

Abstract
A defect in Klotho gene expression in mice accelerates the degeneration of multiple age-sensitive traits. Here, we show that overexpression of Klotho in mice extends life span. Klotho protein functions as a circulating hormone that binds to a cell-surface receptor and represses intracellular signals of insulin and insulin-like growth factor 1 (IGF1), an evolutionarily conserved mechanism for extending life span. Alleviation of aging-like phenotypes in Klotho-deficient mice was observed by perturbing insulin and IGF1 signaling, suggesting that Klotho-mediated inhibition of insulin and IGF1 signaling contributes to its anti-aging properties. Klotho protein may function as an anti-aging hormone in mammals.
Klotho was originally identified as a mutated gene in a mouse strain that accelerates age-dependent loss of function in multiple age-sensitive traits (1). An insertional mutation that disrupts the 5′ promoter region of the Klotho gene resulted in a strong hypomorphic allele. Mice homozygous for the mutated allele (KL−/− mice) appeared normal until 3 to 4 weeks old but then began to manifest multiple age-related disorders observed in humans, including ectopic calcification, skin atrophy, muscle atrophy, osteoporosis, arteriosclerosis, and pulmonary emphysema. KL−/− mice suffered premature death around two months of age.
The Klotho gene encodes a single-pass transmembrane protein that is detectable in limited tissues, particularly the distal convoluted tubules in the kidney and the choroid plexus in the brain. Because a defect in the Klotho gene leads to systemic age-dependent degeneration, the Klotho protein may function through a circulating humoral factor that regulates the development of age-related disorders or natural aging processes (2). Notably, some single-nucleotide polymorphisms in the human KLOTHO gene are associated with altered life span (3) and altered risk for coronary artery disease (4), osteoporosis (5-7), and stroke (8).
Little is known about Klotho protein function and the molecular mechanism by which it suppresses the development of aging-like pheno-types. The extracellular domain of Klotho protein is composed of two internal repeats, KL1 and KL2, that share amino acid sequence homology to β-glucosidases of bacteria and plants (20 to 40% identity) (1). However, glucosidase activity is not present in recombinant Klotho protein (9), and the essential glutamate residue at the β-glucosidase active center is replaced with asparagine and alanine in KL1 and KL2, respectively (10). Here, we demonstrate that Klotho is an aging suppressor gene whose product functions as a hormone that inhibits intracellular insulin and IGF1 signaling.
Klotho overexpression extends life span in mice
We previously generated independent transgenic lines of mice that over-express Klotho under the control of the human elongation factor 1α promoter (1) (EFmKL46 and EFmKL48) (fig. S1). Here, we compared the life span of the transgenic mice with that of wild-type controls that are near-coisogenic by virtue of backcrossing onto the C3H background four times. Each line was previously confirmed to express functional Klotho protein from the transgene (1). Mice carrying the EFmKL46 or EFmKL48 transgenic alleles, fed ad libitum, outlived wild-type controls by 20.0 and 30.8%, respectively, in males (Fig. 1A) and by 18.8 and 19.0%, respectively, in females (Fig. 1B).
Fig. 1.

Klotho overexpression extends life span in the mouse. Kaplan-Meier analysis of survival in (A) males [P = 0.006 in EFmKL46 versus wild-type (WT) mice, and P < 0.0001 in EFmKL48 versus wild type by log-rank test) and in (B) females (P = 0.01 in EFmKL46 versus wild type, and P = 0.01 in EFmKL48 versus wild type by log-rank test). The average life span of male wild-type, EFmKL46, and EFmKL48 mice was 715 ± 44 days, 858 ± 40 days, and 936 ± 47 days (means ± SEM), respectively. The average life span of female wild-type, EFmKL46, and EFmKL48 mice was 697 ± 45 days, 829 ± 32 days, and 830 ± 29 days, respectively. (C) Body weight of wild-type, EFmKL46, and EFmKL48 mice. No significant difference in growth was observed. (D) Klotho overexpression reduces fecundity. Twelve breeding pairs at 12 weeks of age were set up for each genotype. The number of offspring generated during 12 months was recorded for each breeding pair. Although average litter size (pups per birth) of wild-type, EFmKL46, and EFmKL48 pairs was not significantly different (6.6 ± 1.0, 6.1 ± 1.3, and 7.0 ± 1.2, respectively), the number of births (births per pair per 12 months) was fewer in transgenic mice pairs (7.2 ± 1.6, 4.2 ± 0.8, and 4.5 ± 2.2, respectively), resulting in significantly fewer offspring in transgenic pairs. Data are means ± SD. *, P < 0.05; †, P < 0.01 versus wild-type mice by analysis of variance (ANOVA).
Caloric restriction is associated with increased longevity in various species (11). To assess whether mice overexpressing Klotho were restricting their own diets, we monitored food intake and oxygen consumption in transgenic and wild-type mice for 24 hours at 32 to 36 weeks of age. No significant differences in these parameters were observed (table S1). Small body size is also associated with extended longevity in diet-restricted mice and in mice that are mutant for pituitary or growth hormone receptor function (12, 13). However, we did not observe any substantial difference in growth between EFmKL46, EFmKL48, and wild-type mice (Fig. 1C). Both EFmKL46 and EFmKL48 breeding pairs generated fewer offspring than wild-type breeding pairs (Fig. 1D). As expected from the evolutionary theory of longevity, maximum fitness of the organism is a trade-off between life span and fertility (14). These data indicate that Klotho systemically modulates aging through mechanisms independent of food intake and growth, but potentially in association with reproduction.
Klotho increases resistance to insulin and IGF1
Many genetic data demonstrate that inhibited insulin and IGF1 signaling extends life span in animals from C. elegans, to Drosophila, to mice (15-21). Because Klotho must mediate aging through effects of a systemic hormone, we investigated whether the Klotho gene is involved in the inhibition of insulin or IGF1 signaling. Mice defective in Klotho gene expression have reduced blood glucose and insulin levels coupled with enhanced sensitivity to insulin (22).
We compared glucose metabolism in the Klotho-overexpressing transgenic mice with wild-type animals. Blood glucose levels were normal in each transgenic line (Fig. 2A). However, male EFmKL46 and EFmKL48 mice had higher blood insulin levels than did wild-type males (Fig. 2B), suggesting that the male transgenic mice are somewhat insulin resistant. We directly assessed sensitivity to insulin with a hyperinsulinemic euglycemic clamp (23). As expected, male EFmKL46 and EFmKL48 mice required lower glucose infusion rates than did wild-type males to maintain normal blood glucose levels (Fig. 2C). Furthermore, insulin and IGF1 tolerance tests revealed significant attenuation in hypoglycemic response to injected insulin and IGF1 in male transgenic mice (Fig. 2, D and E). Although we were unable to detect insulin resistance in female transgenic mice (Fig. 2, C and D), they were significantly resistant to IGF1 (Fig. 2E). These studies demonstrate that Klotho overexpression induces resistance to insulin and IGF1.
Fig. 2.
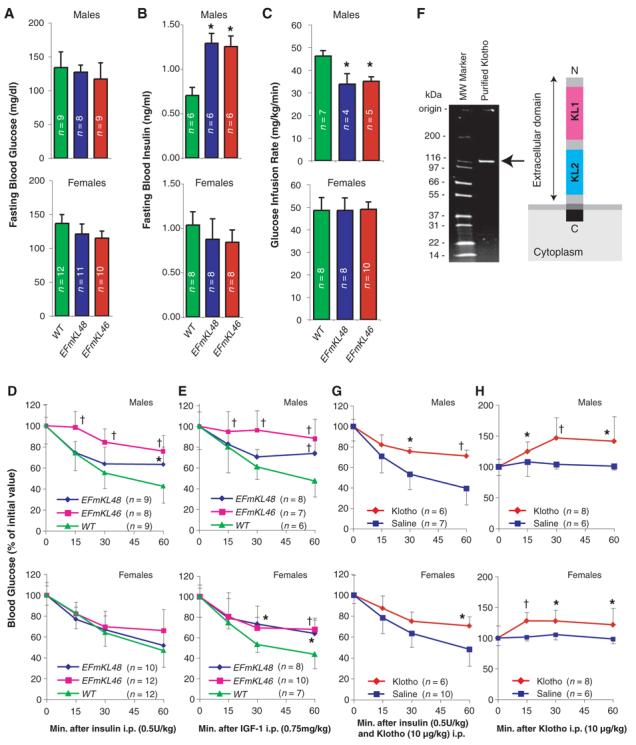
Klotho increases resistance to insulin and IGF1. (A and B) Fasting blood glucose (A) and insulin (B) levels were compared between wild-type (WT), EFmKL48, and EFmKL46 mice. (C) Hyperinsulinemic euglycemic clamp experiments. Glucose infusion rate (mg/kg/min) was compared between wild-type, EFmKL48, and EFmKL46 mice. During the clamp experiments there were no differences in blood glucose concentration between wild-type, EFmKL46, and EFmKL48 mice. The number of animals (n) for each group is indicated in the bars. (D and E) Insulin (D) and IGF1 (E) tolerance tests. Blood glucose levels after injection of insulin (0.5 U/kg) or IGF1 (0.75 mg/kg) were expressed as a percentage change from blood glucose concentration at time zero. Error bars indicate SD. *, P < 0.05 and †, P < 0.01 versus wild-type mice by ANOVA. (F) A schematic representation of Klotho extracellular peptide (right) and analysis of purified recombinant Klotho peptide by SDS-polyacrylamide gel electrophoresis (left). (G) The effect of Klotho injection on insulin tolerance in mice. Insulin tolerance tests were performed with age-matched wild-type mice immediately after intraperitoneal injection with saline or purified recombinant Klotho peptide (10 μg/kg). (H) The effect of Klotho injection on blood glucose levels. Saline or Klotho protein (10 μg/kg) was administered into age-matched wild-type mice by intraperitoneal injection. Error bars indicate SD. *, P < 0.05 and †, P < 0.01 versus saline-injected mice by ANOVA.
Klotho functions as a hormone
The extracellular domain of Klotho is shed on the cell surface and detected in the blood and cerebrospinal fluid in mice and humans (24). Immunoblot analysis of plasma with the use of rabbit anti-Klotho antiserum demonstrated that the extracellular Klotho peptide can be detected in wild-type, EFmKL48, and EFmKL46 mice but not in KL−/− mice (fig. S2). Radioimmunoassay further demonstrated that Klotho peptide is ∼100 pM in wild-type mice and about two times as high in the transgenic overexpression strains (fig. S3). The extra-cellular domain of Klotho may function as a hormone-like substance (2).
To assess the function of the Klotho extracellular peptide, we generated a soluble form of recombinant Klotho protein comprising the 952–amino acid extracellular domain, and determined whether this promoted insulin resistance when injected into mice. Intraperitoneal injection of insulin (0.5 U/kg) and purified Klotho extracellular peptide (10 μg/kg, Fig. 2F) in wild-type male and female mice attenuated the hypoglycemic response expected from insulin alone (Fig. 2G). Klotho peptide alone rapidly increased blood glucose levels in male wild-type mice and to a smaller extent in females (Fig. 2H). However, Klotho peptide injection did not induce significant changes in blood insulin and glucagon levels (fig. S4), suggesting that Klotho peptide inhibits insulin action directly in peripheral tissues.
To test whether Klotho antagonizes insulin at receptive cells, we determined whether recombinant Klotho peptide would reduce glucose uptake by blocking insulin binding to the insulin receptor. We measured cellular glucose uptake in cultured myoblastic cells (L6) incubated with or without insulin in the presence or absence of 100 pM of Klotho extracellular peptide. Klotho peptide suppressed insulin-induced glucose uptake by 55% without reducing the binding of [125I] insulin to the cells (fig. S5). Thus, Klotho does not appear to inhibit ligand-binding to the insulin receptor, suggesting that Klotho may block insulin action by disrupting one or more alternative insulin-dependent intracellular signaling pathways. Accordingly, we measured the potential for [125I]-labeled Klotho to bind directly to the cell surface. Hepatoma cells bound [125I] Klotho in a dose-dependent manner and saturated when the total Klotho concentration exceeded 600 pM, and unlabeled Klotho peptide inhibited the binding of [125I] Klotho (fig. S6). Together, these observations suggest that cells present a receptor at their surface other than the insulin receptor that binds to the Klotho peptide.
Klotho inhibits intracellular insulin and IGF1 signaling
Because membrane-bound Klotho peptide must inhibit ligand activation of the insulin receptor within the cells, we investigated the influence of Klotho on insulin receptor signal transduction (25, 26). We incubated L6 cells or rat hepatoma cells (H4IIE) with recombinant Klotho peptide and insulin (10 nM) or IGF1 (10 nM). Klotho peptide did not inhibit the binding of [125I] insulin or [125I] IGF1 (fig. S5) but suppressed ligand-stimulated autophosphorylation of insulin and IGF1 receptors in a dose-dependent manner (Fig. 3, A and B). Additionally, Klotho reduced activation of signaling events downstream of receptor activation, including tyrosinephosphorylated insulin receptor substrate (IRS) 1 and 2, the association of the subunit of phosphoinositide 3–kinase p85 with IRS proteins (Fig. 3, A and B). Because the inhibitory effect of Klotho on insulin signaling was observed as early as 1 min after insulin stimulation (Fig. 3C), the decline in tyrosine-phosphorylated insulin and IGF1 receptors is unlikely due simply to the loss of receptors. Notably, Klotho peptide can inactivate active insulin receptors that were previously tyrosine phosphorylated by insulin stimulation. In H4IIE cells that were exposed to 10 nM insulin before adding Klotho peptide, Klotho suppressed tyrosine phosphorylation of the insulin receptor (Fig. 3D). We observed a similar effect on IGF1 receptor autophosphorylation in L6 cells with IGF1 before adding Klotho peptide (9). Importantly, the inhibitory effect of Klotho on autophosphorylation of receptor tyrosine kinases is specific. We observed no inhibitory effect of Klotho on the epidermal growth factor receptor and the platelet-derived growth factor receptor (fig. S7). Overall, Klotho appears to inhibit activation of the insulin and IGF1 receptor and to repress activated insulin and IGF1 receptors. Whether Klotho peptide functions by accelerating removal of tyrosine phosphorylation from the activated insulin receptor remains to be determined.
Fig. 3.

Klotho protein inhibits intracellular insulin and IGF1 signaling. (A and B) Effect of Klotho on tyrosine phosphorylation of insulin and IGF1 receptors as well as IRS-1 and IRS-2, association of IRS-1 and IRS-2 with the PI3-kinase regulatory subunit (p85), and phosphorylation of Akt in L6 cells stimulated with 10 nM of insulin (A) or 10 nM of IGF1 (B). Antibodies used for immunoprecipitation (ip) and immunoblotting (ib) were indicated. IR, antibody to insulin receptor β chain; pY20, antibody to phosphotryrosine; IRS-1, antibody to IRS-1; IRS-2, antibody to IRS-2; p85, antibody to PI3-kinase regulatory subunit; IGF-1R, antibody to IGF1 receptor β chain. (C) A time course of the inhibitory effect of Klotho protein (400 pM) on insulin signaling in H4IIE cells. The cells were harvested before (0′) and at the indicated time points after insulin stimulation (10 nM). (D) Inactivation of activated insulin receptor by Klotho protein. H4IIE cells were stimulated with insulin (10 nM) at time 0′ and harvested 15 min later. Klotho (400 pM) or phosphate-buffered saline (PBS) was added at the indicated time points indicated (left panel). The cell lysates were immunoprecipitated with IR and immunoblotted with pY20 or IR (right panel).
Inhibition of insulin and IGF1 signaling rescues KL−/− phenotypes
If the ability of Klotho to inhibit insulin and IGF1 signaling extends survival by retarding senescence, independent manipulations to inhibit insulin and IGF1 signaling may ameliorate some of the aging-like phenotypes in KL−/− mice. Accordingly, we crossed a loss-of-function mutation of IRS-1 into the KL−/− mice (27). Survival was improved in KL−/− mice heterozygous for an IRS-1 null allele (KL−/− IRS-1+/−) relative to KL−/− control mice (Fig. 4, A and B). In addition, KL−/− IRS-1+/− mice ameliorated many age-related pathologies typical of KL−/− mice, including arteriosclerosis, ectopic calcification, skin atrophy, pulmonary emphysema, and hypogonadism (Fig. 4, C to N). Heterozygosity of IRS-1 alone (KL+/+ IRS-1+/− littermates) appears to have no effect on survival and the age-progressive degeneration when compared with those factors in wild-type littermates during these experiments (9).
Fig. 4.
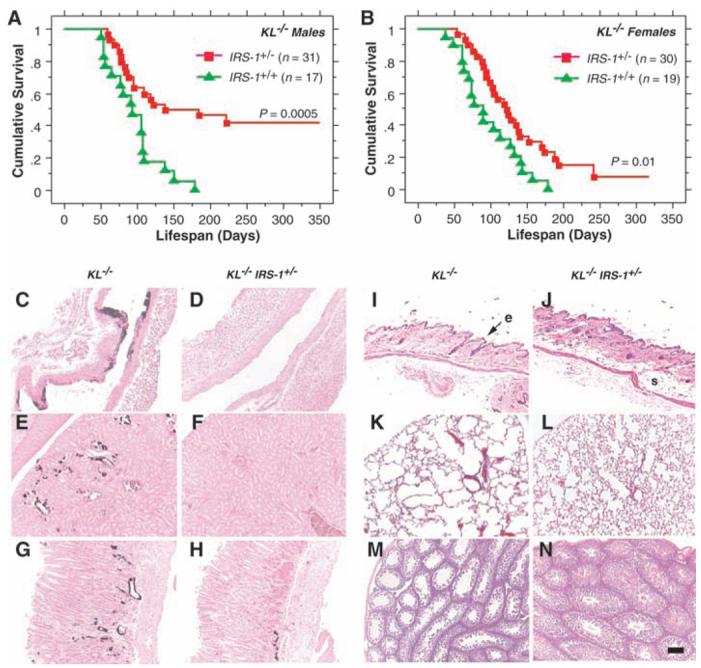
Rescue of aging-like phenotypes in KL−/− mice by genetic intervention in insulin and IGF1 signaling. (A and B) Life-span extension in KL−/− mice by reducing IRS-1 expression. KL−/− mice heterozygous for an IRS-1 null allele (IRS-1+/−) lived longer than those without the mutation (IRS-1+/+) both in males (P = 0.0005 by log-rank test) and females (P = 0.01 by log-rank test). [(C) to (N)] Rescue of aging-like phenotypes in KL−/− IRS-1+/− mice at the histological level. Typical findings from four 8-week-old males of each genotype are shown. (C and D) Aorta (von Kossa staining). Calcification of arterial walls [black deposits in (C)] was decreased in KL−/− IRS-1+/− mice (D). (E and F) Kidney (von Kossa staining). Calcification of small arteries and renal tubules [black deposits in (E)] was decreased in KL−/− IRS-1+/− mice (F). (G and H) Stomach (von Kossa staining). Ectopic calcification in gastric mucosa and small arteries [black deposits in (G)] was alleviated in KL−/− IRS-1+/− mice (H). (I and J) Cross-sections of the skin. Hematoxylin-eosin (HE) staining. Reduction in epidermal layer (e) thickness observed in KL−/− mice (I) was improved and subcutaneous fat (s) was restored in KL−/− IRS-1+/− mice (J). (K and L) Lung (HE staining). Emphysematous changes, including enlargement of air spaces and destruction of the normal alveolar architecture were observed in KL−/− mice (K), but were alleviated in KL−/− IRS-1+/− mice (L). (M and N) Testis (HE staining). Seminiferous tubules were atrophic and no mature sperm was observed in KL−/− mice (M). Spermatogenesis was restored in KL−/− IRS-1+/− mice (N). All panels were shown in the identical magnification (×200). Scale bar, 200 μm.
Conclusion
We previously reported that a defect in Klotho gene expression leads to a syndrome that may resemble premature aging (1). Here, we show that overexpression of Klotho can extend life span, and we suggest that Klotho functions as an aging suppressor gene in mammals. We found that the extra-cellular domain of Klotho protein circulates in the blood and binds to a putative cell-surface receptor. Klotho has marked effects on insulin physiology, apparently because it suppresses tyrosine phosphorylation of insulin and IGF1 receptors, which results in reduced activity of IRS proteins and their association with PI3-kinase, thereby inhibiting insulin and IGF1 signaling. Extended life span upon negative regulation of insulin and IGF1 signaling is an evolutionarily conserved mechanism to suppress aging (28). Klotho appears to be a peptide hormone to modulate such signaling and thereby mediate insulin metabolism and aging.
Supplementary Material
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/1112766/DC1
Materials and Methods
References and Notes
- 1.Kuro-o M, et al. Nature. 1997;390:45. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi Y, Kuro-o M, Ishikawa F. Proc. Natl. Acad. Sci. U.S.A. 2000;97:12407. doi: 10.1073/pnas.210382097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arking DE, et al. Proc. Natl. Acad. Sci. U.S.A. 2002;99:856. [Google Scholar]
- 4.Arking DE, et al. Am. J. Hum. Genet. 2003;72:1154. doi: 10.1086/375035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogata N, et al. Bone. 2002;31:37. doi: 10.1016/s8756-3282(02)00786-x. [DOI] [PubMed] [Google Scholar]
- 6.Kawano K, et al. J. Bone Miner. Res. 2002;17:1744. doi: 10.1359/jbmr.2002.17.10.1744. [DOI] [PubMed] [Google Scholar]
- 7.Yamada Y, Ando F, Niino N, Shimokata H. J. Mol. Med. 2005;83:50. doi: 10.1007/s00109-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 8.Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC. Circ. Res. 2005;96:412. doi: 10.1161/01.RES.0000157171.04054.30. [DOI] [PubMed] [Google Scholar]
- 9.Kuro-o M, et al. data not shown.
- 10.Grabnitz F, Seiss M, Rucknagel KP, Staudenbauer WL. Eur. J. Biochem. 1991;200:301. doi: 10.1111/j.1432-1033.1991.tb16186.x. [DOI] [PubMed] [Google Scholar]
- 11.Weindruch R, Walford RL, Fligiel S, Guthrie D. J. Nutr. 1986;116:641. doi: 10.1093/jn/116.4.641. [DOI] [PubMed] [Google Scholar]
- 12.Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- 13.Miller RA. Sci. Aging Knowledge Environ. 2001;2001:vp6. doi: 10.1126/sageke.2001.9.vp6. [DOI] [PubMed] [Google Scholar]
- 14.Williams GC. Evol. Int. J. Org. Evol. 1957;11:398. [Google Scholar]
- 15.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. Nature. 1993;366:461. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 16.Morris JZ, Tissenbaum HA, Ruvkun G. Nature. 1996;382:536. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- 17.Tatar M, et al. Science. 2001;292:107. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- 18.Clancy DJ, et al. Science. 2001;292:104. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 19.Holzenberger M, et al. Nature. 2003;421:182. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 20.Bluher M, Kahn BB, Kahn CR. Science. 2003;299:572. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 21.Kenyon C. Cell. 2005;120:449. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Utsugi T, et al. Metabolism. 2000;49:1118. doi: 10.1053/meta.2000.8606. [DOI] [PubMed] [Google Scholar]
- 23.Halseth AE, Bracy DP, Wasserman DH. Am. J. Physiol. 1999;276:E70. doi: 10.1152/ajpendo.1999.276.1.E70. [DOI] [PubMed] [Google Scholar]
- 24.Imura A, et al. FEBS Lett. 2004;565:143. doi: 10.1016/j.febslet.2004.03.090. [DOI] [PubMed] [Google Scholar]
- 25.Saltiel AR, Kahn CR. Nature. 2001;414:799. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 26.LeRoith D, Roberts CT., Jr. Cancer Lett. 2003;195:127. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 27.Araki E, et al. Nature. 1994;372:186. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 28.Tatar M, Bartke A, Antebi A. Science. 2003;299:1346. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- 29.We thank D. H. Wasserman and Vanderbilt Mouse Metabolic Phenotyping Center for physiological analysis of the mice; R. L. Dobbins for hyperinsulinemic euglycemic clamp experiments; J. A. Richardson and Molecular Pathology Core Facility at UT Southwestern for histological analysis; D. W. Russell at UT Southwestern for Klotho receptor identification; R. Komuro and H. Kuriyama at UT Southwestern for insulin and IGF1 signaling analysis; Genentech for providing IGF1; H. Masuda, T. Suga, R. Nagai, A. T. Dang, R. Shamlou, P. Bezzera, T. Reed, C. Iucu, W. Lai for earlier contributions and supports to this study; and E. C. Friedberg, M. S. Brown, and K. A. Wharton Jr. at UT Southwestern for critical reading of the manuscript. This work was supported in part by grants from Endowed Scholar Program at UT Southwestern (M.K.), Pew Scholars Program in Biomedical Science (M.K.), Eisai Research Fund (M.K.), High-Impact/High-Risk Research Program at UT Southwestern (M.K.), and NIH (R01AG19712 to M.K. and R01AG25326 to M.K. and K.P.R.). J.H. is supported by the NIH, the Perot Family Foundation, and the Humboldt Foundation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.