

Abstract
The modulation of gene regulation by blocking the interaction between the thyroid receptor (TR) and obligate coregulators (CoRs) has been reported recently with discovery of the lead compound 3-(dimethylamino)-1-(4-hexylphenyl)propan-1-one). Herein we report studies aimed at optimization of this initial hit to determine the basic parameters of the structure–activity relationships (SAR) and clarify the mechanism of action. These studies provided new insights, showing that activity and TRβ isoform selectivity is highly correlated with the structural composition of these covalent inhibitors.
Keywords: Thyroid hormone receptor, T3, coactivator, SRC2, high-throughput screen, protein-protein interaction, structure-activity relationship, β-aminoketone, electrophile, Mannich base, irreversible inhibitor, U2OS, ARO, PAMPA
Introduction
The thyroid hormone receptors (TRs) belong to the superfamily of nuclear receptors (NRs) and are involved in the regulation of development, growth, and metabolism.1, 2, 3 Thyroid hormone T3 induces most of the transcriptional responses mediated by TRs in vivo.4 The TRs have two isoforms, TRα and TRβ, encoded by two genes, with each isoform having two distinct subtypes due to alternative splicing.5 The main functional domains of the TRs are: 1) an amino terminal ligand-independent transcription activation domain (AF-1); 2) a central DNA-binding domain (DBD); and 3) a carboxy terminal ligand-binding domain (LBD) containing a thyroid hormone-inducible activation domain (AF-2).6 TR most often acts in concert with retinoid X receptor (RXR).7 This heterodimer, formed in the in the absence of ligand, associates with an NR corepressor such as NCoR or the silencing mediator of retinoic acid (SMRT) and suppresses basal transcription.8 In the presence of T3, TR undergoes a conformational change, releasing NCoR/SMRT and recruiting coactivator (CoA) proteins to activate gene transcription. The dominant group of CoAs is the p160 or steroid receptor coactivator (SRC) proteins,7 including SRC1,9 SRC2,10, 11 and SRC3.12 The SRCs have variable numbers of conserved LXXLL motifs, called NR boxes, within their nuclear receptor interacting domain (NID). These leucine-rich motifs interact with hydrophobic amino acid residues located on helixes 3, 4, 5, and 12 of the TR LBD, which form the AF-2 pocket.13, 14 Additional hydrogen bonding in close proximity has been observed between charged side chains of the CoAs and lysine 288 and glutamic acid 457 of the TRβ (charge clamp residues).
We have reported that short peptides consisting of the LXXLL motif and flanking amino acid are able to bind to the TR15, 16 and developed inhibitors based on macrolactam-constrained peptides.17, 18 The first small-molecule inhibitors were discovered in a high-throughput screen using fluorescence polarization.19 The most promising hit from this screen was an aromatic β-aminoketone, which, after in situ elimination to form an enone, bound covalently to the TR and inhibited its interaction with SRCs. This compound class, known as Mannich bases, was systematically studied by Carl Mannich in the beginning of the 20th century.20 Several Mannich bases have been marketed as drugs, including pipoctanone21 (antihypertensive), hexacaine22 (antiarrhythmic), propitocaine23 (local anesthetic), and dyclonine24 (local anesthetic). Although the mode of action has not been elucidated, experimental evidence supports their binding to the inner pore of voltage gated sodium channel and blocking current.25 In contrast to other local anesthetics, Mannich base drugs are well known to liberate α,β-unsaturated ketones by internal elimination of the amino group in vitro, a lthough this reaction proceeds very slowly in aqueous buffers at physiological pH.26 Soft electrophiles such as enones can alkylate protein nucleophiles with a strong chemoselectivity for the sulfhydryl of cysteine.27 We showed that certain β-aminophenylketones are very likely to specifically alkylate TRβ in vitro,19 thus inhibiting its hormone-induced gene transcription.
These compounds have a great potential as tools for studying the role of the interaction between TR and its coregulators in vivo and potentially for progression to lead optimization as a preclinical candidate to treat aspects of metabolic syndrome. Here we present a broad study of the structure–activity relationships (SARs) of this lead series governing activities, toxicity, solubility, and permeability studies of β-aminophenylketones and similar electrophilic compounds.
Results
We recently reported the discovery of small molecules that inhibit the interaction between TR and the CoA SRC2-2.19 The most potent molecules identified by the screen were substituted β-aminophenylketones. An initial SAR for this series was established using the relative response of other β-aminophenylketones present in the screening collection. The relative efficacies at a fixed dose of 30 μM are shown in Figure 1.
Figure 1.

Summary of screened β-aminophenylketone. Inhibition (%) of the interaction between coactivator peptide SRC2-2 and TRβ in the presence of 30 μM compound is depicted in bold.
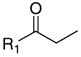
The screened compounds were divided into four groups on the basis of their chemical structures. Group A combined unsubstituted β-aminophenylketones with various nitrogen substituents. These compounds inhibited the interaction between CoA SRC2-2 and TRβ weakly, with efficacies between 2% and 19%, at a 30 μM concentration. Group B consisted of β-aminophenylketones with small alkoxy/hydroxy and/or halogen substituents in a para position to the ketone functionality. The highest efficacy in this group was 9% at a 30 μM concentration. Little SAR was established in these two groups. In contrast, group C combining β-aminophenylketones with larger hydrophobic substituents in a para position to the ketone showed dramatic SAR. The compounds with 4-ethyl cyclohexyl and n-hexyl substituents exhibited inhibition values of 87% and 97%, respectively, whereas only 9% efficacy was found for the p-ethyl β-aminophenylketone. β-Aminonaphthylketones in group D were more active (26–30% inhibition) at 30 μM than were the β-aminophenylketones in group A (12%). This analysis showed that a hydrophobic moiety at the para position of β-aminophenylketone is an important feature of potent inhibitors of the TR-CoA interaction.
To address the importance of nitrogen substituents, analogues of group C, β-aminophenylketones bearing n-hexyl substituents at the para position of the aromatic ring, were synthesized (Scheme 1). 1-Phenylhexane reacted with acryloyl chloride in the presence of aluminum chloride, giving compounds 1 and 2 in a ratio of 8:2 after acidic work-up.28 Treatment of this mixture with secondary amines afforded a focus library of β-aminophenylketones in 85–97% yield (Table 1).
Scheme 1.
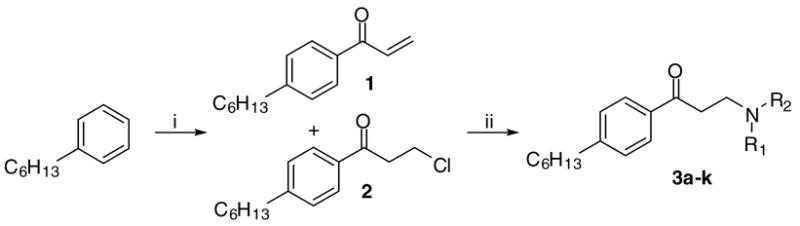
Synthesis of β-aminophenylketones 3a–k.a
aReagents: (i) AlCl3 (2 equiv), acryloyl chloride, DCM, 0°C, 1 h; (ii) HNR1R2, THF, rt, 1 h.
Table 1.
Summary of β-aminophenylketones 3a–k data: Yield, inhibition (IC50) of the interaction between coregulatory peptide SRC2-2 and TRα/TRβ, viability (LD50) of U2OS and ARO cells in the presence of compound, solubility in PBS buffer containing 5% DMSO, and permeability across an artificial membrane (PAMPA).
| In vitro Potency | Cytotoxicity | Pharmacological Properties | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Compound | Yielda (%) | TRαb IC50 (μM) | TRβb IC50 (μM) | TRα: TRβ ratio | AROc LD50 (μM) | U2OSc LD50 (μM) | Solubilityc (μM) | Permeabilityc (10−6 cm/s) |
| 1 | 2-phenylethyl | methyl | 3a | 84 | 3.6 | 1.5 | 2.4 | 2.5 | 18.4 | 15 | 57 |
| 2 | 2-hydroxyethyl | methyl | 3b | 87 | 3.0 | 1.6 | 1.9 | 13.2 | 13.2 | 97 | 965 |
| 3 | isopropyl | isopropyl | 3c | 81 | 4.1 | 2.0 | 2.1 | 15.4 | 20.7 | 55 | 747 |
| 4 | pyrroline | 3d | 85 | 4.3 | 2.7 | 1.6 | 19.0 | 38.4 | 341 | 2144 | |
| 5 | morpholine | 3e | 90 | 4.1 | 2.7 | 1.5 | 7.2 | 20.8 | 110 | 1210 | |
| 6 | methyl carboxylate | piperidine-4- | 3f | 86 | 6.3 | 3.9 | 1.6 | 8.2 | 19.7 | 27 | 145 |
| 7 | n-butyl | n-butyl | 3g | 90 | 6.9 | 4.2 | 1.6 | 5.4 | 25.5 | 152 | 1957 |
| 8 | methyl | methyl | 3h | 94 | 10.6 | 4.3 | 2.5 | 11.0 | 34.9 | 321 | 2323 |
| 9 | furan-2-ylmethyl | methyl | 3i | 92 | 22.6 | 12.1 | 1.9 | 15.5 | 18.6 | 47 | 681 |
| 10 | cyclohexyl | cyclohexyl | 3j | 78 | 16.5 | 12.7 | 1.3 | 25.6 | 27.3 | 32 | 104 |
| 11 | n-propyl | hydrogen | 3k | 93 | 25.8 | 15.2 | 1.7 | 102 | 160 | 354 | 254 |
Isolated yield;
values are means of two independent experiments done in quadruplicate; the general error limits are 10%;
values are means of two independent experiments done in triplicate; the general error limits are 10–15%. Actual error values for each measurement are omitted for clarity but are included in tables in the supplemental materials.
Four major factors come into play in the optimization of compounds for use in biochemical and cellular assays: potency and efficacy against the target, cytotoxicity and other off-target effects, solubility, and cellular permeability. Proper optimization requires seeking the best balance between these factors. The potency and efficacy of the β-aminophenylketones synthesized were investigated using a competitive fluorescence polarization assay. Labeled SRC2-2 peptide, TR binding domain (TR-LBD) protein, and thyroid hormone T3 were incubated with varying concentrations of the β-aminophenylketones 3a–k for 3 h.29 The resulting measured fluorescence polarization reflected the ratio of bound to unbound CoA. Two TR isoforms were investigated: TRα and TRβ. Additionally, these compounds were incubated in various concentrations with cultured human osteosarcoma epithelial cells (U2OS) and human anaplastic thyroid cancer cells (ARO) to determine cell viability after 48 h of exposure. Bone cancer cell lines have a minimal expression of TR, whereas expression of TRβ has been reported for the thyroid cancer cell line ARO.30, 31 A commercially available luminescence cell viability assay, CellTiter-Glo (Promega Corp.) was used to quantify ATP in viable cells. The solubility of each β-aminophenylketone in PBS buffer containing 5% DMSO was determined by measuring the absorption at 280 nm after filtration of the saturated solution. The condition used (PBS containing 5% DMSO) reflected the liquid conditions of the fluorescence polarization assay. Finally, we measured the permeability using a parallel artificial membrane permeation assay (PAMPA). The partition of the β-aminophenylketones between a donor well and acceptor well separated by a lipid layer was measured by UV absorption. The assay was carried out at pH 7.4, imitating absorbance in a cellular system. The results are summarized in Table 1.
All β-aminophenylketones 3a–k were able to inhibit the interaction between the CoA and TR in a low micromolar potency range. Between the TR isoforms, there was a roughly twofold selectivity toward TRβ. In a cell-based assay, these compounds were generally more toxic to the hormone-responsive thyroid cancer cell line ARO than to the T3-insensitive U2OS cells. LD50 ranges were 2.5–25.6 μM for ARO and 13.2–38.4 μM for U2OS, except for compound 3k, which was significantly less toxic (Table 1, entry 11). The solubility of these compounds depended on the nitrogen substituents and varied between 6 μM and 442 μM in PBS containing 5% DMSO. β-Aminophenylketones bearing small nitrogen alkyl substituents, such as 3d, 3h, and 3k, were the most soluble compounds (Table 1, entries 4, 8, and 11). Although solubility is a basic requirement for permeability, we observed different Pe values among the very soluble compounds 3d, 3h, and 3k. In contrast to 3d and 3h, which showed high diffusion rates, 3k poorly penetrated the lipid layer. β-Aminophenylketones 3b, 3e, and 3g, which showed moderate solubilities (97–152 μM in PBS with DMSO), had relatively high permeabilities (965–1957 × 10−6 cm/s) (Table 1, entries 2, 5, and 7).
Previously, we proposed that the β-aminophenylketones function as prodrugs for the true active species, the unsaturated ketones.19 This is supported by the fact that the ability to bind to TR is very similar for all described β-aminophenylketones because all these compounds afford the same elimination product, 1-(4-hexylphenyl)prop-2-en-1-one, regardless of their amino moieties (Table 2, entry 1). To better understand the importance of the electrophilic character of the inhibitors, we investigated various aromatic unsaturated ketones. 1-Phenylhexane was reacted with acid chlorides or anhydrides under Friedel-Crafts reaction conditions to obtain the corresponding ketones (Scheme 2).
Table 2.
Summary of enones 1 and 4a–e and allylic alcohols 5b–e: Yield, IC50 values of the inhibition between coregulatory peptide SRC2-2 and TRα/TRβ, viability (LD50) of U2OS and ARO cells in the presence of compound, solubility in PBS buffer containing 5% DMSO, and permeability across an artificial membrane (PAMPA).
| In vitro Potency | Cytotoxicity | Pharmacological Properties | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 = 4-hexylbenzene and R = 4-heptylbenzene | Compound | Yielda5 (%) | Yielda4 (%) | TRαb IC50 (μM) | TRβb IC50 (μM) | TRα: TRβ ratio | AROc LD50 (μM) | U2OSc LD50 (μM) | Solubilityc (μM) | Permeabilityc (10−6 cm/s) |
| 1 |
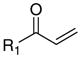
|
1 | - | 55 | 28.1 | 1.5 | 18.7 | 4.5 | 10.1 | 36 | 38 |
| 2 |

|
4a | - | 45 | 19.2 | 17.7 | 1.1 | 5.5 | 57.4 | 252 | 66 |
| 3 |
|
4b | 67 | 57 | 18.9 | 7.6 | 2.5 | 1.4 | 11.6 | 41 | 7 |
| 4 |
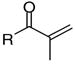
|
4c | 67 | 67 | 52.9 | 23.2 | 2.3 | 10.7 | 18.7 | 37 | 43 |
| 5 |
|
4d | 51 | 64 | >100 | >100 | - | >200 | >200 | 24 | 0 |
| 6 |
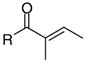
|
4e | 46 | 59 | >100 | >100 | - | >200 | >200 | 36 | 0 |
| 7 |

|
4f | 54 | 77 | >100 | >100 | - | >200 | 57.0 | 30 | 0 |
| 8 |
|
4g | - | 85 | >100 | >100 | - | >200 | >200 | 14 | 0 |
Isolated yield;
values are means of two independent experiments done in quadruplicate; the general error limits are 10%;
values are means of two independent experiments done in triplicate; the general error limits are 10–15%. Actual error values for each measurement are omitted for clarity but are included in tables in the supplemental materials.
Scheme 2.

Synthesis of ketones 1, 4a, and 4g.a
aReagents: (i) AlCl3 (2 equiv), acid chloride (maleic anhydride to form 4a), DCM, 0°C, 1 h.
Compounds 1, 4a, and 4g were obtained in moderate yields (45–85%) (Table 2, entries 1, 2, and 8). The saturated ketone 4a was synthesized as a nonelectrophilic control compound for the TR-CoA competition assay. The use of substituted acryloyl chloride derivatives under Friedel-Crafts reaction conditions resulted in complex reaction mixtures. An alternative two-step synthesis involving a halogen-exchange reaction of 4-bromo-heptylbenzene with n-butyllithium at −78°C followed by addition of substituted acroleins was used to obtain the corresponding allylic alcohols 5b–f. The oxidation to obtain the corresponding unsaturated ketones 4b–f was carried out using 4-methylmorpholine N-oxide in the presence tetrapropylammonium perruthenate.32 The resulting compounds were studied using the methods outlined above, and the results are summarized in Table 2.
The inhibition of the interaction between TR and CoA SRC2-2 depends strongly on the substitution pattern of the unsaturated ketones. Unsubstituted ketone 1 was identified as the most potent compound with the greatest ability to select between TRα and TRα. The IC50 values were 1.5 μM for TRβ and 28.1 μM for TRα (Table 2, entry 1). Derivatives of 1 bearing a methyl substituent in the α or β position (4b and 4c) or a carboxy substituent in the β position (4a) were less active (Table 2, entries 2–4). The dimethyl- and phenyl-substituted unsaturated ketones 4d, 4e, and 4f could not inhibit the interaction between TR and the CoA peptide (Table 2, entries 5–7). The saturated non-electrophilic ketone 4g showed no activity (Table 2, entry 8). A correlation between in vitro activity and toxicity was observed in ARO and U2OS cells. Active compounds 1 and 4a–c were considerably more cytotoxic than the inactive analogues 4d–g. LD50 values below 11 μM were found for compounds 1 and 4a–c in ARO cells. U2OS cells were less sensitive, especially in the case of 4a–b (Table 2, entries 2–3). The solubility of the ketones was limited in PBS containing 5% DMSO except 4a, bearing an acid functionality. All the ketones tested exhibited poor permeability.
We reported previously that aromatic acrylates inhibited the interaction between TR and SRC2-2.19 For this reason, acrylates and acrylamides were explored as a potential scaffolds. Variation of the aromatic ring was the focus of the acrylate library. Thirteen acrylates were synthesized using various phenols, hydroxylcyclohexane, and hydroxyltetrahydronaphthalene. These starting materials were converted into the corresponding acrylates by using acryloyl chloride in presence of triethylamine (Scheme 4).
Scheme 4.
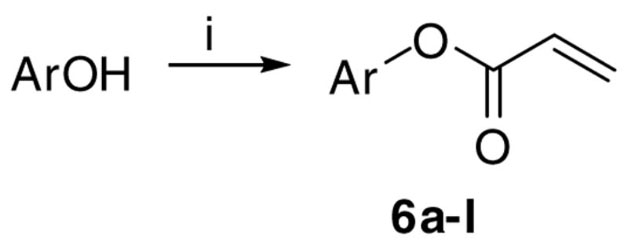
Synthesis of substituted acrylates 6a–l.a
aReagents: (i) acryloyl chloride (1.1 equiv), NEt3 (1.1 equiv), DCM, rt, 1 h.
The unsaturated esters were obtained in 71–94% yield (Table 3). Their ability to inhibit the interaction between TR and CoA was measured using an FP assay with TRα and TRβ. The cytotoxicity was determined in U2OS and ARO cells, and the results are summarized in Table 3.
Table 3.
Summary of IC50 values of 6a–m: Yields; inhibition of coregulatory peptide SRC2-2 and TRα/TRβ binding; viability (LD50 ) of U2OS and ARO cell in the presence of compounds.
| In vitro Potency | Cytotoxicity | |||||||
|---|---|---|---|---|---|---|---|---|
| Entry |
 R= |
Compound | Yielda (%) | TRαb IC50 (μM) | TRβb IC50 (μM) | TRα: TRβ ratio | AROc LD50 (μM) | U2OSc LD50 (μM) |
| 1 | 4- n-propylphenyl | 6a | 85 | >100 | 100 | - | 20.2 | 43.9 |
| 2 | 4- n-butylphenyl | 6b | 86 | 71.0 | 66.0 | 1.1 | 9.9 | 25.6 |
| 3 | 4- n-pentylphenyl | 6c | 88 | 60.9 | 33.9 | 1.8 | 20.0 | 22.7 |
| 4 | 4- tert-pentylphenyl | 6d | 94 | 69.6 | 43.5 | 1.6 | 5.8 | 27.5 |
| 5 | 4-n-hexylphenyl | 6e | 89 | 45.2 | 18.6 | 2.4 | 29.5 | 18.8 |
| 6 | 4-n-heptylphenyl | 6f | 76 | 23.0 | 10.5 | 2.2 | 69.2 | 36.7 |
| 7 | 4-n-octylphenyl | 6g | 81 | 21.4 | 11.4 | 1.9 | 111 | 50.5 |
| 8 | (trans) 4-tert-pentylcyclohexyl | 6h | 62 | 75.6 | 69.1 | 1.1 | 92.8 | 50.2 |
| 9 | 4-n-propylbenzoate | 6i | 75 | >100 | 61.0 | - | 4.8 | 121 |
| 10 | 4-n-pentylbenzoate | 6j | 71 | >100 | 55.8 | - | 21.2 | 36.5 |
| 11 | 2-n-hexylbenzoate | 6k | 85 | 39.1 | 8.2 | 4.8 | 14.6 | 19.4 |
| 12 | 4-n-butyramidophenyl | 6l | 73 | >100 | 85.9 | - | 8.6 | 242 |
| 13 | 5,6,7,8-tetrahydronaphthyl | 6m | 77 | 85.3 | 69.8 | 1.2 | 54.7 | 101 |
Iso lated yield;
values are means of two independent experiments done in quadruplicate; the general error limits are 10%;
values are means of two independent experiments done in triplicate; the general error limits are 10–15%. Actual error values for each measurement are omitted for clarity but are included in tables in the supplemental materials.
Seven para alkyl substituted acrylates were investigated with carbon chain lengths ranging from C3 to C8 (Table 3, entries 1–7). The binding to TR increased with the length of the substituent reaching maximal inhibition (10.5 μM TRβ) for 6f bearing a heptyl substituent (Table 3, entry 6). The selectivity ratios (TRα:TRβ) for these acrylates were about 2:1 in favor of TRβ. Compound 6h, bearing a cyclohexyl ring structure element and a tert-pentyl substituent, was less active than 6d, which had a phenyl ring structure element and the same substituent (Table 3, entries 4 and 8). No selectivity between TRα and TRβ was observed for 6h. The ortho-substituted benzoate 6k was significantly more potent (IC50 = 8.2 μM) than the corresponding para-substituted benzoates 6i and 6j (Table 3, entries 9–11). Compound 6k had the highest TRα:TRβ ratio (5:1) among the compounds in this library. Butyramidophenyl- and 5,6,7,8-tetrahydronaphthyl-substituted acrylates 6l and 6m showed weak activities (Table 3, entries 11 and 12). The measurement of the viability of ARO and U2OS cells in the presence of acrylates revealed that the cytotoxicity of the compounds gradually decreased with increasing length of the n-alkyl para substituent, except in the case of 6a (Table 3, entries 1–7). In general, ARO cells were more sensitive than U2OS cells to the acrylates, except for compounds 6e–h (Table 3, entries 5–8). Interestingly, three of these compounds bear an ester or an amide functionality in the para position (Table 3, entries 9, 10, and 12). The solubility and permeability of the acrylates tested was <10 μM and <5 × 10−6 cm/s, respectively (data not shown).
Variation of the α,β unsaturated system was the focus of the acrylamide library. The synthesis involved the reaction between 4-hexylaniline and various substituted acryloyl chlorides or anhydrides (Scheme 5).
Scheme 5.

Synthesis of substituted acrylamides 7a–l.a
aReagents: (i) acid chloride or anhydride, NEt3 (1.1 equiv, when the acid chloride was used), DCM, rt, 1 h.
The corresponding products 7a–f were obtained in 78–88% isolated yield. Inhibition constants were determined for both TRα and TRβ. Additionally, we determined toxicity in ARO and U2OS cells, solubility in PBS buffer containing 5% DMSO, and artificial membrane permeability. All these results are summarized in Table 4.
Table 4.
Summary of acrylamides 7a–g: Yield, IC50 values of the inhibition between coregulatory peptide SRC2-2 and TRα/TRβ, viability (LD50) of U2OS and ARO cells in the presence of compound, solubility in PBS buffer containing 5% DMSO, and permeability across an artificial membrane (PAMPA).
| In vitro Potency | Cytotoxicity | Pharmacological Properties | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R = 4-hexylbenzene | Compound | Yielda (%) | TRαb IC50 (μM) | TRβb IC50 (μM) | TRα: TRβ ratio | AROc LD50 (μM) | U2OSc LD50 (μM) | Solubility (μM) | Permeability (10−6 cm/s) |
| 1 |

|
7a | 88 | 21.6 | 20.3 | 1.1 | 113 | 66.5 | 47 | 833 |
| 2 |

|
7b | 85 | 17.5 | 17.9 | 1.0 | >200 | 148 | 418 | 29 |
| 3 |

|
7c | 78 | >100 | >100 | - | >200 | >200 | 42 | 432 |
| 4 |
|
7d | 84 | >100 | >100 | - | >200 | >200 | 37 | 98 |
| 5 |
|
7e | 89 | >100 | >100 | - | 174 | 69.4 | 105 | 14 |
| 6 |
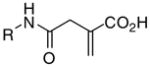
|
7f | 80 | >100 | >100 | - | 183 | 124 | 286 | 57 |
| 7 |
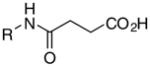
|
7g | 78 | >100 | >100 | - | .>200 | >200 | 372 | 48 |
Isolated yield;
values are means of two independent experiments done in quadruplicate; the general error limits are 10%;
values are means of two independent experiments done in triplicate; the general error limits are 10–15%. Actual error values for each measurement are omitted for clarity but are included in tables in the supplemental materials.
Among the acrylamides tested, only 7a and 7b inhibited the interaction between TR and SRC2-2 with IC50 values (TRβ) of 17.9 μM and 20.3 μM, respectively (Table 4, entries 1 and 2). No TR isoform selectivity was observed. Interestingly, all compounds showed minor toxic effects in ARO cells. Similar results were obtained for U2OS, with the exception of slightly more cytotoxic compounds 7a and 7e (Table 4, entries 1 and 4). High solubilities were measured for carboxylic acid-substituted acrylamides 7b, 7f, and 7g (Table 4, entries 2, 6, and 7). These compounds exhibit limited permeabilities in the PAMPA assay (Pe < 60 × 10−6 cm/s. In contrast, good diffusion rates were observed for the less soluble compounds 7a and 7c (Table 4, entries 1 and 3).
Finally, several other potential electrophilic moieties were explored. First, propiolic acid derivatives 8a and 8b were obtained using diisopropylcarbodiimide as a coupling reagent, starting from the corresponding phenol or aniline (Scheme 6).
Scheme 6.

Synthesis of 8a and 8b.a
aReagents: (i) propionic acid (1.3 equiv), 4-dimethylaminopyridine (0.001 equiv), N,N′-diisopropylcarbodiimide, DCM, rt, 5 h.
The halo-ketones 8c–e were synthesized under Friedel-Crafts conditions (Scheme 2). Fluoroacetyl chloride33 was obtained from the corresponding sodium salt. The keto-epoxide 8f was synthesized for the corresponding α,β-unsaturated ketone 1 obtained after HBr elimination of the β-bromo ketone 8e using DBU as a base (Scheme 7). The yields, inhibition constants for both TRα and TRβ, cytotoxicity in ARO and U2OS cells, solubility in PBS with 5% DMSO, and permeability (PAMPA) are summarized in Table 5.
Scheme 7.

Synthesis of 8a and 8b.a
aReagents: (i) DBU (1.1 equiv), benzene, rt, 2 h; (ii) H2O2 (30%) (5 equiv), NaOH (1M in H2O), MeOH, 20 min.
Table 5.
Summary of electrophilic compounds 8a–f: Yield, IC50 values of the inhibition between coregulatory peptide SRC2-2 and TRα/TRβ, viability (LD50) of U2OS and ARO cells in the presence of compound, solubility in PBS buffer containing 5% DMSO, and permeability across an artificial membrane (PAMPA).
| In vitro Potency | Cytotoxicity | Pharmacological Properties | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R = 4-hexylbenzene | Compound | Yielda (%) | TRαb IC50 (μM) | TRβb IC50 (μM) | TRα: TRβ ratio | AROc LD50 (μM) | U2OSc LD50 (μM) | Solubility (μM) | Permeability (10−6 cm/s) |
| 1 |
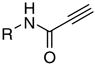
|
8a | 63 | 68.9 | 35.1 | 2.0 | 8.0 | 17.1 | 60 | 621 |
| 2 |

|
8b | 57 | 16.7 | 6.2 | 2.7 | 7.9 | 13.9 | 18 | 21 |
| 3 |

|
8c | 84 | 47.7 | 23.1 | 2.1 | 12.2 | 8.3 | 103 | 1029 |
| 4 |

|
8d | 41 | >100 | >100 | - | 2.4 | 69 | 95 | 974 |
| 5 |
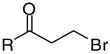
|
8e | 69 | 4.2 | 3.0 | 1.4 | 40.1 | 17 | 85 | 1190 |
| 6 |

|
8f | 95 | >100 | >100 | - | 28.7 | 25.3 | 26 | 58 |
| 7 |
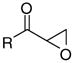
|
8g | 74 | 33.2 | 19.1 | 1.7 | >200 | 58 | 204 | 1117 |
Isolated yield;
values are means of two independent experiments done in quadruplicate; the general error limits are 10%;
values are means of two independent experiments done in triplicate; the general error limits are 10–15%. Actual error values for each measurement are omitted for clarity but are included in tables in the supplemental materials.
Propiolic acid derivatives 8a and 8b were able to inhibit the interaction between TR and SCR2-2 with IC50 values of 35.1 μM and 6.2 μM for TRβ (Table 5, entries 1 and 2). A general IC50 ratio of 1:2 (TRβ:TRβ) was observed for all active electrophiles. Two α-halogen ketones, 8c and 8d, were investigated. The α-chloro ketone 8c was able to inhibit the interaction between SRC2-2 and TR, but α-fluoro ketone 8d was not active (Table 5, entries 3 and 4). The β-bromo ketone 8e was the most potent compound in this group (Table 5, entry 5) but had poorer selectivity, exhibiting IC50 values of 4.2 μM and 3.0 μM for TRα and TRβ, respectively. In contrast, the γ-bromo ketone 8f was not active. The racemic epoxy-ketone 8f inhibited the interaction between TR and the CoA SRC2-2 with an IC50 value of 47.7 μM for TRα and 23.1 μM for TRβ (Table 6, entry 7). The viability of ARO and U2OS cells in the presence of these electrophiles was limited. Major differences in sensitivities between the two cell lines were measured for α-fluoro ketone 8d and epoxy ketone 8g (Table 5, entries 4 and 7). Although most of the electrophiles were not very soluble in PBS buffer containing 5% DMSO, we observed high permeabilities for 8c–e and 8g.
The relationships between biochemical activities, cellular activities, and pharmacological properties are important in the identification of potential drug candidates. To assess such relationships, scatter plots based on measured data were constructed. First, we visualized the relationship between their physical properties, solubility and permeability (Figure 2).
Figure 2.
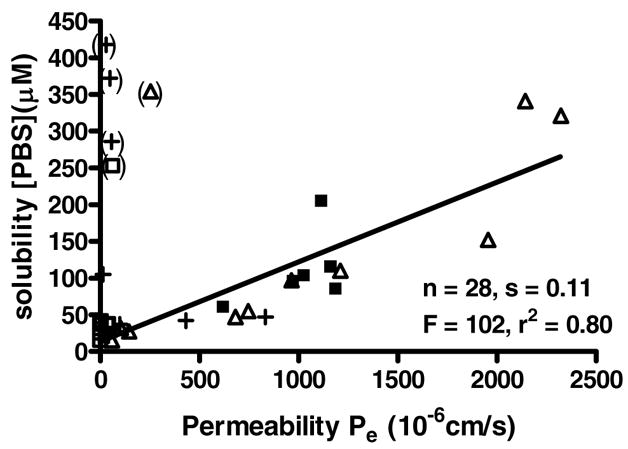
Scatter plot of solubility and permeability data for all compounds except acrylates.a
aΔ Aminophenylketones (Table 1), □ enones (Table 2), + acrylamides (Table 4), ■ electrophiles (Table 5); n = number of X values, s = slope, F = F-values, r2 = square correlation coefficient, () excluded data points.
The scatter plot revealed a significant relationship between both properties. The linear regression resulted in a positive slope of 0.11, indicating a positive correlation between permeability and solubility. In contrast to the general trend, we observed low permeability for highly soluble carboxylic acids (4a, 7b, 7f, and 7g) and the secondary amine 3k. The compounds with the highest solubility and permeability were β-aminophenylketones 3d and 3h. Compound 3h was selected for further stability studies. Over a period of 24 h, we monitored the stability of an aqueous and saline solution (150 mM NaCl) of 3h at pH 7 using liquid chromatography-mass spectrometry (LCMS). The results are given in Figure 3.
Figure 3.

LCMS-PDA analysis of 3h in water, saline (150 mM NaCl), and human blood plasma (all pH 7).a
aIntegrated absorption area at 254 nm (PDA); black: ratio between peak area of 3h and peak area of internal standard (salicylic acid) measured in blood plasma (left y-axis); grey: area of 3h measured in water (right axis); white: area of 3h measured in saline (right y-axis).
No degradation of 3h was detected in water or saline at pH 7 during this time course. Additionally, we performed a stability study in human blood plasma using 5 μM 3h. Degradation with a half-life of 1.4–2.4 h was determined during a 24-h experiment.
The in vitro cytotoxicity data revealed a good correlation between the LD50 in ARO cells and U2OS cells (Figure 4).
Figure 4.

Scatter plot of LD50 values measured in U2OS and ARO cells for all compounds.a
aΔ Aminophenylketones (Table 1), □ enones (Table 2), * acrylates (Table 3), + acrylamides (Table 4), ■ electrophiles (Table 5), () excluded data points.
The slope (0.52) indicated a general twofold higher cytotoxicity in thyroid cancer cells (ARO) than in bone cancer cells (U2OS). Compounds 4a, 6i, 6l, and 8d were highly selective between the two cell lines (Figure 4, data point in parentheses). ARO:U2OS LD50 ratios up to 1:28 were observed. The selectivity was not scaffold related.
Given the general utility of the wide range of the electrophiles and the variance in the specific nature of inhibition, we wished to understand the relationship between three-dimensional structure and activity. Pharmacophore modeling is an important tool for elucidating a general molecular structure underlying the active compounds in a bioactive screen. The study was carried out using Molecular Operating Environment (MOE) software.34 The different compound libraries we analyzed separately anticipating the same binding mode for all compounds. The results are illustrated in Figure 5.
Figure 5.

Pharmacophore elucidation using MOE software.a
aMolecules are illustrated in stick form: gray = carbon, red = oxygen, blue = nitrogen; green mesh balls = hydrophobic center (Uns = unsaturated, Aro = aromatic, Ali = aliphatic); blue mesh balls = possible electron acceptor site; A: enones (active in FP assay), B: acrylates (IC50 < 30 μM), C: acrylamides (active in FP assay).
The pharmacophore models for all three libraries (enones A, acrylates B, and acrylamides C) were very similar (Figure 5). All had an electrophilic head group (Hyd/Uns), an aromatic core structure (Hyd/Aro), and a hydrophobic center (Hyd/Ali) represented by green mesh balls. The distance between Hyd/Uns and Hyd/Aro differed for A, B, and C between 4.65 and 5.69 Ǻ, depending on the linkage C(O), OC(O), or NHC(O). The distance between Hyd/Ali and Hyd/Aro varied between 3.89 and 5.32 Ǻ. The analysis of a possible electron acceptor interaction revealed an electron acceptor site (Acc) for all pharmacophores. The average distance between Hyd/Uns and Acc for all models was 4.31 ± 0.37 Ǻ, and the distance between Hyd/Aro and Acc was 4.39 ± 0.25 Ǻ.
To elucidate the mode of binding of these molecules to the TR-CoA binding site, we modeled compound 1 with a covalent bond to C309 (Figure 6). Point mutation analysis showed significantly less binding affinity for the TR C309A mutant than for the wild type, as determined by two independent assays.19 We observed a ridged binding site for the electrophilic head group and a possible activation of a carbonyl functionality by K306. The distance between the carbonyl oxygen and the K306 amide hydrogen was 3.3 Ǻ. A relatively spacious hydrophobic groove surrounded the aromatic core structure, and a hydrophobic curved surface accommodated the alkyl substituent.
Figure 6.
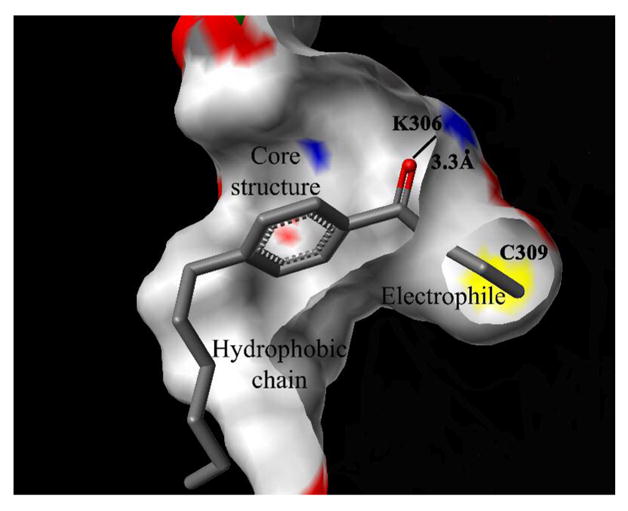
Modeling study of the bonding of compound 1 to the TR coactivator binding pocket.a
aSurface gray: hydrophobic; blue: nitrogen; red: oxygen; yellow: sulfur.
The AF-2 domain of TRβ has a unique architecture in comparison the other NR -- containing three cysteine residues (Cys309, Cys298, and Cys294) not present in any other NR AF-2. Two of the sulfhydryl functionalities of these unique cysteines (Cys309 and Cys298) are exposed to the solvent. To confirm that this aspect of the TRβ structure affords selectivity of the inhibitors for TRβ, we carried out fluorescence polarization competition assays employing the following nuclear receptors: the androgen receptor, the estrogen receptor α, and the peroxisome proliferator activated receptor γ. No inhibition of coregulator recruitment to any of these NR could be detected after 3h of exposure to 3h at concentrations up to 100 μM (data not shown). Thus, this class of inhibitors appears to be intrinsically selective for TR relative to other NR due to the presence of unique cysteine residues in the AF-2 binding site.
Discussion
These studies of TR CoA modulators provided new structural insight into the mode of action of these compounds. First, within a given aromatic substitution pattern, all β-aminophenylketones tested showed similar activities in the competition binding assay, despite the wide variation in structural and electronic properties of the amine substituents. This behavior is consistent with the β-aminophenylketones acting as prodrugs due to a deamination reaction liberating the active electrophilic unsaturated ketones. Stability studies simulating assay conditions showed no detectable elimination of the β-aminophenylketones in the absence of protein. Moderate stability was observed in blood plasma, resulting in a half-life of about 2 h.
The investigations applying unsaturated ketones bearing different substituents showed that activity is highly dependent on the steric nature of the electrophile. Enones with a α-methyl, trans-methyl, or cis-carboxy substituent (4a–c) were able to inhibit the interaction between TR and the CoA SRC2-2. In contrast, enones with two methyl substituents or a single β-phenyl substituent were not tolerated (4d–f). This correlated with the modeled complex of TR-LBD–exhibiting compound 1, which had a well defined and rigid binding site unsuitable for highly substituted enones. Additional binding data were observed for the acrylamides. In this series, unsubstituted and cis-carboxy acrylamides (7a and 7b) were able to interrupt the TR-CoA interaction, whereas trans-methyl substituted acrylamide 7c was not active.
Another important feature of functional inhibitors is the distance between the electrophilic head group and the aromatic core structure. In contrast to the inactive trans-methylated unsaturated amide 7c, the trans-methylated unsaturated ketone 4b was active, although both unsubstituted amide 7a and the unsaturated ketone 1 were also active. A similar dependency was observed in compounds 7b and 7f. The pharmacophore model showed a distance between the electrophilic group (Hyd/Uns) and core structure (Hyd/Aro) of 4.65 Ǻ for enones and 5.68 Ǻ for acrylamides. The difference of 1.03 Ǻ is likely to limit substitution of active acrylamides at the α-position, assuming the same mode of binding for enones and acrylamides. Electrophilic effects might support this result as well. Enones are more reactive Michael acceptors than acrylamides, and substituents in the β-position have a strong inductive effect on the polarization of double bonds. Decreased Michael acceptor activity from 1 (enone) to 4b (β-methylketone) to 7a (acrylamide) and 7c (β-methylacrylamide) could be supported by calculations of differences in the lowest unoccupied and highest occupied molecular orbitals (ΔE = 9.175 > 9.164 > 8.683 > 8.487).
One of the most important structural requirements for TR-CoA inhibitors is the hydrophobic substituent in the para position relative to the electrophilic head group. Table 3 shows that a medium chain length of six to seven carbon atoms is most favorable for binding. Although ortho alkyl-substituted β-aminophenylketones are not functional inhibitors (data not shown), 2-hexyl benzoate 6k was one of the most potent inhibitors. The carboxy functionality increases the rigidity of the substituent and alters the orientation of the alkyl chain. In the case of 6k, this resulted in an increase in binding affinity. In contrast, para-substituted benzoates 6i–j were less potent than compounds 6c and 6f bearing an n-alkyl chain in the para position. This might imply that the presence of carboxy functionality could alter hydrophobic interactions as well, resulting in weaker binding affinities for 6i–j.
Interestingly, the core structure of the TR-CoA inhibitors, the phenyl ring, can be exchanged with a cyclohexyl ring without a major loss of potency (see 6h and 6d). In general, the cyclohexyl ring is more space-filling, more hydrophobic, and unable to π-stack with other aromatic systems. The tolerance of a core structure with a similar size but different electronic properties implies that the contribution of this group to binding is solely hydrophobic in nature.
The pharmacophore modeling study (Figure 5) revealed a possible electron acceptor/hydrogen donor (Acc) in close proximity to Hyd/Uns and Hyd/Aro. Examining the crystal structure of the AF-2 pocket for possible hydrogen-donating amino acids, we identified K306 as a likely residue (Figure 6). The small molecule modeling study showed a possible moderate interaction (3.3 Å) between K306 and the carbonyl/carboxyl oxygen conserved in all compounds.
Finally, we showed that nine 4-hexylphenyl compounds with different electrophilic head groups inhibited the interaction between TR and CoA (1, 3a–k, 4a, 7a and 8a–g). The most active compounds were enone 1, β-aminophenylketones 3a–k, and β-bromo ketone 8e. We assumed that, like the β-aminoketones, 8e underwent an elimination reaction to yield the corresponding enone 1. This was supported by the fact that γ-bromo ketone 8f, missing a β-positioned leaving group, was not active. α-Fluoro ketones were, in general, weak electrophiles exhibiting no binding affinity in our study (8d). All other electrophilic compounds, involving the acrylic acid and propiolic acid derivates, keto-epoxide, and α-halo ketones, showed comparable activity in our SAR study.
The toxicity, solubility, and permeability studies revealed the drug like character of the compounds investigated. Among the sub-series studied, we found strong variations in drug like properties. All members of the acrylate group exhibited limited solubility and permeability and moderate activity. Among the acrylamide group, the only active compounds identified were 7a and 7b. These compounds were either not very soluble (7a) or not very permeable (7b), as reflected by the weak cytotoxic effects in the ARO and U2OS cell lines. Four of the eight enones tested were not active. The active enones 1 and 4a–c showed limited permeability but elevated cytotoxicity, especially in the ARO cell line. In general, we observed high solubility but low permeability for all compounds bearing a carboxylic acid functionality (4a, 7b, 7f, and 7g). Compound 4a showed higher cytotoxicity in ARO cells than 7b, 7f, and 7g did. Among the active compounds of the electrophile group, three compounds exhibited Pe values > 1000 × 10−6 cm/s (8c, 8e, and 8g). Compound 8g was the most soluble compound, and 8e was the most active one.
We observed the best permeability values within the group of aminophenylketones (3d, 3e, 3g, and 3h). Compounds 3d and 3h also were very soluble in PBS with 5% DMSO and were able to inhibit the interaction between TRβ and SRC2-2 in concentrations less than 5 μM. Stability studies confirmed a half-life of 1.4–2.4 h in plasma and no degradation in aqueous solution over a period of 24 h.
Conclusion
This SAR study of small molecule TR-CoA inhibitors revealed compounds with a defined structure consisting of an electrophilic head group, a hydrophobic core structure, and a hydrophobic alkyl substituent. The most active compounds were those bearing an n-heptyl or n-hexyl para substituent and having a distance between the core structure and the electrophilic center of 6.0 Ǻ or less. Electronic properties of the Michael acceptor have an important influence on potency, with highly reactive enones being the most potent inhibitors. Among the compounds we investigated, we identified potent inhibitors with promising properties in terms of toxicity, solubility, and permeability: for example, 3-(dimethylamino)-1-(4-hexylphenyl)propan-1-one.
These and similar compounds have been distributed as local anesthetics and antiarrhythmics, altering ion-channel activity. The ion-channel activity of these compounds is governed strongly by the pKa of the nitrogen. In our case, we discovered elimination of the amine liberated the active enone inhibiting TR. By altering the pKa of the nitrogen we might be able to decouple the undesired ion channel activity of this class of compounds from the desired activity against TR. The development of these prodrugs might establish new treatments for metabolic syndrome and metabolic disease based on coregulator dysfunction. In addition, these irreversible inhibitors represent valuable research tools in vivo and in vitro to study distribution and expression levels of the TR, to map potential active sites on the protein surface, and to elucidate signal pathways mediated by the TR.
Experimental section
General Considerations. Unless otherwise noted, all materials were obtained from commercial suppliers and used without further purification. All solvents used were dried using an aluminum oxide column. Thin-layer chromatography was performed on pre-coated silica gel 60 F254 plates. Purification of compounds was done by reverse-phase high-performance liquid chromatography (HPLC; Flex [Biotage] and RP-C18 Xterra column 5μm, 19 mm × 50 mm [Waters]) or by normal phase column chromatography (SP1 [Biotage], Silica gel 230–400 mesh) followed by evaporation (HT-4X evaporator [Genevac]). The analysis (photodiode array, total ion count, and expected mass [m/z]) was performed using a HPLC-MS (Alliance HT, Micromass ZQ 4000 and RP-C18 Xterra column 5 μm, 6 mm × 50 mm [Waters], flow rate: 1 mL/min; gradient: 30:70 [methanol: water (0.05% TFA)]) to 100% MeOH over 10 min. NMR spectra were recorded on a Bruker 400 MHz and referenced internally to the residual resonance in CDCl3 (δ 7.26 ppm) for hydrogen and (δ = 77 ppm) for carbon atoms.
General procedure for 3a–k
To a suspension of AlCl3 (26.6 g, 200 mmol) in CH2Cl2 (200 mL) was added acryloyl chloride (9.78 mL, 100 mmol) at 0°C. The resulting clear solution was treated with 1-phenylhexane (18.9 mL, 100 mmol) and stirred for 2 h at room temperature. The reaction was poured into a mixture of concentrated HCl (40 mL) and ice (200 g). The organic layer was separated, the aqueous layer was extracted with CH2Cl2 (100 mL), and the combined organic layers were washed with saturated aqueous NaHCO3 (100 mL) and brine (100 mL) and dried over MgSO4. The filtered solution was concentrated in vacuuo to yield compounds 1 and 2 (15.6 g, 70%) as a yellow oil in a ratio of 4:1. Next, 9 g (45 mmol) of the crude product was dissolved in THF (30 mL) to obtain a 1.5 M stock solution. The corresponding amines (3 mmol) were dissolved in THF (8 mL) and treated with 2 mL of the stock solution (1.5 M solution of 1 and 2) and stirred at room temperature overnight. The reaction mixtures were evaporated, and the residual amines were removed by repeated azeotropic evaporations using ethanol to yield the corresponding β-aminophenylketones in 85–97% yield with a purity of 85–95%, as determined by HPLC-MS. Subsequent purification by RP-HPLC (flow rate: 20 mL/min; gradient: 0.05% formic acid in water to 0.05% formic acid in 80% acetonitrile over 15 min) resulted in desired product (> 99% purity). (Compound 3a is given as an example. For characterization of compounds 3b–k: see supplemental material.)
(3a) 1-(4-Hexylphenyl)-3-(methyl(phenethyl)amino)propan-1-one
White solid, mp 147–149°C, yield 84%; LCMS RT = 5.02 min; 1H-NMR (400 MHz) δ = 7.73 (d, J = 8.3 Hz, 2H), 7.19–7.06 (m, 8H), 3.56–3.51 (m, 1H), 3.45–3.33 (m, 3H), 3.08–3.25 (m, 1H), 3.13–3.08 (m, 1H), 2.98–2.94 (m, 2H), 2.75 (s, 3H), 1.49–1.46 (m, 2H), 1.20–1.11 (m, 6H), 0.73 (t, J = 6.9 Hz, 3H); 13C-NMR (100 MHz) δ = 195.32, 150.25, 135.37, 133.11, 129.10, 128.97, 128.66, 128.32, 127.51, 58.13, 51.36, 40.47, 36.04, 33.18, 31.61, 30.95, 30.43, 28.86, 22.53, 14.04; MS calcd for C24H33NO (H+) 352.26, found 352.32.
General procedure for 1, 4a, and 4g: see general procedure for 3a–k. 1 is given as an example. For characterization of compounds 4a and 4g: see supplemental material.
(1) 1-(4-hexylphenyl)prop-2-en-1-one
Colorless liquid, yield 55%; LCMS RT = 6.90 min; 1H-NMR (400 MHz) δ = 7.88 (d, J = 8.2 Hz, 2H), 7.28 (d, J = 8.2 Hz, 2H), 7.17 (dd, J = 17.1 Hz, J = 10.5 Hz, 1H), 6.43 (dd, J = 17.1 Hz, J = 1.6 Hz, 1H), 5.90 (dd, J = 10.5 Hz, J = 1.6 Hz, 1H), 2.67 (t, J = 7.8 Hz, 2H), 1.63 (p, J = 7.1 Hz, 2H), 1.36–1.27 (m, 6H), 0.88 (t, J = 6,8 Hz, 3H); 13C-NMR (100 MHz) δ = 190.55, 148.82, 134.94, 132.41, 129.62, 128.86, 128.67, 36.02, 31.65, 31.06, 28.92, 22.56, 14.05; MS calcd for C15H20O (H+) 217.15, found 217.82.
General procedure for 5b–f
To a solution of 1-bromo-4-heptylbenzene (408 μL, 2 mmol) in THF (3 mL) was added n-BuLi (2M in pentane, 1.1 mL, 1.1 equiv.) at −78°C. After stirring for 20 min, aldehyde (2 mmol, 1 equiv.) was added and the solution was stirred for an additional 2 h at −78°C. The reaction mixture was quenched at −78°C with NH4Cl (aq) (2 mL) and allowed to warm to room temperature. The organic layer was separated. The aqueous layer was extracted with ethyl acetate (3 mL, twice) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuuo. Purification was done by flash column chromatography (SP1, 25M (SiO2), flow rate: 25 mL/min; gradient: 1% to 30% ethyl acetate in hexanes over 20 CV). 5b is given as an example. For characterization of compounds 5c–f: see supplemental material.
(5b) (E)-1-(4-heptylphenyl)but-2-en-1-ol
Colorless liquid, yield 51%; LCMS (E/Z) RT = 7.75/8.28 min; 1H-NMR (400 MHz) δ = 7.28 (d, J = 7.9 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 5.82–5.66 (m, 2H), 5.15–5.11 (m, 1H), 2.60 (t, J = 7.5 Hz, 2H), 1.95 (m, 1H (OH)), 1.71 (m, 3H), 1.62 (m, 2H), 1.36–1.27 (m, 8H), 0.90 (t, J = 6.9 Hz, 3H); 13C-NMR (100 MHz) δ = 142.25, 140.61, 133.68, 128.46, 127.07, 126.02, 75.04, 35.61, 31.79, 31.48, 29.27, 29.15, 22.64, 17.66, 14.07, MS calcd for C17H26O(H+) 247.20, found 228.80 (-OH).
General procedure for 4b–f
To a solution of alcohol (0.36 mmol), 4-methylmorpholine N-oxide (65 mg, 0.54 mmol, 1.5 equiv.), molecular sieves (200 mg) in acetonitrile (3 mL) was added tetrapropylammonium perruthenate (6.5 mg, 0.018 mmol, 0.05 equiv). The reaction mixture was stirred for 1–4 h, absorbed onto silica gel and purified by column chromatography (SP1, 25M (SiO2), flow rate: 25 mL/min; gradient: 1–10% ethyl acetate in hexanes over 18 CV). 4b is given as an example. For characterization of compounds 4c–f: see supplemental material.
(4b) (E)-1-(4-heptylphenyl)but-2-en-1-one
Colorless liquid, yield 67%; LCMS RT = 8.48 min; 1H-NMR (400 MHz) δ = 7.85 (d, J = 8.3 Hz, 2H), 7.26 (d, J = 8.4 Hz, 2H), 7.06 (m, 1H), 6.91 (m, 1H), 2.65 (t, J = 8.0 Hz, 2H), 1.99 (d, J = 6.8 Hz, 3H), 1.62 (m, 2H), 1.34–1.24 (m, 8H), 0.87 (t, J = 7.0 Hz, 3H); 13C-NMR (100 MHz) δ = 190.30, 148.35, 144.42, 135.50, 128.65, 128.55, 127.48, 35.99, 31.76, 31.14, 29.22, 29.12, 22.63, 18.57, 14.7; MS calcd for C17H24O(H+) 245.77, found 245.77.
General procedure for 6a–m
A solution of triethylamine (83 μL, 0.6 mmol, 1.1 equiv) and alcohol (0.5 mmol) in DCM (2 mL) was treated with acryloyl chloride (49 μL, 0.6 mmol, 1.1 equiv.) and stirred for 1 h at rt. The reaction mixture absorbed on silica cartridge and purified by flash column chromatography (SP1, 25S (SiO2), flow rate: 25 mL/min; gradient: 1% to 20% ethyl acetate in hexanes over 20 CV). (6a is given as an example. For characterization of compounds 6b–m: see supplemental material)
(6a) 4-propylphenyl acrylate
Colorless liquid, yield 85%; LCMS RT = 6.18 min; 1H-NMR (400 MHz) δ = 7.18 (d, J = 8.6 Hz, 2H), 7.03 (d, J = 8.5 Hz, 2H), 6.59 (dd, J = 17.3 Hz, J = 1.3 Hz, 1H), 6.31 (dd, J = 17.3 Hz, J = 10.4 Hz, 1H), 6.00 (dd, J = 10.4 Hz, J = 1.3 Hz, 1H), 2.58 (t, J = 7.4 Hz, 2H), 1.64 (h, J = 7.4 Hz, 2H), 0.94 (t, J = 7.3 Hz, 3H); 13C-NMR (100 MHz) δ = 164.71, 148.51, 140.26, 132.26, 129.31, 128.08, 121.10, 39.44, 24.51, 13.78; MS calcd for C12H14O2 (H+) 191.10, found 191.37.
General procedure for 7a–g
To a solution of 4-hexylaniline (216 μL, 1 mmol), triethylamine (209 μL, 1.5 mmol, 1.5 equiv.) in DCM (5 mL) was added acid chloride (1.1 mmol) or acid anhydride (1 mmol). The reaction mixture was stirred for 1 h, poured into NH4Cl (aq) (10 mL), diluted with ethyl acetate (10 mL), and the organic layer was separated. The aqueous layer was extracted with ethyl acetate (10 mL, twice) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuuo. Purification was done by flash column chromatography (SP1, 25M (SiO2), flow rate: 25 mL/min; gradient: 1–30% ethyl acetate in hexanes over 20 CV). 7a is given as an example. For characterization of compounds 7b–g: see supplemental material.
(7a) N-(4-hexylphenyl)acrylamide
White solid, mp 91–92°C, yield 88%; LCMS RT = 8.15 min; 1H-NMR (400 MHz) δ = 7.37 (d, J = 8.1 Hz, 2H), 7.30 (s, NH), 7.02 (d, J = 8.1 Hz, 2H), 6.30 (dd, J = 16.8 Hz, J = 1.2 Hz, 1H), 6.13 (dd, J = 16.8 Hz, J = 10.4 Hz, 1H), 5.62 (dd, J = 10.6 Hz, J = 1.2 Hz, 1H), 2.46 (t, J = 7.6 Hz, 2H), 1.47 (p, J = 7.1 Hz, 2H), 1.22–1.16 (m, 6H), 0.76 (t, J = 6.8 Hz, 3H); 13C-NMR (100 MHz) δ = 163.44, 139.31, 135.30, 131.26, 128.86, 127.44, 120.00, 35.36, 31.69, 31.42, 28.89, 22.58, 14.06 MS calcd for C15H21NO (H+) 231.33, found 231.16.
General procedure for 8a–b
To a solution of 4-hexylaniline (178 μL, 1 mmol) or 4-hexylphenol (179 μL, 1 mmol), propiolic acid (70 μL, 1.3 mmol, 1.3 equiv.), 4-dimethylaminopyridine (0.01 mmol, 1.2 mg, 0.01 equiv) in DCM (5 mL) was added N,N′-diisopropylcarbodiimide (126 μL, 1.3 mmol, 1.3 equiv.). The resulting reaction mixture was stirred at rt for 5 h, poured into NH4Cl (aq) (10 mL), diluted with ethyl acetate (10 mL) and the organic layer was separated. The aqueous layer was extracted with ethyl acetate (10 mL, twice) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuuo. Purification was done by flash column chromatography (SP1, 25M (SiO2), flow rate: 25 mL/min; gradient: 1% to 15% ethyl acetate in hexanes over 15 CV). 8a is given as an example. For characterization of compounds 8b: see supplemental material.
(8a) N-(4-hexylphenyl)propiolamide
Brown solid, mp 62–64°C, yield 79%; LCMS RT = 8.43 min; 1H-NMR (400 MHz) δ = 7.53 (s (br), NH), 7.41 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 2.91 (s, 1H), 3.57 (t, J = 7.5 Hz, 2H), 1.58 (p, J = 7.4 Hz, 2H), 1.35–1.23 (m, 6H), 0.88 (t, J = 6.7 Hz, 3H); 13C-NMR (100 MHz) δ = 154.70, 140.19, 134.38, 129.00, 120.07, 75.64, 74.02, 35.38, 31.68, 31.37, 28.87, 22.58, 14.07; MS calcd for C15H19O (H+) 229.15, found 229.80.
General procedure for 8c and 8e: see general procedure for 3a–k. 8c is given as an example. For characterization of compounds 8e: see supplemental material.
(8c) 2-chloro-1-(4-hexylphenyl)ethanone
White solid, mp 42–44°C, yield 84%; LCMS RT = 6.72 min; 1H-NMR (400 MHz) δ = 7.88 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.3 Hz, 2H), 4.69 (s, 2H), 2.67 (t, J = 7.8 Hz, 2H), 1.63 (p, J = 7.7 Hz, 2H), 1.36–1.27 (m, 6H), 0.88 (t, J = 7.0 Hz, 3H); 13C-NMR (100 MHz) δ = 190.69, 149.99, 131.93, 128.92, 128.66, 45.95, 36.07, 31.62, 30.98, 28.90, 22.55, 14.05; MS calcd for C14H19ClO (H+) 238.11, found 238.80.
General procedure for 8f
To a solution of 8e (296 mg, 1 mmol) in benzene (3 mL) was added DBU (1,8-Diazabicyclo[5.4.0]undec-7-ene) (150 μL, 1.1 mmol, 1.1 equiv.) and the reaction mixture was stirred 2 h at room temperature, filtered, and evaporated to yield crude 1. The crude product was resuspended in MeOH (5 mL) and treated with H2O2 (30% in H2O) (0.3 mL, 3 mmol, 3 equiv.) and NaOH (1M in H2O) (1 mL). The reaction mixture was evaporated, dissolved in water, and extracted with ethyl acetate (20 mL, twice) and the combined organic layers were dried over MgSO4, filtered, and concentrated in vacuuo. Purification was done by flash column chromatography (SP1, 25M (SiO2), flow rate: 25 mL/min; gradient: 1% to 15% ethyl acetate in hexanes over 15 CV).
(8f) (4-hexylphenyl)(oxiran-2-yl)methanone
Colorless liquid, yield 74%; LCMS RT = 7.20 min; 1H-NMR (400 MHz) δ = 7.97 (d, J = 8.1 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 4.23 (m, 1H), 3.11 (m, 1H), 2.97 (m, 1H), 2.67 (t, J = 7.9 Hz, 2H), 1.63 (p, J = 7.1 Hz, 2H), 1.36–1.27 (m, 6H), 0.88 (t, J = 6.9 Hz, 3H); 13C-NMR (100 MHz) δ = 194.12, 149.91, 133.14, 128.85, 128.46, 50.97, 47.49, 36.06, 31.60, 30.97, 28.87, 22.57, 14.03; MS calcd for C15H20O2 (H+) 233.15, found 233.15.
TR-SRC2-2 competition assay
This assay has been described in detail previously.19, 29 The concentration of each compound required to inhibit 50% of the binding between TRβ-LBD and SRC2-2 peptide is presented in Tables 1–5. hTRβ1 LBD (His6 T209-D461) was expressed in BL21 (DE3) (Invitrogen) and the peptide SRC2-2 (CLKEKHKILHRLLQDSSSPV) was labeled with 5-iodoacetamidofluorescien (Molecular Probes). The competition binding experiments were evaluated using PrismPad and the IC50 values were obtain by fitting data to equation: Sigmoidal dose-response (variable slope) or four parameter logistic equation; Y=Bottom + (Top−Bottom)/(1+10^((LogIC50−X)*HillSlope)); X is the logarithm of concentration; Y is the response. Two independent experiments were done in quadruplicate and values are given as the mean values with 95% confidence interval.
Toxicity study
Human bone osteosarcoma epithelial cells (U2OS; ATCC HTB-96) were grown to 80% confluency at 37°C in McCoy’s 5a medium with 1.5 mM L-glutamine adjusted to contain 2.2 g/L sodium bicarbonate, 10% fetal bovine serum (ATCC), 50 units/mL penicillin, and 50 μg/mL streptomycin. Human anaplastic thyroid cancer cells ARO (kindly provided by Dr. M. D. Ringel) were maintained in RPMI 1640 culture medium supplemented with 10% fetal bovine serum (heat inactivated), 1× nonessential amino acid (ATCC), 10% fetal bovine serum (ATCC), 50 units/mL penicillin, and 50 μg/mL streptomycin. Cells were grown to 80% confluency, collected, and resuspended at a concentration of 57000 cells/mL dispensed in 384-well plates at 35 μL per well (2000 cells). The plates were incubated for 48 h at 37°C. The small molecules were serially diluted from 50000 μM to 2.5 μM in DMSO into a 96-well plate (Costar 3365), and 0.5 μL was transferred into the 384-well cell culture plates (Corning 3917) yielding a final compound concentration range of 714–0.036 μM. After 48 h incubation at 37°C, 25 μL of CellTiter-Glo was added using a Wellmate (Matrix), followed by agitation using an MTS 2/4 digital microtiter shaker (IKA). Luminescence was measured after 10 min using a EnVision (PerkinElmer). The concentration of each compound required for 50% cell survival (LD50) is presented in Tables 1–5. These LD50 values were obtained by fitting data to the equation: Sigmoidal dose-response (variable slope) or four parameter logistic equation; Y=Bottom + (Top−Bottom)/(1+10^((Log LD50−X)*HillSlope)); X is the logarithm of concentration; Y is the response (PrismPad). Two independent experiments were done in triplicate, and values are given as the mean values with 95% confidence interval.
Solubility study
The assay was done using the MultiScreen Solubility assay developed by Millipore. Briefly, 16 test compounds per 96-well polypropylene plate (Costar 3365) were serial diluted from 10000 μM to 625 μM in DMSO starting from columns 1–5 and 7–11. Columns 6 and 12 were filled with DMSO. From each well, 5 μL was transferred into the 96-well disposable UV-Star (Greiner Bio-One). Acetonitrile (97.5 μL) and PBS buffer (97.5 μL) were added to each well, and the plate was agitated for 30 min (IKA microtiter plate shaker). The UV spectra from 200–500 nm was measured for all wells and subtracted from the background (DMSO). The correlations between concentrations and absorbance at 260, 280, and 300 nm were determined as slopes. Then, 5 μL from each well of the polypropylene plate was added to a MultiScreen Solubility Filter Plate (Millipore) and diluted with 195 μL of PBS. The plate was agitated for 2 h and filtered into a 96-well disposable UV-Star plate, and the UV absorbance at 260, 280, and 300 nm was measured. The aqueous solubility (Amax filtrate/slope) was determined for all three wavelengths, and values are given as the means with 95% confidence intervals.
PAMPA procedure
The PAMPA procedure was conducted using a published method.35, 36 Briefly, all liquid-handling steps for the PAMPA assay were performed on a Biomek FX Laboratory Automation Workstation (Beckman-Coulter) and analyzed by pION’s (London, UK) PAMPA Evolution 96 Command Software. The PAMPA Evolution 96 Permeability Assay Kit includes the Acceptor Sink Buffer (ASB), Double-Sink Lipid Solution, and a PAMPA sandwich plate, preloaded with magnetic disks. For each experiment, 4 μL of lipid was transferred onto the support membrane in the acceptor well, followed by addition of 200 μL of ASB (pH 7.4). Then, 180 μL of diluted test compound (50 μM in system buffer at pH 7.4 starting from a 10 mM DMSO solution) was added to the donor wells. The PAMPA sandwich plate was assembled and placed on the Gut-Box and stirred for 30 min. The distribution of the compounds in the donor and acceptor buffers (100 μL aliquot) was determined by measuring the UV spectra from 200 to 500 nm using the SpectraMax reader (Molecular Devices). The permeability coefficient was determined using the maximum absorbance from 200 to 500 nm using the following formula:
VD is the donor well volume (cm3); A is the filter area (cm2); CD (0) is the sample concentration in the donor well at time 0 (mole/cm3); CD (t) is the sample concentration in the donor well at time t (mole/cm3); t is the interval of time (sec); tLAG is the lag time needed to reach steady state conditions (sec); and R is the membrane retention (related to the membrane/water partition coefficient). Standards used were verapamil (Pe = 1505 × 10−6 cm/s) as a high permeability standard, carbamazepine (Pe = 150 × 10−6 cm/s) as medium permeability standard and ranitidine (Pe = 2.3 × 10−6 cm/s) as low permeability standard. The compounds were measured in triplicate, and values are given as the mean values with 95% confidence intervals.
Supplementary Material
Experimental procedures and characterization data for 3b–k, 4a–g, 5c–f, 6c–f, 6b–m, 7b–g, 8b, and 8e. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 3.

Synthesis of unsaturated ketones 4b–f.a
aReagents: (i) BuLi, THF, −78°C, 20 min; (ii) RCHO, −78°C, 2 h; (iii) tetrapropylammonium perruthenate (0.05 equiv), 4-methylmorpholine N-oxide (1.5 equiv), molecular sieves, CH3CN, rt, 1–4 h.
Acknowledgments
This work was supported by the NIH (R01 No. DK58080) and the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children’s Research Hospital (SJCRH). We thank Naoaki Fuji for suggestions and comments, David Galloway in Scientific Editing for review of the manuscript, and the Hartwell Center for assistance in the preparation of peptides.
Footnotes
Abbreviations: AF-1, activation domain; ARO, human anaplastic thyroid cell line; ATCC, American type culture collection; ATP, adenosine triphosphate; CoA, coactivator; CoR, coregulator; DBD, DNA-binding domain; DMSO, dimethyl sulfoxide; LBD, ligand-binding domain; LCMS, liquid chromatography-mass spectrometry; MOE, molecular operating environment software; NCoR, nuclear receptor corepressor; NID, nuclear receptor interacting domain; NR, nuclear hormone receptor; PAMPA, parallel artificial membrane permeation assay; PBS, phosphate buffered saline; PDA: photodiode array; RXR, retinoid X receptor; SAR, structure-activity relationship; SMRT, silencing mediator of retinoic acid; SRC, steroid receptor coactivator; SRC2-2, steroid receptor coactivator 2 peptide representing the second NID; TR, thyroid receptor; U2OS, human osteosarcoma epithelial cell line.
References
- 1.Yen PM. Physiological and molecular basis of thyroid hormone action. Physiol Rev. 2001;81(3):1097–1142. doi: 10.1152/physrev.2001.81.3.1097. [DOI] [PubMed] [Google Scholar]
- 2.Malm J. Thyroid hormone ligands and metabolic diseases. Curr Pharm Des. 2004;10(28):3525–3532. doi: 10.2174/1381612043382873. [DOI] [PubMed] [Google Scholar]
- 3.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81(3):1269–1304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 4.Harvey CB, Williams GR. Mechanism of thyroid hormone action. Thyroid. 2002;12(6):441–446. doi: 10.1089/105072502760143791. [DOI] [PubMed] [Google Scholar]
- 5.Williams GR. Cloning and characterization of two novel thyroid hormone receptor beta isoforms. Mol Cell Biol. 2000;20(22):8329–8342. doi: 10.1128/mcb.20.22.8329-8342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The Nuclear Receptor Superfamily - the 2Nd Decade. Cell. 1995;83(6):835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu JM, Li QT. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol. 2003;17(9):1681–1692. doi: 10.1210/me.2003-0116. [DOI] [PubMed] [Google Scholar]
- 8.Hu X, Lazar MA. Transcriptional repression by nuclear hormone receptors. Trends Endorinol Metab. 2000;11(1):6–10. doi: 10.1016/s1043-2760(99)00215-5. [DOI] [PubMed] [Google Scholar]
- 9.Onate SA, Tsai SY, Tsai MJ, Omalley BW. Sequence and Characterization of a Coactivator for the Steroid-Hormone Receptor Superfamily. Science. 1995;270(5240):1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 10.Voegel JJ, Heine MJS, Zechel C, Chambon P, Gronemeyer H. TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J. 1996;15(14):3667–3675. [PMC free article] [PubMed] [Google Scholar]
- 11.Hong H, Kohli K, Garabedian MJ, Stallcup MR. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol Cell Biol. 1997;17(5):2735–2744. doi: 10.1128/mcb.17.5.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suen CS, Berrodin TJ, Mastroeni R, Cheskis BJ, Lyttle CR, Frail DE. A transcriptional coactivator, steroid receptor coactivator-3, selectively augments steroid receptor transcriptional activity. J Biol Chem. 1998;273(42):27645–27653. doi: 10.1074/jbc.273.42.27645. [DOI] [PubMed] [Google Scholar]
- 13.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes & Dev. 1998;12(21):3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng WJ, Ribeiro RCJ, Wagner RL, Nguyen H, Apriletti JW, Fletterick RJ, Baxter JD, Kushner PJ, West BL. Hormone-dependent coactivator binding to a hydrophobic cleft on nuclear receptors. Science. 1998;280(5370):1747–1749. doi: 10.1126/science.280.5370.1747. [DOI] [PubMed] [Google Scholar]
- 15.Moore JMR, Galicia SJ, McReynolds AC, Nguyen NH, Scanlan TS, Guy RK. Quantitative proteomics of the thyroid hormone receptor-coregulator interactions. J Biol Chem. 2004;279(26):27584–27590. doi: 10.1074/jbc.M403453200. [DOI] [PubMed] [Google Scholar]
- 16.Moore JMR, Guy RK. Coregulator interactions with the thyroid hormone receptor. Mol Cell Proteomics. 2005;4(4):475–482. doi: 10.1074/mcp.R500001-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Geistlinger TR, Guy RK. An inhibitor of the interaction of thyroid hormone receptor beta and glucocorticoid interacting protein 1. J Am Chem Soc. 2001;123(7):1525–1526. doi: 10.1021/ja005549c. [DOI] [PubMed] [Google Scholar]
- 18.Geistlinger TR, Guy RK. Novel selective inhibitors of the interaction of individual nuclear hormone receptors with a mutually shared steroid receptor coactivator 2. J Am Chem Soc. 2003;125(23):6852–6853. doi: 10.1021/ja0348391. [DOI] [PubMed] [Google Scholar]
- 19.Arnold LA, Estébanez-Perpiñá E, Togashi M, Jouravel N, Shelat A, McReynolds AC, Mar E, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J Biol Chem. 2005;280(52):43048–55. doi: 10.1074/jbc.M506693200. [DOI] [PubMed] [Google Scholar]
- 20.Arend M, Westermann B, Risch N. Modern variants of the Mannich reaction. Angew Chem Int Ed. 1998;37(8):1045–1070. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 21.Karpati E, Domok L, Szporny L. 1-piperidino-3-(4′-octylphenyl)-propane-3-one-hydrochloride, a new peripheral and coronary vasodilator. Arzneimittelforschung. 1969;19(6):1011–5. [PubMed] [Google Scholar]
- 22.Kudrin AN, Aslaniants ZK, Polevoi LG. Combined administration of quinidine with the beta-adrenolytic agent inderal and the local anaesthetic of the beta-aminoketone group, hexacaine, in various experimental models of arrhythmias. Cor Vasa. 1975;17(2):133–44. [PubMed] [Google Scholar]
- 23.Profft E. The production of new, highly effective anesthetics with great latitude of efficacy. Chem Tech (Leibzig) 1951;3:210–13. [Google Scholar]
- 24.Shelmire B, Gastineau FM, Shields TL. Evaluation of a new topical anesthetic, dyclonine hydrochloride. AMA Arch Derm. 1955;71(6):728–30. doi: 10.1001/archderm.1955.01540300050013. [DOI] [PubMed] [Google Scholar]
- 25.Armstrong CM, Hille B. Voltage-gated ion channels and electrical excitability. Neuron. 1998;20(3):371–80. doi: 10.1016/s0896-6273(00)80981-2. [DOI] [PubMed] [Google Scholar]
- 26.Gul M, Gul HI, Das U, Hanninen O. Biological evaluation and structure-activity relationships of bis-(3-aryl-3-oxo-propyl)-methylamine hydrochlorides and 4-aryl-3-arylcarbonyl-1-methyl-4-piperidinol hydrochlorides as potential cytotoxic agents and their alkylating ability towards cellular glutathione in human leukemic T cells. Arzneimittelforschung. 2005;55(6):332–7. doi: 10.1055/s-0031-1296868. [DOI] [PubMed] [Google Scholar]
- 27.Davioud-Charvet E, McLeish MJ, Veine DM, Giegel D, Arscott LD, Andricopulo AD, Becker K, Muller S, Schirmer RH, Williams CH, Kenyon GL. Mechanism-based inactivation of thioredoxin reductase from Plasmodium falciparum by Mannich bases. Implication for cytotoxicity. Biochemistry. 2003;42(45):13319–13330. doi: 10.1021/bi0353629. [DOI] [PubMed] [Google Scholar]
- 28.Ward RS, Davies J, Hodges G, Roberts DW. Synthesis of quaternary alkylammonium sulfobetaines. Synthesis-Stuttgart. 2002;(16):2431–2439. [Google Scholar]
- 29.Arnold LA, Estébanez-Perpiñá E, Togashi M, Shelat A, Ocasio CA, McReynolds AC, Nguyen P, Baxter JD, Fletterick RJ, Webb P, Guy RK. A high-throughput screening method to identify small molecule inhibitors of thyroid hormone receptor coactivator binding. Sci STKE. 2006;2006(341):pl3. doi: 10.1126/stke.3412006pl3. [DOI] [PubMed] [Google Scholar]
- 30.Chen ST, Shieh HY, Lin JD, Chang KS, Lin KH. Overexpression of thyroid hormone receptor beta1 is associated with thyrotropin receptor gene expression and proliferation in a human thyroid carcinoma cell line. J Endocrinol. 2000;165(2):379–89. doi: 10.1677/joe.0.1650379. [DOI] [PubMed] [Google Scholar]
- 31.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–99. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ley SV, Norman J, Griffith WP, Marsden SP. Tetrapropylammonium Perruthenate, Pr4N+Ruo4-, Tpap - a Catalytic Oxidant for Organic-Synthesis. Synthesis-Stuttgart. 1994;(7):639–666. [Google Scholar]
- 33.Truce WE. The Preparation of Fluoroacetyl Chloride. Journal of the American Chemical Society. 1948;70(8):2828–2828. [Google Scholar]
- 34.Molecular Operating Environment (MOE) 2003.02, C. C. G. Quebec, Canada (2003).
- 35.Ruell JA, Tsinman KL, Avdeef A. PAMPA - a drug absorption in vitro model 5. Unstirred water layer in iso-pH mapping assays and pK(a)(flux) - optimized design (pOD-PAMPA) European Journal of Pharmaceutical Sciences. 2003;20(4–5):393–402. doi: 10.1016/j.ejps.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 36.Yu S, Konstantin T, Zhu Z, Graham T. Automation of a Double-Sink PAMPA Permeability Assay on the Biomek® FX Laboratory Automation Workstation. Pharmaceutical Discovery. 2005 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for 3b–k, 4a–g, 5c–f, 6c–f, 6b–m, 7b–g, 8b, and 8e. This material is available free of charge via the Internet at http://pubs.acs.org.