14 PrP. Thus, Tg(PG14-EGFP) mice, like the original Tg(PG14) animals, model key features of the corresponding human prion disease.
Tg(PG14-EGFP+/+) mice (X4 line) develop disease at ~390 days of age, much later than Tg(PG14+/+) or Tg(PG14+/−) mice (65 or 240 days, respectively, for the A2 and A3 lines) (Chiesa et al., 1998). This difference is most likely attributable to the significantly lower transgene expression level in the Tg(PG14-EGFP) X4 line compared to the Tg(PG14) lines (0.3X vs. 2X endogenous PrP levels when the transgene arrays are homozygous). We have previously observed a strong inverse correlation between protein expression level and age at disease onset in Tg(PG14) mice (Chiesa et al., 1998). Thus far, we have not recovered lines of Tg(PG14-EGFP) mice with higher transgene expression levels, but it is uncertain whether this reflects a particular toxicity of the PG14-EGFP molecule or other factors.
The relatively low transgene expression level in Tg(PG14-EGFP) mice is also likely to explain why these animals did not exhibit granule cell degeneration in the cerebellum, in contrast to Tg(PG14) mice which show dramatic granule cell apoptosis (Chiesa et al., 2000; Chiesa et al., 1998). In a previous study, we found that deletion of the Bax gene rescued granule cell death without altering clinical symptoms or synaptic degeneration in Tg(PG14) mice (Chiesa et al., 2005). We thus concluded that synaptic loss makes an important contribution to the Tg(PG14) phenotype that can account for the persistence of neurological symptoms in the absence of granule cell death. We hypothesize that in Tg(PG14-PrP) mice, which display a much more indolent clinical course compared to Tg(PG14) mice, the low expression level of the mutant protein produces synaptic degeneration before granule cell loss can ensue.
PG14-EGFP forms aggregates in axons
A major conclusion of our study is that PG14-EGFP forms prominent intra-axonal aggregates. These aggregates were visible in axon-rich areas of the brain such as the molecular layer of the cerebellum, striatum, corpus callosum, and mossy fibers of the hippocampus. They were also prominent in peripheral axons, in particular those of the sciatic nerve. In cultured cerebellar granule neurons, PG14-EGFP aggregates were evident within neurites, where they did not colocalize with a marker for the plasma membrane, demonstrating that the deposits are intracellular. The fluorescent aggregates visible microscopically presumably correspond to those that are defined biochemically by detergent insolubility, protease and PIPLC resistance, and 15B3 reactivity. In brain sections from Tg(PG14-EGFP) mice, we observed, in addition to aggregates, a more uniform, but weaker fluorescence pattern similar to the one seen in Tg(WT-EGFP) mice. This fluorescence signal presumably corresponds to the proportion (50–75%) of PG14-EGFP that is soluble (see Fig. 3B). We have referred to the soluble form of PG14 PrP as PG14Sol, and have shown that it possesses all of the biochemical properties of PrPC (Biasini et al., manuscript submitted).
It is likely that PG14-EGFP aggregates contribute to the disease phenotype, although the precise relationship requires further investigation. We observed that the number of PG14-EGFP aggregates is positively correlated with transgene expression level: Tg(PG14-EGFP+/+) mice, most of which become ill, displayed more aggregates than Tg(PG14-EGFP+/−) mice, most of which remain healthy. However, aggregation of PG14 PrP occurs long before the onset of neuropathology or clinical disease, as indicated by the existence of fluorescent aggregates in neonatal neurons (Fig. 7) and by the presence of detergent-insoluble protein in neonatal brain tissue (Chiesa et al., 1998). These observations suggest that the pathological consequences of PrP aggregation may take an extended time to evolve. Alternatively, aggregates may increase in size or number over time until a critical threshold level is reached for induction of disease. When we compared young and old animals, we did not observe dramatic differences in the number, size, or distribution of aggregates (data not shown), although careful quantitation will be required to test the possibility that subtle alterations may occur slowly with aging.
Because PrP is a GPI-linked membrane protein, the intra-axonal deposits of PrP-EGFP we observe presumably reside in the lumen of intracellular transport vesicles. These deposits may represent aggregates of the mutant protein within individual vesicles, or possibly collections of multiple vesicles. The axonal localization of PG14-EGFP aggregates is consistent with evidence from immunolocalization studies demonstrating that endogenous PrP is present on axons and pre-synaptic nerve terminals (Moya et al., 2000; Salès et al., 1998), and that it is subject to both anterograde and retrograde fast axonal transport (Borchelt et al., 1994; Moya et al., 2004; Rodolfo et al., 1999).
New insights into mutant PrP localization and trafficking
The picture of PG14 PrP localization in brain provided here using the intrinsic fluorescence of an EGFP fusion protein differs markedly from the one suggested by previous studies of Tg(PG14) mice, all of which relied upon immunostaining following application of antigen retrieval techniques. These earlier studies revealed punctate deposits of the mutant protein in numerous brain regions, including the cerebellum, hippocampus and neocortex. The deposits, which were present primarily in neuropil regions and were largely absent from white matter, were characterized as “synaptic-like”, since they had a distribution reminiscent of synaptic terminals (Chiesa et al., 2000; Chiesa et al., 1998). Recent electron microscopic studies indicate that these deposits are primarily extracellular (M. Jeffrey, A.Z. Medrano, S. Barmada, and D.A. Harris, unpublished data).
Although intracellular deposits of misfolded forms of PrP, including PrPSc, have been described in a few conventional immunohistochemical studies of brain (Kovacs et al., 2005; Laszlo et al., 1992), such deposits may be particularly susceptible to loss or redistribution induced by antigen retrieval methods, explaining why most studies have emphasized extracellular aggregates. Thus, we believe that PrP-EGFP fusion proteins provide a more accurate representation of the distribution of PrP aggregates, particularly those localized to intracellular compartments, than conventional immunocytochemistry. Recently, we have identified prominent intraneuronal deposits of PrPSc in scrapie-infected Tg(WT-EGFP) mice (Barmada and Harris, 2005). Some of these deposits were localized to the Golgi apparatus in neuronal cell bodies, and some were also present along axons. Thus, intra-axonal aggregation may be common to both PrPSc and mutant forms of PrP.
The findings reported here also significantly extend our previous studies of mutant PrP molecules in non-neuronal cell lines, which indicated altered localization and trafficking of these proteins. Consistent with the results presented here for neurons, immunostaining of transfected BHK and CHO cells revealed markedly reduced levels of PG14 and other mutant PrPs at the plasma membrane (Ivanova et al., 2001). This phenomenon is correlated with delayed maturation of mutant PrP molecules in biosynthetic labeling experiments (Drisaldi et al., 2003), and with evidence that mutant PrPs begin to aggregate very soon after synthesis in the ER (Daude et al., 1997). In contrast to our observations in neurons, however, the only abnormal intracellular accumulations of mutant PrP identified by immunostaining of CHO and BHK cells were localized to the ER (Ivanova et al., 2001). We think it likely that, since the immunostaining experiments using transfected cells did not employ antigen retrieval techniques, they detected primarily soluble forms of mutant PrP in transit through the secretory pathway, and missed more highly aggregated deposits such as those visualized here using EGFP fusion proteins. The same limitation may apply to a previous immunolocalization study of PG14 PrP in cultured neurons (Fioriti et al., 2005).
Taken together, the available data suggest that, although PG14 PrP molecules may transit the secretory pathway more slowly than WT PrP, in neurons they are eventually delivered to axonal transport vesicles which are thought to bud from the trans-Golgi (Calakos and Scheller, 1996). Since PG14-EGFP aggregates can be observed arrayed along the length of axons in vivo and in culture, axonal transport of the mutant protein is not completely blocked. However, there is clearly a defect in delivery of PG14 PrP molecules to the surface membrane of axons and nerve terminals, perhaps due to retarded axonal transport or to failure of transport vesicles to fuse with axonal or synaptic target membranes.
A novel pathogenic mechanism
The results reported here suggest the novel hypothesis that PG14 and other aggregation-prone PrP molecules induce pathology by blocking or altering normal axonal transport processes. For example, vesicles laden with PG14 aggregates may fail to reach nerve terminals, or they may cause “traffic jams” of other axonally transported organelles, thereby preventing delivery of essential cargo molecules to synapses. As a consequence of these abnormalities, structural or functional abnormalities in axons or synapses may ensue. Our study of Bax-deficient Tg(PG14) mice, highlighting the importance of synaptic loss in the neurodegenerative process (Chiesa et al., 2005), is consistent with this model, as are reports demonstrating a role for PrP in axon outgrowth and synaptic function (Herms et al., 1999; Kanaani et al., 2005; Moya et al., 2005; Salès et al., 2002). Interestingly, deficiencies in axonal transport have been associated with several other neurodegenerative diseases caused by protein aggregation, including Huntington’s and Alzheimer’s diseases (Goldstein, 2003; Gunawardena et al., 2003; Roy et al., 2005; Stokin et al., 2005). The availability of Tg(PG14-EGFP) mice will now make it possible to perform real-time, fluorescence imaging of the axonal transport of mutant PrP to determine whether abnormalities in cellular trafficking contribute to the disease phenotype.
Supplementary Material
Vibratome sections from the brains of Tg(WT) mice (A, D, G), Tg(PG14) mice (B, E, H), or Prn-p0/0 mice (C, F, I) were immunostained using the following anti-PrP antibodies: P45-66 (which recognizes residues 45-66; (Lehmann and Harris, 1995)) (A–C); 3F4 (which recognizes residues 105–111; (Lund et al., 2007)) (D–F); or 8H4 (which recognizes residues 147–200; (Zanusso et al., 1998)) (G–I). The transgenically encoded PrP expressed by both Tg(WT) and Tg(PG14) mice carries an epitope tag for the 3F4 antibody (Chiesa et al., 1998). Sections were derived from the dentate gyrus (A–C), or the cerebellar cortex (DI). Tg(WT) sections stain strongly with all three antibodies. Tg(PG14) sections stain weakly, but above background levels seen in Prn-p0/0 mice. Abbreviations are: MF, mossy fibers; DGL, dentate granule cell layer; ML, molecular layer; PC, Purkinje cell layer; CGL, cerebellar granule cell layer. The scale bar in A (applicable to all panels) is 20μm.
Vibratome sections were cut from the brains of Tg(WT-PrP) mice (A, D, H, L), as well as the following lines of Tg(PG14-EGFP) mice: line D (B); line X4 (C, E, I, M); line X1 (F, J, N); line X3 (G, K, O). The panels show the whole hippocampus (A–C), the dentate gyrus (D–G), the CA1 sub-field of the hippocampus (H–K), and the cerebellar cortex (L–O). Due to low transgene expression, it was necessary to stain sections from the X1 and X3 lines with anti-GFP antibody in order to detect PG14-EGFP. The abbreviations are: LAC, stratum lacunosum moleculare; AL, alveus; MF, mossy fibers; DGL, dentate granule cell layer; OR, stratum oriens; PYR, pyramidal cells; RA; stratum radiatum; PC, Purkinje cell layer; CGL, cerebellar granule cell layer; ML, molecular layer. The scale bar in A (applicable to A–C) is 200 μm and in D (applicable to D–O) is 20μm.
Acknowledgments
We thank Man-Sun Sy for 8H4 antibody; Richard Kascsak for 3F4 and 6D11 antibodies; and Alex Raeber and Bruno Oesch (Prionics, Zurich, Switzerland) for 15B3 antibody. We also acknowledge Charles Weissmann for supplying Prn-p0/0 mice. We are grateful to Cheryl Adles and Su Deng for mouse colony maintenance and genotyping. This work was supported by a grant from the NIH to D.A.H. (NS040975). A.Z.M. was supported by the Lucille P. Markey Pathway at Washington University, S.J.B. by the Medical Scientist Training Program at Washington University (NIH Grant T32GM07200), and E.B. by a fellowship from Telethon-Italy (GFP04007).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barmada S, et al. GFP-tagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol Dis. 2004;16:527–537. doi: 10.1016/j.nbd.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Barmada SJ, Harris DA. Visualization of prion infection in transgenic mice expressing green fluorescent protein-tagged prion protein. J Neurosci. 2005;25:5824–5832. doi: 10.1523/JNEUROSCI.1192-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, et al. Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci. 1992;12:363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton DC, et al. Molecular location of a species-specific epitope on the hamster scrapie agent protein. J Virol. 1991;65:3667–3675. doi: 10.1128/jvi.65.7.3667-3675.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, et al. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal Biomol Eng. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, et al. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J Biol Chem. 1994;269:14711–14714. [PubMed] [Google Scholar]
- Büeler H, et al. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Calakos N, Scheller RH. Synaptic vesicle biogenesis, docking, and fusion: a molecular description. Physiol Rev. 1996;76:1–29. doi: 10.1152/physrev.1996.76.1.1. [DOI] [PubMed] [Google Scholar]
- Chiesa R, et al. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc Natl Acad Sci USA. 2000;97:5574–5579. doi: 10.1073/pnas.97.10.5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R, et al. Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proc Natl Acad Sci USA. 2005;102:238–243. doi: 10.1073/pnas.0406173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R, et al. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- Chiesa R, et al. Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol. 2003;77:7611–7622. doi: 10.1128/JVI.77.13.7611-7622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daude N, et al. Identification of intermediate steps in the conversion of a mutant prion protein to a scrapie-like form in cultured cells. J Biol Chem. 1997;272:11604–11612. doi: 10.1074/jbc.272.17.11604. [DOI] [PubMed] [Google Scholar]
- Drisaldi B, et al. Mutant PrP is delayed in its exit from the endoplasmic reticulum, but neither wild-type nor mutant PrP undergoes retrotranslocation prior to proteasomal degradation. J Biol Chem. 2003;278:21732–21743. doi: 10.1074/jbc.M213247200. [DOI] [PubMed] [Google Scholar]
- Duchen LW, et al. Dementia associated with a 216 base pair insertion in the prion protein gene: clinical and neuropathological features. Brain. 1993;116:555–567. doi: 10.1093/brain/116.3.555. [DOI] [PubMed] [Google Scholar]
- Fioriti L, et al. Cytosolic prion protein (PrP) is not toxic in N2a cells and primary neurons expressing pathogenic PrP mutations. J Biol Chem. 2005;280:11320–8. doi: 10.1074/jbc.M412441200. [DOI] [PubMed] [Google Scholar]
- Fischer M, et al. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- Goldstein LS. Do disorders of movement cause movement disorders and dementia? Neuron. 2003;40:415–425. doi: 10.1016/s0896-6273(03)00630-5. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Harris DA. Trafficking, turnover and membrane topology of PrP. Br Med Bull. 2003;66:71–85. doi: 10.1093/bmb/66.1.71. [DOI] [PubMed] [Google Scholar]
- Herms J, et al. Evidence of presynaptic location and function of the prion protein. J Neurosci. 1999;19:8866–8875. doi: 10.1523/JNEUROSCI.19-20-08866.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova L, et al. Mutant prion proteins are partially retained in the endoplasmic reticulum. J Biol Chem. 2001;276:42409–42421. doi: 10.1074/jbc.M106928200. [DOI] [PubMed] [Google Scholar]
- Kanaani J, et al. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J Neurochem. 2005;95:1373–1386. doi: 10.1111/j.1471-4159.2005.03469.x. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, et al. Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest. 1987;57:230–236. [PubMed] [Google Scholar]
- Kitamoto T, et al. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Path. 1992;140:1285–1294. [PMC free article] [PubMed] [Google Scholar]
- Kong Q, et al. Inherited prion diseases. In: Prusiner SB, editor. Prion Biology and Diseases. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2004. pp. 673–775. [Google Scholar]
- Korth C, et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature. 1997;390:74–77. doi: 10.1038/36337. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, et al. Subcellular localization of disease-associated prion protein in the human brain. Am J Pathol. 2005;166:287–294. doi: 10.1016/S0002-9440(10)62252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, et al. Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Mol Brain Res. 1995;34:173–176. doi: 10.1016/0169-328x(95)00175-r. [DOI] [PubMed] [Google Scholar]
- Laszlo L, et al. Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J Pathol. 1992;166:333–341. doi: 10.1002/path.1711660404. [DOI] [PubMed] [Google Scholar]
- Lehmann S, Harris DA. A mutant prion protein displays an aberrant membrane association when expressed in cultured cells. J Biol Chem. 1995;270:24589–24597. doi: 10.1074/jbc.270.41.24589. [DOI] [PubMed] [Google Scholar]
- Lund C, et al. Characterization of the prion protein 3F4 epitope and its use as a molecular tag. J Neurosci Methods. 2007;165:183–190. doi: 10.1016/j.jneumeth.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Miller TM, Johnson EM., Jr Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule cells. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya KL, et al. Axonal transport of the cellular prion protein is increased during axon regeneration. J Neurochem. 2005;92:1044–1053. doi: 10.1111/j.1471-4159.2004.02940.x. [DOI] [PubMed] [Google Scholar]
- Moya KL, et al. Enhanced detection and retrograde axonal transport of PrPC in peripheral nerve. J Neurochem. 2004;88:155–160. doi: 10.1046/j.1471-4159.2003.02150.x. [DOI] [PubMed] [Google Scholar]
- Moya KL, et al. Immunolocalization of the cellular prion protein in normal brain. Microsc Res Tech. 2000;50:58–65. doi: 10.1002/1097-0029(20000701)50:1<58::AID-JEMT9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Narwa R, Harris DA. Prion proteins carrying pathogenic mutations are resistant to phospholipase cleavage of their glycolipid anchors. Biochemistry. 1999;38:8770–8777. doi: 10.1021/bi990736c. [DOI] [PubMed] [Google Scholar]
- Owen F, et al. A dementing illness associated with a novel insertion in the prion protein gene. Mol Brain Res. 1992;13:155–157. doi: 10.1016/0169-328x(92)90056-h. [DOI] [PubMed] [Google Scholar]
- Pankiewicz J, et al. Clearance and prevention of prion infection in cell culture by anti-PrP antibodies. Eur J Neurosci. 2006;23:2635–2647. doi: 10.1111/j.1460-9568.2006.04805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SK, et al. Development of polarity in cerebellar granule neurons. J Neurobiol. 1997;32:223–236. doi: 10.1002/(sici)1097-4695(199702)32:2<223::aid-neu7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, editor. Prion Biology and Diseases. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2004. [Google Scholar]
- Rodolfo K, et al. A novel cellular prion protein isoform present in rapid anterograde axonal transport. Neuroreport. 1999;10:3639–3644. doi: 10.1097/00001756-199911260-00032. [DOI] [PubMed] [Google Scholar]
- Roy S, et al. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol (Berl) 2005;109:5–13. doi: 10.1007/s00401-004-0952-x. [DOI] [PubMed] [Google Scholar]
- Salès N, et al. Developmental expression of the cellular prion protein in elongating axons. Eur J Neurosci. 2002;15:1163–1177. doi: 10.1046/j.1460-9568.2002.01953.x. [DOI] [PubMed] [Google Scholar]
- Salès N, et al. Cellular prion protein localization in rodent and primate brain. Eur J Neurosci. 1998;10:2464–2471. doi: 10.1046/j.1460-9568.1998.00258.x. [DOI] [PubMed] [Google Scholar]
- Shyng SL, et al. The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J Biol Chem. 1995;270:14793–14800. doi: 10.1074/jbc.270.24.14793. [DOI] [PubMed] [Google Scholar]
- Stokin GB, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Van Everbroeck B, et al. Antigen retrieval in prion protein immunohistochemistry. J Histochem Cytochem. 1999;47:1465–1470. doi: 10.1177/002215549904701112. [DOI] [PubMed] [Google Scholar]
- Zanusso G, et al. Prion protein expression in different species: analysis with a panel of new mAbs. Proc Natl Acad Sci USA. 1998;95:8812–8816. doi: 10.1073/pnas.95.15.8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Vibratome sections from the brains of Tg(WT) mice (A, D, G), Tg(PG14) mice (B, E, H), or Prn-p0/0 mice (C, F, I) were immunostained using the following anti-PrP antibodies: P45-66 (which recognizes residues 45-66; (Lehmann and Harris, 1995)) (A–C); 3F4 (which recognizes residues 105–111; (Lund et al., 2007)) (D–F); or 8H4 (which recognizes residues 147–200; (Zanusso et al., 1998)) (G–I). The transgenically encoded PrP expressed by both Tg(WT) and Tg(PG14) mice carries an epitope tag for the 3F4 antibody (Chiesa et al., 1998). Sections were derived from the dentate gyrus (A–C), or the cerebellar cortex (DI). Tg(WT) sections stain strongly with all three antibodies. Tg(PG14) sections stain weakly, but above background levels seen in Prn-p0/0 mice. Abbreviations are: MF, mossy fibers; DGL, dentate granule cell layer; ML, molecular layer; PC, Purkinje cell layer; CGL, cerebellar granule cell layer. The scale bar in A (applicable to all panels) is 20μm.
Vibratome sections were cut from the brains of Tg(WT-PrP) mice (A, D, H, L), as well as the following lines of Tg(PG14-EGFP) mice: line D (B); line X4 (C, E, I, M); line X1 (F, J, N); line X3 (G, K, O). The panels show the whole hippocampus (A–C), the dentate gyrus (D–G), the CA1 sub-field of the hippocampus (H–K), and the cerebellar cortex (L–O). Due to low transgene expression, it was necessary to stain sections from the X1 and X3 lines with anti-GFP antibody in order to detect PG14-EGFP. The abbreviations are: LAC, stratum lacunosum moleculare; AL, alveus; MF, mossy fibers; DGL, dentate granule cell layer; OR, stratum oriens; PYR, pyramidal cells; RA; stratum radiatum; PC, Purkinje cell layer; CGL, cerebellar granule cell layer; ML, molecular layer. The scale bar in A (applicable to A–C) is 200 μm and in D (applicable to D–O) is 20μm.