

Abstract
Enterobacter sakazakii (ES) causes neonatal meningitis and necrotizing enterocolitis with case-fatality rates among infected infants ranging from 40 to 80%. Very little is known about the mechanisms by which these organisms cause disease. Here, we demonstrate that ES invades human brain microvascular endothelial cells (HBMEC) with higher frequency when compared with epithelial cells and endothelial cells from different origins. The entry of ES into HBMEC requires the expression of outer membrane protein A (OmpA), as the OmpA-deletion mutant was sevenfold less invasive than the wild type ES and the bacterium does not multiply inside HBMEC. Anti-OmpA antibodies generated against the OmpA of Escherichia coli K1, which also recognize the OmpA of ES, did not prevent the invasion of ES in HBMEC. ES invasion depends on microtubule condensation in HBMEC and is independent of actin filament reorganization. Both PI3-kinase and PKC-α were activated during ES entry into HBMEC between 15 min and 30 min of infection. Concomitantly, overexpression of dominant negative forms of PI3-kinase and PKC-α significantly inhibited the invasion of ES into HBMEC. In summary, ES invasion of HBMEC is dependent on the expression of OmpA similar to that of E. coli K1; however, the epitopes involved in the interaction with HBMEC appears to be different.
Keywords: Enterobacter sakazakii, Outer membrane protein A, Endothelial cells, Protein kinase C-α, 1-Phosphatidylinositol 3-kinase, Microtubules
1. Introduction
Enterobacter sakazakii (ES) is a gram-negative rod-shaped bacterium within the family Enterobacteriaceae. ES is a food borne pathogen that can cause severe illness and death in newborns [1–4]. The majority of cases of ES infection occurs in neonates, and may result in sepsis, meningitis, and necrotizing enterocolitis. The case-fatality rates among infected infants have been reported to be as high as 40–80% [4]. In addition to neonatal infection, ES has been associated with adult bacteremia and osteomyelitis [5]. Premature infants and those with underlying medical conditions may be at highest risk for developing ES infection. A growing number of outbreaks of infection among neonates have provided compelling evidence that milk-based powdered infant formula are the source of infection [1]. Gram-negative bacterial meningitis is typically treated with third generation cephalosporins. Unfortunately, ES commonly contains chromosomally mediated, depressed type 1 β-lactamases, which renders cephalosporins ineffective [6,7]. Therefore, improved understanding of the pathogenesis of ES infection is crucial to the development of innovative and effective therapeutic strategies.
Several pathogenic bacteria that cause central nervous system infections such as Escherichia coli K1, Citrobacter freundii, Listeria monocytogenes and Group B streptococcus invade human brain microvascular endothelial cells (HBMEC), an in vitro model of the blood–brain barrier [8–11]. E. coli K1 has also been shown to traverse across the blood–brain barrier in the experimental newborn rat model of hematogenous meningitis [12]. Interestingly, these bacteria invade HBMEC with lower frequency (0.05–1%) when compared to the invasion frequency of other bacteria such as Salmonella serovar Typhimurium in epithelial cells. However, entry of very few bacteria is sufficient to induce meningitis in animal model. In addition, the meningitis causing pathogens also use contrasting mechanisms to invade HBMEC. To assess the role of ES in human disease, evaluation of potential virulence factors is required. However, at this time very little is known about the mechanisms of ES pathogenicity in meningitis. It is a common trait of microbial pathogens to express adherence factors responsible for recognizing and binding to specific receptor moieties of cells, thereby enabling the bacteria to resist host strategies that would impede colonization. Recently, two distinctive adherence patterns including a diffuse adhesion pattern and the formation of localized clusters of bacteria on the cell surface could be distinguished on epithelial cells and brain endothelial cells [13]. Adherence was maximal during late exponential phase, and increased with higher inoculum sizes. Mannose, hemagglutination, trypsin digestion experiments and transmission electron microscopy suggest that the adhesion of ES to the epithelial and endothelial cells is predominantly non-fimbrial based. Invasion of ES in Caco-2 cells has been shown to require microtubules and induced the disruption of tight junctions [14].
Venkitanarayanan et al. reported that the outer membrane protein A (OmpA) of ES is 88% similar at the protein level to that of E. coli K1 and plays a critical role in invasion of human intestinal epithelial cells, which has been shown to depend on both microfilaments and microtubules [15,16]. Of note, our studies with E. coli K1, another meningitis causing gram-negative bacterium, also require OmpA for its invasion in HBMEC [8]. E. coli K1 interacts with a receptor on HBMEC and induces actin condensation at the sites of bacterial entry [17]. The entry of E. coli is specific to only HBMEC but not of other non-brain endothelial or epithelial cells [12]. We further demonstrated that activation of protein kinase C-α, PI3-kinase, and caveolin-1 is required for the condensation of actin filaments during E. coli invasion [18–20]. However, mechanisms involved in the entry of ES into HBMEC are not clearly known. In an effort to understand the pathogenesis of meningitis caused by ES, we have used HBMEC to study the interaction between ES and HBMEC. Here we report that ES efficiently invades HBMEC in vitro, when compared to other cell types and that the OmpA expression in ES is required for invasion of HBMEC. Additionally, we attempt to elucidate the eukaryotic mechanisms triggered by ES invasion through the use of different inhibitors, and by overexpressing dominant negative forms of PKC-α and PI3-kinase. The data obtained in this study were compared throughout with the mechanisms utilized by E. coli K1. A better understanding of the molecular underpinnings of ES pathogenesis will allow the development of targeted and novel therapeutic strategies.
2. Results
2.1. ES invades HBMEC in a dose- and time-dependent manner
The ability of ES to enter HBMEC was assessed via cell culture invasion assays (gentamicin protection assay) as previously described [8]. E. coli K1 and HB101 strains were used as positive and negative controls, respectively. Initially, we examined the effect of inoculum size (104–107 cfu/well) on bacterial adhesion and invasion of HBMEC. Approximately 9% (8.5 ± 1.5 ×104 cfu/well) of ES added to the monolayers bound at inoculum size of 106 cfu/well and further increase in the inoculum resulted in a small increase in binding (9.5 ± 1.3 ×104 cfu/well) after 2 h incubation (Fig. 1A). E. coli K1 showed increased binding as the inoculum size increases (7% at inoculum size 106), whereas only 1% of HB101 bound to HBMEC. The invasion of ES was found to be approximately 0.5% (8450 ± 2500 cfu/well) at a multiplicity of infection of 10 (bacteria-to-cell ratio 10:1, i.e. 106 cfu/well) whereas lower inoculum sizes show negligible invasion. Of note, greater number of bacteria entered the cells at an inoculum size of 107 cfu/well compared to inoculum size of 106 cfu/well although the percent of invasion was dropped to 0.05–0.09 (5. 1 ± 0.9 × 103 cfu/well) (Fig. 1B). E. coli K1 invaded HBMEC at a frequency of 0.35% at an MOI of 10. In contrast, very small number of HB101 invaded the cells. Next, we examined whether increasing incubation times had any effect on the bacterial invasion with an inoculum size of 106. The binding and invasion frequency increased by twofold at 4 h post-incubation when compared to the frequency at 2 h (Fig. 1C and D). However, the invasion frequency in HBMEC at any given point appears to be between 0.5 and 1.0% after considering the growth of the bacteria in the medium. Similarly, both binding and invasion of E. coli K1 in HBMEC increased over time. Neither binding nor invasion of HB101 changed significantly with increased incubation time. These results suggest that both ES and E. coli K1 invade HBMEC with a similar frequency. Hereafter, the invasion experiments are performed by incubating ES at an MOI of 10 for 4 h unless otherwise stated.
Fig. 1.

Invasion of ES in HBMEC is dose- and time-dependent and significantly higher than entry into epithelial cells and non-brain endothelial cells. (A and B) Confluent monolayers of HBMEC were infected with different concentrations of ES, E. coli K1 or HB101 for 2 h or (C and D) with 106 cfu/well for different time points, followed by gentamicin treatment (100 μg/ml) for 60 min. The cells were then treated with 0.5% Triton X-100 to release the intracellular bacteria and enumerated by plating the dilutions on blood agar (B and D). Total cell associated bacteria were determined as described for invasion except that the gentamicin step was omitted (A and C). The data represent the number of bacteria bound or invaded per well in 24 well plates. The error bars represent means ± SD from three independent experiments performed in triplicate. (E) Human umbilical vein endothelial cells (HUVEC), rat intestinal epithelial cells (IEC-6), human conjunctival epithelial cells (CEC), Chinese hamster ovary cells (CHO) or HBMEC were grown in 24-well cell culture plates to 95% confluence and were incubated with 106 cfu/well of ES for 4 h. Total cell associated (binding) and intracellular bacteria (invasion) were determined. The data represent relative values considering the binding to and invasion of HBMEC as 100%. The error bars indicate standard deviations for means of four separate experiments performed in triplicate. The invasion of ES was significantly lower in epithelial cells or HUVEC compared to the invasion of HBMEC (*p < 0.001 and **p < 0.05 by Student’s t test).
2.2. ES exhibits greater invasion frequency in HBMEC compared to human umbilical vein endothelial cells or epithelial cells
Our previous studies have shown that BMEC differ from other endothelial cells in their cell-to-cell “tightness” and expression of certain receptors [21,22]. In addition, E. coli K1 specifically invades HBMEC compared to other non-brain endothelial cells and epithelial cells [12]. Therefore, we examined the ability of ES to enter various cells or cell lines such as human umbilical vein endothelial cells (HUVEC), rat intestinal epithelial cells (IEC-6), conjunctival epithelial cells (CEC) or Chinese hamster ovary cells (CHO) using gentamicin protection assays. ES was found to be non-invasive in CEC and IEC cell lines (0.03% and 0.04%) and invades with lower frequency into CHO cells and HUVEC (0.2% and 0.3%, respectively) as compared to 0.7% in HBMEC (Fig. 1E). Of note, the total cell associated bacteria to these cells were similar, suggesting that HBMEC are more susceptible to ES invasion. A longer time period of incubation of these cells with ES showed further increase in the invasion, however, the percent of invasion remains same when considered the growth of the bacteria in the medium (data not shown).
2.3. ES does not multiply inside HBMEC
To ascertain whether ES survives and replicates within HBMEC, invasion assays were performed as previously described except with a prolonged gentamicin treatment time. The number of intracellular bacteria remained constant event after 8 h post-infection when the inoculum size used was 106 cfu/well, suggesting that the invaded bacteria did not multiply inside HBMEC (Fig. 2A). To confirm that ES is still surviving inside HBMEC, GFP labeled ES was added to the endothelial cell monolayers at an MOI of 10 and incubated for 4 h. The monolayers were washed and further incubated in the presence of gentamicin for additional 4 h. The fixed monolayers were observed under confocal laser microscope and optical sectioning was performed from top of the cell to the bottom. As shown in Fig. 2B, several ES were attached as clusters to HBMEC (Fig. 2Ba). z-Stack sectioning at the middle of the cell revealed that the bacteria were still intact (round shape, Fig. 2Be and f). Transmission electron microscopy (TEM) was performed to further confirm this phenomenon and also characterize the interaction of ES with HBMEC. TEM photographs revealed various stages of ES entry into HBMEC, including an initial attachment followed by an intimate interaction between the bacteria and the cell surface (Fig. 2Ca and b). Visible condensation of electron-dense particles accumulating within the vicinity of contact was observed. ES was found to penetrate HBMEC by inducing HBMEC pseudopods during this time. After extended incubation for a total of 4 h, ES was observed in single membrane vacuole-like structures (Fig. 2Bc). However, after 8 h, several bacteria invaded HBMEC still remained in the vacuoles without any significant multiplication. Rarely were bacteria found to be multiplying in a single phagosome or in between two endothelial cells. These results suggest that ES enters through the cell membrane but not via the tight junctions, and does not efficiently multiply in HBMEC.
Fig. 2.

Internalized ES does not multiply inside HBMEC. (A) HBMEC were infected with ES for 4 h followed incubation with gentamicin containing medium for additional 4 h and 8 h. The number of intracellular bacteria was determined as described in Fig. 1. The data represent means ± SD from three independent experiments performed in triplicate. (B) HBMEC monolayers were infected with GFP+ ES for 4 h, washed, and fixed with 2% paraformaldehyde. z-Stacks of confocal images were acquired using a Leica confocal laser-scanning microscope and Leica confocal software equipped with 63× oil lens. (C) In separate experiments, HBMEC monolayers were infected with 106 cfu/well for varying periods. Samples were taken at 1 h (a), 2 h (b), and 8 h (c) post-infection and were processed for transmission electron microscopy as described in Section 4. Arrows indicate the vacuoles containing ES. Magnification for the included photographs: (a, b) 12,000; (c) 13,000 and (d) 8500.
2.4. OmpA expression is required for ES invasion of HBMEC
Studies from our lab have demonstrated that OmpA contributes to E. coli K1 invasion of HBMEC by interacting with an HBMEC specific receptor, Ecgp [23]. The OmpA–Ecgp interaction was blocked by the anti-OmpA antibodies, which mainly recognizes external loops of E. coli OmpA that was generated in our lab, and thereby the invasion of E. coli K1. Since the OmpA of ES has 88% sequence similarity to that of OmpA of E. coli K1 at the protein level, we examined whether the anti-OmpA antibody recognizes the OmpA of ES by dot-blot analysis. As shown in Fig. 3A, both ES and E. coli K1 were reacted to the anti-OmpA antibodies but not OmpA – E. coli. However, neither adhesion nor invasion was significantly affected by prior treatment of ES with anti-OmpA antibodies (8.5 × 103 ± 1.0 × 103 cfu/well invaded in controls versus 7.9 × 103 ± 1.8 × 103 cfu/well invaded in anti-OmpA treated cells, Fig. 3B). To examine whether OmpA expression is required for the invasion of ES into HBMEC, an OmpA deletion mutant was generated. Three independently isolated OmpA− ES colonies were examined by PCR with ompA specific primers for the deletion of ompA in ES51R, which data showed no amplification of 1.7 kb ompA gene in all the mutants (data not shown). The expression of OmpA by Western blotting with anti-OmpA antibodies revealed that the wild type ES and E. coli K1 reacted to a 35 kDa protein, whereas the OmpA−ES showed no reactivity (Fig. 3C), suggesting that the OmpA− ES strains are devoid of OmpA. These strains when used in the invasion assays have shown significantly lower invasion in HBMEC than the wild type ES despite binding to the cells similar to that of OmpA+ ES (Fig. 3D), indicating that OmpA was required for the invasion. Taken together these results suggest that the other epitopes of OmpA distinct than those involved in E. coli K1 interaction are required for interaction with HBMEC surface structures for invasion of ES.
Fig. 3.

OmpA expression in ES is required for the invasion of HBMEC. (A) Approximately 103 cfu of ES were loaded onto nitrocellulose along with OmpA+ and OmpA− E. coli (as positive and negative controls) and immunoblotted with anti-OmpA antibodies or control isotype matched antibodies followed by horseradish peroxidase conjugated secondary antibodies. The color was developed using diaminobenzidine and hydrogen peroxide. (B) Various concentrations (25 μg, 50 μg, and 100 μg) of purified anti-OmpA antibodies or control antibodies (cAb) were incubated with ES (106 cfu) for 1 h on ice, then added to confluent monolayers of HBMEC and incubated for 4 h. Both total cell associated and intracellular bacteria were determined and the results expressed as relative binding to or invasion being taken the parameters for untreated HBMEC as 100%. The data represent means ± SD of three separate experiments performed in triplicate. (C) Total lysates of OmpA+ E. coli, ES resistant to rifampicin (ES51R), or OmpA− ES strains were subjected to immunoblotting with anti-OmpA antibody. (D) HBMEC monolayers were infected with either ES resistant to rifampicin or various OmpA− ES strains at an MOI of 10 for 4 h. Total cell associated or intracellular bacteria were determined as described in Section 4. The data represent means ± SD from at least three different experiments performed in triplicate and expressed as relative binding or invasion being taken the total cell associated and intracellular bacteria of HBMEC as 100%. The invasion of OmpA− ES was significantly lower than that of wild type ES, p < 0.001 by Student’s t test.
2.5. Microtubules, PKC-α and PI3-kinase are required for ES invasion of HBMEC
To determine whether cytoskeleton and/or signaling molecules of HBMEC are necessary for ES invasion, the effects of various eukaryotic inhibitors on the invasion process were assessed. The role of actin-based cytoskeleton in ES invasion was examined by using cytochalasin D (CD), an agent that causes microfilament depolymerization in eukaryotic cells. HBMEC were pretreated with various concentrations of CD for 30 min prior to invasion assays. As shown in Fig. 4A, CD did not show any profound effect on the ability of ES to invade HBMEC even at a concentration of 10 μg. In contrast, pretreatment of HBMEC with nocodazole, which is a microtubule-depolymerizing agent, led to inhibition of ES invasion of HBMEC (Fig. 4B). Cells pretreated with 1.0 μM nocodazole demonstrated 50% decrease in invasion, whereas 10 μM concentration showed ~80% inhibition. In addition, inhibitors to other signaling molecules such as PKC (GFx), PI3-kinase (wortmannin), and filipin (caveolae formation) were also used. The data suggested that PI3-kinase inhibitor wortmannin at a concentration of 400 nM significantly reduced the ES invasion (Fig. 4C). The general PKC inhibitor GFx showed only marginal inhibition of ES invasion, whereas filipin had no effect. Of note, none of these inhibitors significantly affected the total cell associated ES to HBMEC. These results suggest that microtubule polymerization, PKC-α, and PI3-kinase play an important role in the invasion process, but not microfilaments and caveolae formation.
Fig. 4.
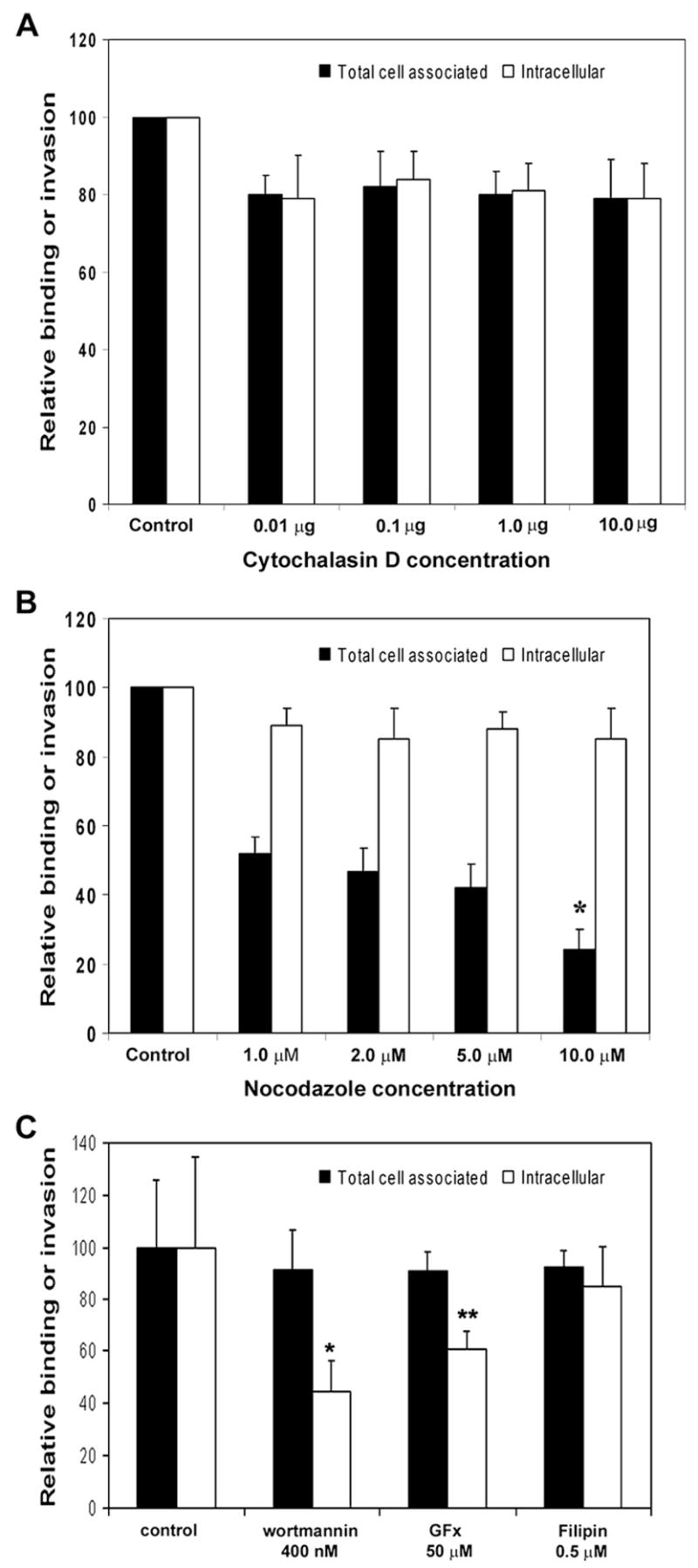
Effect of various inhibitors on ES invasion of HBMEC. Different concentrations of cytochalasin D (A), nocodazole (B) or wortmannin, GFx and filipin (C) were added to confluent monolayers of HBMEC for 30 min at 37 °C prior to the addition of ES. Total cell associated and intracellular ES was determined as described earlier. The experiments were performed at least 3 times in triplicate and are expressed as means ± SD. *p < 0.001 and **p < 0.05 by Student’s t test.
2.6. Microtubule aggregation is associated with ES invasion of HBMEC
To further examine the role of microtubules or actin filaments in ES invasion of HBMEC, we examined by fluorescence microscopy whether there were changes in the microtubule or actin network. HBMEC monolayers were infected with either GFP-OmpA+ ES or GFP-OmpA− ES for varying periods, fixed and stained with either rhodamine phalloidin or anti-α tubulin antibody. Staining of non-infected HBMEC showed uniform distribution of actin filaments throughout the cell with occasional stress fibers, whereas no significant background green fluorescence (for GFP) was observed (Fig. 4A and B). The OmpA+ ES infected cells showed that the bacteria attached in cluster to the cells surface (represents both adhered and invaded) at 60 min and no actin condensation underneath or away from the bacteria was observed, providing further evidence that actin filament reorganization is not involved in the entry of ES into HBMEC. On the other hand, microtubule condensation beneath the bacterial attachment site was prominently observed with OmpA+ ES infected cells (Fig. 5G). Overlay of green and red fluorescence images suggested clear co-localization of ES and microtubules (note the yellow color) (Fig. 5I). In contrast, OmpA− ES despite binding to the cells in good numbers did not show microtubule condensation (Fig. 5J–L) suggesting that OmpA mediated microtubule reorganization is required for ES invasion of HBMEC.
Fig. 5.

Microtubule aggregation is associated with ES invasion of HBMEC. Confluent monolayers of HBMEC in eight-well chamber slides were either uninfected (A–C) or infected with ES (D–I) or OmpA− ES for 60 min. The cells were then washed, fixed with 2% paraformaldehyde for 15 min, and stained with rhodamine phalloidin (A–F) or anti-α-tubulin antibody followed by incubation with Cy3-conjugated secondary antibody (G–L). Overlay images of red, green, and blue (for nucleus) were also shown (C, F, I, and L). Magnification: 100×.
2.7. Activation of PI3-kinase and PKC-α during ES invasion of HBMEC
As shown, the general PKC inhibitor (GFx) marginally blocked ES invasion into HBMEC, whereas PI3-kinase inhibitor significantly abrogated the invasion. Our previous studies showed that E. coli K1 invasion of HBMEC requires activation of both PI3-kinase and PKC-α [18,19]. Therefore, we examined the activation of these molecules in HBMEC following infection with ES. Confluent monolayers of HBMEC were treated with ES for 0–90 min and the total cell lysates were examined for the activation of PI3-kinase and PKC-α. Since Akt is the effector protein of PI3-kinase, we examined the phosphorylation of Akt to evaluate the PI3-kinase activation. As shown in Fig. 6A, the phosphorylation of Akt was increased by 15 min post-infection, which was gradually reduced by 90 min when compared to control cells. The blot was stripped and reprobed with an antibody to non-phosphorylated form of Akt to verify equal loading of the proteins. Similarly, the total cell lysates were probed with anti-phospho-PKC-α antibody, which demonstrated activation between 15 min and 30 min post-infection, when compared to untreated control cells (Fig. 6B). Differences in PKC-α phosphorylation level are not due to unequal loading of the proteins, since the antibodies to the non-phosphorylated form of PKC-α showed similar protein levels. Together, these results suggest that the entry of ES into HBMEC induces both PI3-kinase and PKC-α activation.
Fig. 6.

ES incubation of HBMEC activates PI3-kinase and PKC-α. Confluent monolayers of HBMEC were infected with ES (106 cfu/well) for 0–90 min, total cell lysates prepared, and subjected to Western blotting with anti-phospho-Akt or anti-Akt antibodies (A). Similar blot was also subjected to Western blotting with anti-phospho-PKC-α or anti-PKC-α antibody (B). In addition, HBMEC were transfected with dominant negative mutants of either p85 subunit of PI3-kinase or PKC-α. Total cell associated (binding) and intracellular bacteria (invasion) were determined relative to the binding and invasion of HBMEC being taken as 100%. The experiments were performed at least 3 times in triplicate and the data are expressed as means ± SD (C). The invasion of ES into HBMEC transfectants was significantly lower than into HBMEC transfected with pcDNA3, *p < 0.001 by Student’s t test. (D) HBMEC, non-transfected (a–d), transfected with plasmid alone (e–h) or with DN-PKC-α were either non-infected (a–d) or infected with GFP+ ES for 90 min (e–l). The cells were washed, fixed, and stained with anti-phospho-PKC-α antibody (c, g, and k). The bright light images were taken to show the morphology and boundaries of the cells (a, e, and i). The overlay images showed that ES entry induced the accumulation of phospho-PKC-α (yellow color) in HBMEC.
2.8. Overexpression of dominant negative forms of PKC-α and PI3-kinase subunit p85 prevents the invasion of ES into HBMEC
To further confirm the role of PKC-α in ES invasion, HBMEC were transfected with mammalian expression vectors containing hemagglutinin tagged PKC-CAT/KR. The resulting cell lines were designated as PKC-CAT/KR/HBMEC, which confer G418 resistance. The PKC-CAT/KR construct encodes a truncated protein in which the catalytic domain (CAT) containing amino acids 326–672 of PKC is preserved, with a point mutation that abolishes ATP binding ability, while the regulatory N-terminal domain is deleted [24]. The PKC-CAT/KR construct has been shown to dominantly inhibit PKC-α activation. HBMEC transfected with pcDNA3 vector alone were used as a control. We have previously demonstrated that the cell lysates from PKC-CAT/KR/HBMEC show greater levels of PKC-α protein when compared with either non-transfected or pcDNA3-transfected HBMEC [18]. Invasion assays using these transfectants reveal that ES entry was significantly inhibited into PKC-CAT/KR/HBMEC when compared to pcDNA3/HBMEC (1275 ± 275 cfu/well in DN transfected cells versus 7525 ± 480 cfu/well in pcDNA3-transfected cells, p < 0.001) (Fig. 6C), suggesting that PKC-α is required for ES entry into HBMEC.
In addition to PKC-α, PI3-kinase appears important in ES invasion, as wortmannin significantly inhibited the entry. To further demonstrate the role of PI3-kinase, HBMEC were transfected with dominant negative mutants (DNs) of either p85 (p85), or p110 (p110K) subunits of PI3-kinase. The cDNAs of both mutants were cloned under the control of cytomegalovirus promoter with an amino-terminal FLAG epitope tag in the eukaryotic expression vector pcDNA3, which also confers G418 resistance [19]. The DN/p85 mutant contains a defective iSH2 region, fails to bind to PI3-kinase catalytic subunit p110, and dominantly inhibits the activation of PI3-kinase by titrating out the signaling molecules that interact with PI3-kinase. Whereas, DN/p110K is a kinase inactive mutant, has a CAAX motif, and is constitutively translocated to the membrane. HBMEC colonies resistant to G418 were pooled and used in further experiments. Previous studies from our lab revealed that both DN/p110 and DN/p85 were overexpressed only in HBMEC transfected with appropriate vector but not in cells transfected with pcDNA3 [19]. HBMEC transfected with pcDNA3 or PI3-kinase mutants when used in ES invasion assays revealed that ES invasion of HBMEC was blocked by 85% and 50%, respectively, in cells expressing DN/p85 and DN/p110K (7525 ± 489 cfu/well with pcDNA3 versus 1860 ± 468 cfu/well and 3570 ± 251 cfu/well in HBMEC transfected with DN/p85 and DN/p110K, respectively, p < 0.002) (Fig. 6C). However, the total cell associated bacteria was not significantly different between these two transfectants when compared to control cells. These results clearly support that both PKC-α and PI3-kinase are crucial for ES entry into HBMEC.
2.9. Overexpression of dominant negative forms of PKC-α and PI3-kinase subunit p85 prevents the microtubule condensation beneath the ES attachment site
To examine whether the activation of PKC-α and PI3-kinase is upstream or downstream of microtubule condensation immuno-cytochemistry was performed using GFP-ES and HBMEC transfectants. Either PKC-CAT/KR/HBMEC or DN/p85/HBMEC were infected with GFP-ES for 2 h, fixed, and examined under a confocal laser microscope. As shown in Fig. 6D, the uninfected pcDNA3/HBMEC showed no green fluorescence (for ES) or very diffused red fluorescence for microtubules (a–d). In contrast, infected pcDNA3/HBMEC showed attached ES under which microtubule condensation was visible. Overlay of both green and red images show that the condensation of microtubules was directly underneath the ES attachment site. Of note, ES infected PKC-CAT/KR/HBMEC despite showing the attachment of bacteria did not reveal the condensation of microtubules (I–l). Similar results were also observed with DN/p85/HBMEC (data not shown). These data suggest that activation of both PKC-α and PI3-kinase could be upstream of microtubule condensation.
3. Discussion
E. sakazakii is an opportunistic pathogen associated with contaminated powdered infant formula and a rare cause of gram-negative sepsis that can develop into meningitis and brain abscess formation in neonates. The pathogenesis of ES meningitis and brain abscess formation is not clearly understood. However, it is widely believed that like other meningitis causing bacteria, ES must also penetrate the blood–brain barrier, an intricate structure that consists of the brain capillary endothelium, to cause disease. Several genotypically distinct ES strains were shown to invade rat capillary endothelial brain cells in vitro [25], however, the mechanisms involved in the invasion process remain unknown. In this report, we have utilized HBMEC to gain insights into the pathogenic mechanisms utilized by ES during the invasion.
Our data suggest that ES invades HBMEC with higher frequency than that of IEC-6, CEC and CHO cells. These results are similar to the invasion characteristics of E. coli K1, which specifically invades HBMEC with a 10-fold higher frequency than HUVEC [12]. In contrast, ES showed the capability of invading HUVEC, although at 50% reduced frequency. We also demonstrated that E. coli K1 invasion requires the interaction of OmpA with its receptor, Ecgp, a gp96 homologue and that the anti-OmpA antibodies significantly inhibited the invasion [22]. Interestingly, the invasion of ES into HBMEC also depends on the expression of OmpA although the antibodies to OmpA did not block the invasion, indicating that the interaction of ES may be different from that of E. coli K1 interaction with HBMEC. It is possible that different epitopes of OmpA are involved in the interaction of ES when compared to E. coli K1 with HBMEC. Our studies on the interaction of E. coli K1 OmpA with GlcNAc1–4GlcNAc epitopes by computer simulation revealed that OmpA is highly mobile structure and needs to be stabilized by other structures such as IbeA on the bacterial surface to acquire a special three dimensional conformation [26]. Thus, the OmpA of ES is probably acquiring an entirely different orientation to interact with its cognate receptors on HBMEC to invade the cells. In support of this notion, ES invasion was dependent on microtubule reorganization whereas E. coli invasion has been shown to depend on microfilament condensation [17]. Similar to these studies ES invasion of INT407 cells has also been shown to be dependent on OmpA expression in ES, however, requires both microfilaments and microtubules for invasion [16]. This observation is in contrast to our study that intestinal epithelial cells (IEC-6) are not susceptible for invasion by ES [27]. This discrepancy could be due to the methods used for invasion assays. During the interaction of ES with INT407 cells, the authors centrifuged the bacteria against the cells to increase the chances of binding to the cells, while we just performed stand-alone experiments. Kim and Loessner also demonstrated that ES ATCC 29544 invades Caco-2 cells with a frequency of 0.5–1.0% and requires microtubules for invasion, although cytochalasin D treatment increased the invasion [14]. These experimental discrepancies from different studies could be due to the use of different cell types and/or ES strains. For example, studies using the same cells demonstrated that the entry of C. freundii is similar to that of ES in HBMEC, as it depends on the microtubule reorganization. The microtubule aggregation is directly beneath the ES attachment site whereas C. freundii induces the condensation away from the attachment site [9]. Microtubules have been previously shown to be required for invasion of many pathogens (e.g., Neisserria gonorrhoeae, Haemophilus influenzae, Klebsiella pneumoniae, enteropathogenic and enterohemorrhagic E. coli, and Campylobacter jejuni) [28–32].
Cytoskeletal rearrangements during the invasion of bacteria require cross-talk between varieties of host-signaling molecules. Our previous studies demonstrated that HBMEC invasion by E. coli requires PKC-α and PI3-kinase for actin rearrangements [18,19]. ES invasion showed the involvement of both PKC-α and PI3-kinase, but was dependent upon microtubules. Overexpression of DN-PKC-α significantly prevented the entry of ES into HBMEC although a general inhibitor of PKC showed a marginal effect. Of note, since filipin could not block the invasion, caveolae formation is not required for ES entry, which contrasts with the mechanisms of E. coli invasion. Upon activation of PI3-kinase, the catalytic subunit p110 catalyzes the formation of phosphatidyl 3,4,5-trisphosphate [33]. Binding of phosphatidyl 3,4,5-trisphosphate to pleckstrin homology domains of cellular and cytoskeletal proteins mediates membrane recruitment of several kinases, including PDK1 [34]. PDK1 has been reported to form a complex with PKC, which subsequently phosphorylates the activation loop of most members of the PKC family. Activation of PKC has also been shown in EPEC infection [35,36]. It has been shown that both PKC-α and PI3-kinase stabilize the microtubules during human herpes virus entry of dermal microvascular endothelial cells [35]. Therefore it is possible that microtubule stabilization underneath the ES attachment site 6 may be provided by these two molecules for subsequent movement of bacteria containing endosomes along the microtubules. Taken together, we have shown that OmpA expression in ES is critical for invasion of HBMEC and requires microtubule condensation, PI3-kinase and PKC-α activation. Identification of ES structures that interact with HBMEC for invasion would provide novel strategies in the prevention of infection by this emerging pathogen. Currently, we are investigating whether OmpA receptor of E. coli is also responsible for binding to and entry of ES into HBMEC.
4. Materials and methods
4.1. Bacterial strains and cell lines
ES 51329 was obtained from ATCC and was grown in LB or Tryptic Soy Broth medium without any antibiotics. ES was transformed with the plasmid pUC13 containing the gfp gene. Transformants were selected by ampicillin (100 μg/ml), and assessed for GFP expression by viewing under ultraviolet light. The ompA deletion mutant of ES was constructed by replacing ompA with a kanamycin (Km) cassette. Briefly, a spontaneous rifampicin-resistant mutant was isolated and named ES51R. A 1.77-kb DNA containing ompA was amplified from ES with primers: 5′-GTGAG CTCCGGGCTAAAAATTCACTCAA (containing a SacI site), and 5′-CA GGTACCATCGTGCAGCTGATTGA (containing a KpnI site). The DNA was cloned into pEP185.2 [37] at the same sites, and the internal 876-bp NruI–BglII fragment was replaced with a 1.2-kb Km cassette from pUC-4K (Pharmacia). The recombinant plasmid was transferred from E. coli to ES51R by conjugation, and double-crossover mutants were selected. Chinese hamster ovary cells (CHO), intestinal epithelial cells (IEC-6), and conjunctival epithelial cells (CEC) were obtained from ATCC. CEC and IEC-6 were maintained at 37 °C in a humidified atmosphere of 5% CO2 in medium containing M-199/Ham F-12 (1:1) supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, and 2 mM glutamine. CHO cells were maintained as above described, but with modified DMEM as the growth medium. Human umbilical vein endothelial cells (HUVEC) were kindly provided by Dr. Scott Filler, Harbor-UCLA Medical Center, Torrance, CA.
4.2. HBMEC culture and transfections
HBMEC were isolated and cultured as described earlier [38,39]. HBMEC cultures were maintained in RPMI 1640 containing 10% heat inactivated fetal bovine serum, 2 mM glutamine,1 mM sodium pyruvate, streptomycin (100 μg/ml), penicillin (100 units/ml). Dominant negative (DN) constructs of p85 and PKC-α have been described previously [18,19]. HBMEC were transfected with mammalian expression vectors using LipofectAMINE-plus (Invitrogen, Carlsbad, CA). Briefly, DNA-LipofectAMINE-plus in RPMI 1640 was added to 50% confluent HBMEC monolayers. After 6 h of incubation at 37 °C, the cells were washed with RPMI 1640, and complete medium was added. After 48 h, the complete medium was replaced with medium containing G418 (400 μg/ml) and maintained at least for 4 weeks before performing invasion assays.
4.3. Bacterial binding and invasion assays
The cells grown in 24-well cell culture plates to 95% confluence were infected with 106 cfu of ES in experimental medium (1:1 mixture of Hams F-12 and M-199 containing 5% heat inactivated fetal bovine serum) and incubated for various time points at 37 °C in an atmosphere containing 5% CO2. The monolayers were then washed 3 times with RPMI 1640, medium containing gentamicin (100 μg/ml) was added to the wells, and further incubated for 1 h at 37 °C. Subsequently, the cells were washed 3 times with RPMI 1640 and lysed with 0.5% of Triton X-100. The released bacteria were diluted with saline and enumerated by plating on blood agar. In duplicate experiments, the total cell associated bacteria were determined as described for invasion, except that the gentamicin step was omitted. Results were expressed as an average of bacterial colony forming units recovered per well from three or more independent determinations ± SD of the means. For inhibition studies, cytochalasin D (CD), nocodazole, colchicine, filipin, wort-mannin, GFx or dimethyl sulfoxide (DMSO as a control) were incubated with the cells for 30–60 min before the monolayers were infected with bacteria. The chemicals were added at different concentrations and were present throughout the experiment.
4.4. Immunofluorescence staining
HBMEC were grown in eight-well chamber slides coated with collagen and infected with ES at an MOI of 10, as described above. The monolayers were washed 4 times with RPMI and fixed with 2% paraformaldehyde for 15 min. The monolayers were then permeabilized by incubating with 0.1% Triton X-100 in 3% normal goat serum in phosphate-buffered saline (NGS-PBST) for 1 h followed by incubation with primary antibody in NGS-PBST or rhodamine phalloidin for 30 min at room temperature. The primary antibody was probed with Cy2-conjugated secondary antibody. The cells were washed again, the chambers removed, and the slides were mounted in an antifade solution containing DAPI. The cells were viewed with a Leica (Wetzlar, Germany) DMRA microscope with Plan-Apochromat 40×/1.25 NA and 100×/1.40 NA oil immersion objective lenses. Images were acquired with a SkyVision-2/VDS digital charge-coupled device camera (12-bit; 1280 × 1024 pixels) in unbinned or 2 × 2 binned models into the EasyFISH software, saved as 16-bit monochrome images, and merged as 24-bit RGB TIFF images (Applied Spectral Imaging, Inc., Carlsbad, CA). The images were assembled and labeled by Adobe Photoshop Version 6.0. z-Stacks of confocal images were acquired using a Leica TCS SP confocal laser-scanning microscope with Leica confocal software. A 63× oil lens (NA 1.4) was used and the Pinhole was set at Airy1. The gain and offset were adjusted with the brightest position of the z-stack to avoid over saturation. The start and end points for each z-stack were set with adjustment of z position manually. The total numbers of image in each z-stack were automatically set with the program in order to reach the optimal resolution. Each acquired image was averaged 4 times to reduce noise. After acquisition, z-stack images were processed with the Leica confocal program for projection.
4.5. Transmission electron microscopy (TEM)
ES was allowed to invade HBMEC as described above for varying periods (1 h, 2 h, 6 h, and 8 h), the cells were washed 4 times with pre warmed RPMI, and fixed with 2% glutaraldehyde in 0.1 M phosphate-buffered saline (pH 7.4) for 1 h. After rinsing 4 times with phosphate-buffered saline, the cells were then stained with methylene blue for 15 min. The cells were rinsed, post-fixed with 2% OsO4 for 1 h, rinsed again, dehydrated with graded ethanol solutions, and embedded in polypropylene oxide. Ultrathin sections were cut at right angles to the culture cell layer, mounted on collodion one-hole grids, stained with uranyl acetate and lead citrate, and examined with a Phillips CM12 transmission electron microscope.
4.6. Reagents
Fluorescein isothiocyanate (FITC)-conjugated secondary antibodies, anti-α tubulin antibodies, and rhodamine phalloidin were obtained from Molecular Probes (San Diego, CA). Cy3-conjugated secondary antibody was obtained from Zymed Laboratories (San Francisco, CA). Normal goat serum and the Vectashield mounting medium with 4′,6′-diamidino-2-phenyindole (DAPI) were obtained from Vector Laboratories, Inc. (Burlingame, CA). Polyclonal anti-OmpA antibodies were described previously [7]. IgG purification kit was obtained from Pierce Co. (Milford, IL). All other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO).
4.7. Western blotting and dot-blot analysis
Bacterial lysates were prepared by sonicating equal number of cfu in PBS containing 0.5% Triton X-100 and centrifuged to remove cell debris at 3000 rpm in a microfuge. For Western blots, equal amounts of bacterial total proteins were transferred onto a polyvinylidene difluoride nitrocellulose sheet with a Bio-Rad semi-dry transblot apparatus. After transfer, the blots were washed with PBS 2 times and blocked with 5% nonfat dry milk in PBS for 1 h at room temperature. Then, the blots were incubated with anti-OmpA antibody (1:3000) for 2 h, washed 4 times with PBS, and incubated with peroxidase-labeled goat anti-rabbit immunoglobulin G at a dilution of 1:2000 in milk for 2 h at room temperature. The blots were washed with PBS containing 0.05% Tween 20 for 15 min with five changes of buffer, and the bands were developed with ECL reagent (Pierce Co.). Control blotting was carried out by omitting anti-OmpA antibodies. For dot-blots, equal number of bacteria were spotted on a nitrocellulose sheet and dried under vacuum. The nitrocellulose sheet was subjected to immunoblotting similar to that of Western blotting except that diaminobenzidine and 0.01% hydrogen peroxide (substrate) was used to develop the color.
Acknowledgments
We thank Anatoly Grishin for providing us IEC-6 cells and Cathie Hunter for critical reading of this manuscript. This work was supported by grants from PHS (AI40567 to NVP).
Footnotes
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- 1.Biering G, Karlsson S, Clark NC, Jonsdottir KE, Ludvigsson P, Steingrimsson O. Three cases of neonatal meningitis caused by Enterobacter sakazakii in powdered milk. J Clin Microbiol. 1989;27:2054–6. doi: 10.1128/jcm.27.9.2054-2056.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clark NC, Hill BC, O’Hara CM, Steingrimsson O, Cooksey RC. Epidemiologic typing of Enterobacter sakazakii in two neonatal nosocomial outbreaks. Diagn Microbiol Infect Dis. 1990;13:467–72. doi: 10.1016/0732-8893(90)90078-a. [DOI] [PubMed] [Google Scholar]
- 3.Muytjens HL, Zanen HC, Sonderkamp HJ, Kollee LA, Wachsmuth IK, Farmer JJ., III Analysis of eight cases of neonatal meningitis and sepsis due to Enterobacter sakazakii. J Clin Microbiol. 1983;18:115–20. doi: 10.1128/jcm.18.1.115-120.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noriega FR, Kotloff KL, Martin MA, Schwalbe RS. Nosocomial bacteremia caused by Enterobacter sakazakii and Leuconostoc mesenteroides resulting from extrinsic contamination of infant formula. Pediatr Infect Dis J. 1990;9:447–9. [PubMed] [Google Scholar]
- 5.Corti G, Panunzi I, Losco M, Buzzi R. Postsurgical osteomyelitis caused by Enterobacter sakazakii in a healthy young man. J Chemother. 2007;19:94–6. doi: 10.1179/joc.2007.19.1.94. [DOI] [PubMed] [Google Scholar]
- 6.Pitout JD, Moland ES, Sanders CC, Thomson KS, Fitzsimmons SR. Beta-lactamases and detection of beta-lactam resistance in Enterobacter spp. Antimicrob Agents Chemother. 1997;41:35–9. doi: 10.1128/aac.41.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drudy D, Mullane NR, Quinn T, Wall PG, Fannin S. Enterobacter sakazakii: an emerging pathogen in powdered infant formula. Food Safety. 2006;42:996–1002. doi: 10.1086/501019. [DOI] [PubMed] [Google Scholar]
- 8.Prasadarao NV, Wass CA, Weiser JN, Stins MF, Huang SH, Kim KS. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect Immun. 1996;64:146–53. doi: 10.1128/iai.64.1.146-153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badger LJ, Stins M, Kim KS. Citrobacter freundii invades and replicates in human brain microvascular endothelial cells. Infect Immun. 1999;67:4208–15. doi: 10.1128/iai.67.8.4208-4215.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nizet V, Kim KS, Stins M, Jonas M, Chi EY, Nguyen D, et al. Invasion of brain microvascular endothelial cells by group B streptococci. Infect Immun. 1997;65:5074–81. doi: 10.1128/iai.65.12.5074-5081.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greiffenberg L, Goebel W, Kim KS, Daniels J, Kuhn M. Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: an electron microscopic study. Infect Immun. 2000;68:3275–9. doi: 10.1128/iai.68.6.3275-3279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prasadarao NV, Wass CA, Kim KS. Endothelial cell GlcNAc beta 1–4GlcNAc epitopes for outer membrane protein A enhance traversal of Escherichia coli across the blood–brain barrier. Infect Immun. 1996;64:154–60. doi: 10.1128/iai.64.1.154-160.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mange JP, Stephan R, Borel N, Wild P, Kim KS, Pospischil A, et al. Adhesive properties of Enterobacter sakazakii to human epithelial and brain microvascular endothelial cells. BMC Microbiol. 2006;26:58–62. doi: 10.1186/1471-2180-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KP, Loessner MJ. Enterobacter sakazakii invasion in human intestinal Caco-2 cells requires the host cell cytoskeleton and is enhanced by disruption of tight junctions. Infect Immun. 2008;76:562–70. doi: 10.1128/IAI.00937-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohan Nair MK, Venkitanarayanan KS. Cloning and sequencing of the ompA gene of Enterobacter sakazakii and development of an ompA-targeted PCR for rapid detection of Enterobacter sakazakii in infant formula. Appl Environ Microbiol. 2006;72:2539–46. doi: 10.1128/AEM.72.4.2539-2546.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohan Nair MK, Venkitanarayanan K. Role of bacterial OmpA and host cytoskeleton in the invasion of human intestinal epithelial cells by Enterobacter sakazakii. Pediatr Res. 2007;62:664–9. doi: 10.1203/PDR.0b013e3181587864. [DOI] [PubMed] [Google Scholar]
- 17.Prasadarao NV, Wass CA, Stins MF, Shimada H, Kim KS. Outer membrane protein A-promoted actin condensation of brain microvascular endothelial cells is required for Escherichia coli invasion. Infect Immun. 1999;67:5775–83. doi: 10.1128/iai.67.11.5775-5783.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sukumaran SK, Prasadarao NV. Regulation of protein kinase C in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem. 2002;277:12253–62. doi: 10.1074/jbc.M110740200. [DOI] [PubMed] [Google Scholar]
- 19.Reddy MA, Prasadarao NV, Wass CA, Kim KS. Phosphatidylinositol 3-kinase activation and interaction with focal adhesion kinase in Escherichia coli K1 invasion of human brain microvascular endothelial cells. J Biol Chem. 2000;275:36769–74. doi: 10.1074/jbc.M007382200. [DOI] [PubMed] [Google Scholar]
- 20.Sukumaran SK, Quon MJ, Prasadarao NV. Escherichia coli K1 internalization via caveolae requires caveolin-1 and protein kinase C-alpha interaction in human brain microvascular endothelial cells. J Biol Chem. 2002;277:50716–24. doi: 10.1074/jbc.M208830200. [DOI] [PubMed] [Google Scholar]
- 21.Sukumaran SK, Prasadarao NV. Escherichia coli K1 invasion increases human brain microvascular endothelial cell monolayer permeability by disassembling vascular-endothelial cadherins at tight junctions. J Infect Dis. 2003;188:1295–309. doi: 10.1086/379042. [DOI] [PubMed] [Google Scholar]
- 22.Prasadarao NV, Srivastava PK, Rudrabhatla RS, Kim KS, Huang SH, Sukumaran SK. Cloning and expression of the Escherichia coli K1 outer membrane protein A receptor, a gp96 homologue. Infect Immun. 2003;71:1680–8. doi: 10.1128/IAI.71.4.1680-1688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prasadarao NV. Identification of Escherichia coli outer membrane protein A receptor on human brain microvascular endothelial cells. Infect Immun. 2002;70:4556–63. doi: 10.1128/IAI.70.8.4556-4563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soh JW, Lee EH, Prywes R, Weistein IB. Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum response element. Mol Cell Biol. 1999;19:1313–24. doi: 10.1128/mcb.19.2.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Townsend SM, Hurrell E, Gonzalez-Gomez I, Lowe J, Frye JG, Forsythe S, et al. Enterobacter sakazakii invades brain capillary endothelial cells, persists in human macrophages influencing cytokine secretion and induces severe brain pathology in the neonatal rat. Microbiology. 2007;153:3538–47. doi: 10.1099/mic.0.2007/009316-0. [DOI] [PubMed] [Google Scholar]
- 26.Datta D, Vaidehi N, Floriano WB, Kim KS, Prasadarao NV, Goddard WA., 3rd Interaction of E. coli outer-membrane protein A with sugars on the receptors of the brain microvascular endothelial cells. Proteins. 2003;50:213–21. doi: 10.1002/prot.10257. [DOI] [PubMed] [Google Scholar]
- 27.Hunter CJ, Singamsetty VK, Chokshi NK, Boyle P, Camerini V, Grishin AV. Enterobacter sakazakii enhances epithelial cell injury by inducing apoptosis in a rat model of necrotizing enterocolitis. J Infect Dis. doi: 10.1086/590186. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oelschlaeger TA, Guerry P, Kopecko DJ. Unusual microtubule-dependent endocytosis mechanisms triggered by Campylobacter jejuni and Citrobacter freundii. Proc Natl Acad Sci U S A. 1993;90:6884–8. doi: 10.1073/pnas.90.14.6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oelschlaeger TA, Tall BD. Invasion of cultured human epithelial cells by Klebsiella pneumoniae isolated from the urinary tract. Infect Immun. 1997;65:2950–8. doi: 10.1128/iai.65.7.2950-2958.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finlay BB, Falkow S. Common themes in microbial pathogenicity revisited. Microbiol Mol Biol Rev. 1997;61:136–69. doi: 10.1128/mmbr.61.2.136-169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.St Geme JW, Falkow S. Haemophilus influenzae adheres to and enters cultured human epithelial cells. Infect Immun. 1990;58:4036–44. doi: 10.1128/iai.58.12.4036-4044.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida S, Sasakawa C. Exploiting host microtubule dynamics: a new aspect of bacterial invasion. Trends in Microbiol. 2003;11:139–43. doi: 10.1016/s0966-842x(03)00023-4. [DOI] [PubMed] [Google Scholar]
- 33.Okkenhaug K, Vanhaesebroeck B. New responsibilities for the PI3 K regulatory subunit p85 alpha. Sci STKE. 2001;65:PE1. doi: 10.1126/stke.2001.65.pe1. [DOI] [PubMed] [Google Scholar]
- 34.Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–5. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 35.Crane JK, Oh JS. Activation of host cell protein kinase C by enteropathogenic Escherichia coli. Infect Immun. 1997;65:3277–85. doi: 10.1128/iai.65.8.3277-3285.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raghu H, Sharma-Walia N, Veettil MV, Sadagopan S, Caballero A, Sivakumar R, et al. Lipid rafts of primary endothelial cells are essential for Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8-induced phosphatidylinositol 3-kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J Virol. 2007;81:7941–59. doi: 10.1128/JVI.02848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heusipp G, Nelson KM, Schmidt MA, Miller VL. Regulation of htrA expression in Yersinia enterocolitica. FEMS Microbiol Lett. 2004;231:227–35. doi: 10.1016/S0378-1097(03)00962-5. [DOI] [PubMed] [Google Scholar]
- 38.Stins MF, Prasadarao NV, Zhou J, Arditi M, Kim KS. Bovine brain microvascular endothelial cells transfected with SV40-large T antigen: development of an immortalized cell line to study pathophysiology of CNS disease. In Vitro Cell Dev Biol Anim. 1997;33:243–7. doi: 10.1007/s11626-997-0042-1. [DOI] [PubMed] [Google Scholar]
- 39.Stins MF, Prasadarao NV, Wass CA, Kim KS. Escherichia coli binding to and invasion of brain microvascular endothelial cells derived from humans and rats of different ages. Infect Immun. 1999;67:5522–5. doi: 10.1128/iai.67.10.5522-5525.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]