

Abstract
Background
Osteopontin (OPN)-transgenic mice exhibit increased carotid artery intima-media thickness (CIMT), smooth muscle cell proliferation, and atheroma formation.
Methods
An association of the human T-66G promoter variant with CIMT was examined in Caucasian adults grouped according to metabolic syndrome (MetS) criteria: Present (+MetS, n=70) and Absent (−MetS, n=70).
Results
The G allele frequency was 22%. For the entire cohort, the G group (TG and GG) was associated with significantly lower age/gender-adjusted CIMT compared to the TT group (p=0.008); similar analysis by MetS group found a significant difference only in the −MetS group (p=0.018). Stepwise multivariable regression showed that after age and waist circumference, the T-66G variant was predictive of CIMT (p=0.007). These data suggest that in a normoglycemic environment, human vascular OPN gene expression contributes to arterial structure, an effect diminished in dysmetabolic states.
Conclusions
Humans with the OPN −66 TT genotype, particularly those without MetS, exhibit thicker CIMT.
Introduction
Osteopontin (OPN) is a phosphorylated sialoprotein secreted by osteoblasts, vascular smooth muscle cells (VSMC), activated T-cells, and cells of the monocyte/macrophage lineage.1,2 First identified as Arg-Gly-Asp (RGD)-dependent adhesion molecule in bone, OPN has emerged as an important paracrine regulator of not only cell migration, but cellular proliferation, T Helper 1 cell cytokine production, extracellular matrix calcium deposition, gelatinase-dependent matrix remodeling, neovascularization, and neointima formation.1,3-7 Elegant murine models have demonstrated that OPN controls aortic VSMC proliferation and extracellular matrix remodeling.7 Transgenic mice overexpressing OPN exhibit increased intima media thickness, medial smooth muscle cell proliferation, aortic matrix metalloproteinase 2 (MMP2) and 9 (MMP9) activation, and extensive atheroma formation. Conversely, mice deficient in OPN are resistant to angiotensin II-induced aortic aneurysm formation.8 Recently, OPN deficiency was shown to reduce diet-induced aortic superoxide formation, vascular inflammation, and pro-MMP9 activation in a murine model of type II diabetes.5 Known metabolic regulators of vascular structure and function, such as glucose and oxysterols, control OPN expression.9-11 Indeed, these specific signals control OPN expression in VSMCs and macrophages via a critical CCTCATGAC motif located between −72 and −80 relative to the transcription start site.9 Upstream stimulatory factor (USF1), activator protein-1 (AP1; c-Fos:c-Jun), and liver X receptor (LXR) multi-component transcription factor complexes assemble at this proximal promoter region.9,11 Thus, mouse OPN gene and murine disease models have robustly established the significance of vascular OPN expression in arterial physiology and disease pathobiology.5,7,8,12-14
By comparison, relatively little is known concerning the role of OPN in human vascular structure and remodeling. Seminal studies have established that OPN protein and mRNA expression are upregulated in rodent arterial neointima and in human atherosclerotic plaques;3 others extended on these studies, demonstrating expression of OPN by human aortic valve myofibroblasts in calcific aortic stenosis.15-18 OPN is expressed by monocytes, macrophages, and T-cells; indeed, OPN was previously called early T-cell antigen 1 (Eta1), reflecting its and play a role as a pro-inflammatory T-cell cytokine.1 Thus, the paracrine actions of mural OPN in vascular disease likely reflect contributions from cells of vascular smooth muscle and immunocyte lineages.1,5,8,14,19
Population studies have found that metabolic syndrome is strongly associated with increased carotid artery intima-media thickness (CIMT), which is a robust intermediate phenotype of early atherosclerosis.20-27 However, the interplay between genetic and metabolic risk factors of cardiovascular disease has not been well characterized. To assess whether OPN expression influences human arterial structure and function, we examined the relationship between the OPN proximal promoter T-66G variant genotype and CIMT in a cohort with and without metabolic syndrome. This specific single nucleotide polymorphism (SNP) was emphasized because it conveys up to 4-fold differences in human OPN gene transcription in human epithelial and fibroblast cell lines and alters OPN DNA-protein interactions;28 basal OPN promoter activity is reduced when the less common −66G allele (which has a minor allele frequency ∼25%) is compared to the activity of the more prevalent −66T allele.28 This occurs because Sp1-dependent protein-DNA interactions that support basal OPN gene expression are reduced by the −66G variant. Moreover, this SNP is immediately adjacent to the phylogenetically conserved element that entrains OPN transcription to mechanical and metabolic signals that perturb vascular structure.9,11,29 Thus, the hypothesis of this study was that this common −66T OPN promoter variant is associated with increased CIMT in humans and that the metabolic environment would modulate this association.
Methods
Study population
The study population consisted of 140 Caucasian adults, 70 with metabolic syndrome (+MetS) and 70 without metabolic syndrome (−MetS), who volunteered for a cardiovascular genetics study. All study subjects underwent a complete cardiovascular evaluation after a minimum 8-hour fast, including: 1) history and physical examination; 2) heart rate and systolic and diastolic blood pressures; 3) fasting serum glucose and insulin levels; 4) fasting plasma lipids (i.e., triglyceride, high-density lipoprotein cholesterol [HDL-C], total cholesterol, and low-density lipoprotein cholesterol [LDL-C] concentrations); and 5) carotid artery ultrasound for measurement of CIMT. This study has been reviewed by the Human Research Protection Office at Washington University; informed consent was obtained prior to study enrollment. Participants were excluded for the following: 1) pregnancy or 2) incomplete characterization of metabolic syndrome criteria.
Assessment of Metabolic Syndrome and Cardiovascular Risk Factors
MetS was diagnosed according to the amended National Cholesterol Education Program's Adult Treatment Panel III guidelines.1,30 Type 2 diabetes mellitus was defined as a fasting serum glucose level ≥ 126 mg/dL or current medical therapy with an oral hypoglycemic agent and/or insulin. Serum glucose levels, insulin levels, and plasma lipid levels were obtained after a minimum 8-hour fast; glucose and insulin levels were collected only in subjects not receiving insulin and/or oral hypoglycemic agents. The Homeostasis Model Assessment of Insulin Resistance (HOMA-IR) was calculated as a marker of insulin resistance.31 LDL-C was calculated according to Friedewald's equation when triglycerides was ≤500 mg/dL, otherwise it was directly measured by ultracentrifugation.32 Hypertension was defined as a blood pressure of ≥ 140/90 mm Hg and/or current medical therapy with an antihypertensive medication.
Carotid artery ultrasound
Carotid ultrasound was performed using a 7-MHz linear array transducer; a single vascular sonographer acquired B-mode images of both carotid arteries in the longitudinal axis. A 1-cm region of the common carotid artery just proximal to the bifurcation was identified and enlarged to highest resolution prior to export for offline digital analysis with Prosolv software (Problem Solving Concepts, Indianapolis, IN). CIMT measures were expressed as the average of the far-wall intima media thickness from both right and left common carotid arteries, excluding raised lesions and plaques when present.33 Each site represents the average of three separate measurements, obtained by a single observer blinded to clinical parameters. In our laboratory, the intraobserver intraclass correlation coefficient for repeated measures of the CIMT is 0.91.
Extraction of deoxyribonucleic acid and OPN genotyping
Genomic DNA was extracted from peripheral blood leukocytes of unrelated Caucasian individuals using standard procedures. The OPN −66 variant (Human Gene Ontology name: SPP1, rs28357094) was genotyped by direct sequencing of the sense and antisense strands following polymerase chain reaction amplification of the promoter regulatory region −469 to +155; (forward primer: tgtcactagtgccattg; reverse primer: caaacgccgaccaaggtaca), as previously described.9,34
Biomarker Assays
Plasma OPN and total plasma MMP9, and serum high-sensitivity assay C-reactive protein (hsCRP) were measured in an unselected subpopulation (n=26 −MetS and n=46 +MetS), since an OPN–MMP9 signaling axis has been implicated in CIMT in murine models of arterial remodeling.7,8 Plasma OPN levels were measured using the commercially available ELISA from Assay Designs (#900−142). Total plasma MMP9 was measured using the Amersham Biotrak Assay (Amersham/GE Healthcare) following methods outlined in the manufacturer's product booklet (RPN2614), with p-aminophenylmercuric acetate treatment to activate pro-MMP9 complexes. High sensitivity CRP assays were performed using a commercial ELISA kit (Life Diagnostics, Cat. #2210), following the methods specified by the manufacturer.
Statistical analysis
Statistics were performed using SAS software (Version 9.1, SAS Institute, Cary, NC). The study cohort was grouped according to the presence or absence of metabolic syndrome (+MetS and −MetS). Each group was further grouped according to a dominant model for the −66 genotype; individuals with G alleles (TG and GG genotypes) were combined for analysis. Values of continuous data were presented as the mean ± standard deviation. Chi-square (χ2) or Fisher's Exact statistics were used to determine differences in proportions. Groups were compared via Analysis of Covariance (ANCOVA) with age- and gender-adjustment and Tukey-Kramer post-hoc analysis. Conformation to Hardy Weinberg proportions was tested by the Chi-square (χ2) Goodness of Fit test. To determine the predictive nature of the OPN gene variant for CIMT in the context of MetS criteria, a stepwise multivariable regression model was tested in the whole cohort, and then separately in the −MetS and +MetS groups. Model variables included the following: individual metabolic syndrome criteria expressed as continuous variables (i.e, waist circumference, triglyceride, HDL-C, systolic and diastolic blood pressures, and fasting glucose), metabolic syndrome as a dichotomous variable, and the OPN T-66G genotype group. Variables not normally distributed were log-transformed for analysis. All statistical tests were two-sided. Variable entry into the stepwise regression models required a p value < 0.1; a p value ≤ 0.05 was considered statistically significant.
Results
Clinical characteristics
Based on the grouping method, anticipated differences were noted in the demographic and clinical characteristics between the MetS groups. As expected, the +MetS group was significantly older and had significantly higher body mass index, waist circumference, systolic and diastolic blood pressure, triglyceride, glucose, and insulin levels compared with the −MetS group; HDL-C and LDL-C levels were significantly lower (the later likely a reflections of statin use in the +MetS group). Hypertension, coronary artery disease, type 2 diabetes mellitus, prior smoking history, and statin drug therapy were all more prevalent among the +MetS group.
OPN genotype distribution and biomarker assays
The distribution of OPN −66 genotypes was consistent with Hardy Weinberg proportions. The G allele frequency in the entire cohort was 22% and was similar between MetS groups. Within each MetS group, the subject demographics and characteristics of the genotype groups (i.e., TT versus TG and GG genotypes) were similar except for increased prevalence of type 2 diabetes mellitus (p=.05) and lower LDL-C (p=.01) in the TG and GG +MetS group (Table 1). MMP9 and hsCRP levels were significantly higher in the +MetS group compared to the −MetS group; serum OPN levels were similar between MetS groups. There were no significant differences in the levels of OPN, MMP9, or hsCRP between the genotype groups (Table 2).
Table 1.
Demographics and clinical characteristics of the study population by metabolic syndrome criteria and genotype
|
-MetS |
+MetS |
|||
|---|---|---|---|---|
|
TT (n = 41) |
TG & GG (n = 29) |
TT (n = 45) |
TG & GG (n = 25) |
|
| Age, years | 46±13 | 47±12 | 54±12 | 52±11 |
| Female, n (%) | 24 (59%) | 15 (52%) | 18 (40%) | 16 (64%) |
| Body mass index, kg/m2 | 25±3 | 24±3 | 35±6 | 34±4 |
| Waist circumference, cm | 80±10 | 81±12 | 107±16 | 103±11 |
| Systolic BP, mmHg | 110±9 | 112±11 | 131±16 | 127±17 |
| Diastolic BP, mmHg | 72±7 | 74±8 | 83±8 | 81±9 |
| Triglyceride, mg/dL | 78±24 | 85±29 | 226±102 | 183±69 |
| HDL cholesterol, mg/dL | 60±12 | 64±19 | 41±10 | 41±8 |
| LDL cholesterol, mg/dL | 120±31 | 120±27 | 110±33 | 103±27 |
| Total cholesterol, mg/dL | 196±33 | 201±32 | 195±34 | 181±30* |
| Past medical history and medical therapy, n (%) | ||||
| Hypertension | — | 2 (7) | 30 (67) | 20 (80) |
| Coronary artery disease | — | — | 2 (4) | 3 (12) |
| Type 2 diabetes mellitus | — | 1 (3) | 16 (36) | 15 (60)* |
| Current smoker | 4 (14) | 3 (13) | 4 (16) | 3 (20) |
| Prior smoker | 12 (32) | 6 (23) | 20 (49) | 10 (45) |
| Statin | 1 (2) | 1 (3) | 12 (27) | 11 (44) |
| † | n = 41 | n = 29 | n = 35 | n = 13 |
|---|---|---|---|---|
| Insulin, mU/mL | 5.7±2.7 | 5.2±2.4 | 17.9±13.3 | 15.0±10.2 |
| Glucose, mg/dL | 83±11 | 86±12 | 100±19 | 105±37 |
| HOMA-IR | 1.1±0.7 | 1.1±0.7 | 4.5±3.5 | 3.9±2.7 |
Data are expressed as mean ± standard deviation or number (percentage) of subjects; p values reflect significance after age- and gender-adjustment within each group.
p=.05 vs. TT group.
Insulin, glucose, and HOMA-IR are in fasting subjects, excluding diabetics receiving insulin or oral hypoglycemic agents.
BP, blood pressure; HDL, high-density lipoprotein; HOMA-IR, homeostasis model assessment of insulin resistance; LDL, low-density lipoprotein; MetS, metabolic syndrome.
Table 2.
Plasma OPN and MMP9, and serum hsCRP by metabolic syndrome criteria and by genotype
|
-MetS (n = 26) |
+MetS (n = 46) |
|||
|---|---|---|---|---|
| OPN, ug/L | 63±40 | 70±42 | ||
| MMP9, ug/L | 6.2±3.0 | 7.9±3.2† | ||
| hsCRP, mg/L | 2.5±4.2 | 8.9±10.0* | ||
|
TT (n = 15) |
TG & GG (n=11) |
TT (n = 28) |
TG & GG (n = 18) |
|
|---|---|---|---|---|
| OPN, ug/L | 58±26 | 69±54 | 68±38 | 74±49 |
| MMP9, ug/L | 5.9±2.6 | 6.5±3.6 | 7.8±3.5 | 8.2±2.7 |
| hsCRP, mg/L | 3.1±4.9 | 1.7±2.9 | 7.8±8.6 | 10.6±12.1 |
Data are expressed as mean±standard deviation. p values reflect significance after age- and gender-adjustment.
p ≤ 0.0001
p < 0.005 vs. -MetS.
hsCRP, high-sensitivity C-reactive protein; MMP9, matrix metalloproteinase 9; OPN, Osteopontin.
OPN T-66G promoter SNP and CIMT
Based on the distribution of the CIMT measurements in the combined cohort, the present study has an expected power of >95% to detect a modest effect (heritability estimates ≥10%) on CIMT assuming a dominant model. After age- and gender-adjustment, the −66G OPN promoter allele was associated with significantly decreased CIMT in the entire cohort, as well as in the −MetS group; however, similar differences were not noted in the +MetS group (Figure 1). Stepwise multivariable regression analysis in the entire cohort showed that after age and waist circumference, the T-66G variant was the next most predictive variable of CIMT (model r2=0.36, p=0.007 for −66 T>G). Although the OPN variant did not contribute significantly to the same model when applied to the +MetS group, it was an independent predictor in the −MetS group (after age and HDL-C, model r2=0.038, p=0.05 for T-66G).
Figure 1. OPN T-66G genotype contributes to CIMT phenotype.
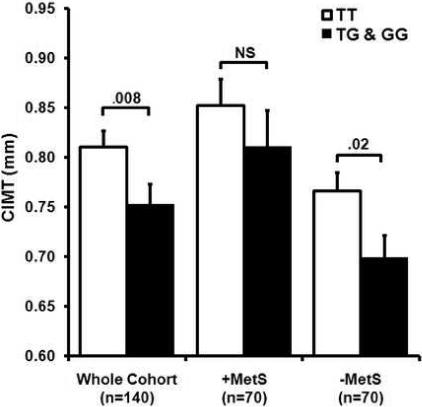
For the entire cohort, age and gender-adjusted values of CIMT were significantly higher for those with the TT genotype compared to the combined TG and GG genotypes. For the metabolic syndrome group (+MetS), there were no significant differences in the adjusted CIMT measurements between the TT and the TG and GG groups. For those without metabolic syndrome (−MetS), adjusted CIMT measurements were significantly higher for the TT genotype. NS = not significant.
Discussion
The arterial vasculature is a dynamic structure, constantly changing in response to morphogenetic, metabolic, mechanical, inflammatory, and endocrine demands. With age, enhanced vascular matrix remodeling increases arterial intima-media thickness and promotes vascular calcium accumulation, which are important structural predictors of increased cardiovascular morbidity and mortality.25,35 As sophisticated as current working models are for macrovascular pathobiology, relatively little is known regarding the interaction between common genetic variants and the metabolic milieu in modifying vascular structure and/or function. In the metabolic syndrome, abdominal obesity, atherogenic dyslipidemia (increased triglycerides, decreased HDL-C), elevated blood pressure, insulin resistance (and/or glucose intolerance) are key determinants of vascular remodeling.36-40 Work from our group and others has identified in mice that OPN is a glucose-inducible vascular matrix cytokine that is: a) upregulated in diabetes via a well-defined bipartite USF/AP1 element;9,19 b) required in vivo for the metabolic and endocrine stressors that promote arterial MMP9 activation;5,8 and c) involved in the regulation of murine arterial intima-media thickness.7 In the present study, we show in humans an association between the common OPN gene promoter T-66G variant and CIMT. Stepwise multivariable regression showed that the OPN promoter polymorphism was the third most predictive variable in the model, which along with age and waist circumference, accounted for 40% of the variance. Thus, via molecular genetics and genotype-phenotype characterization, our data is the first to establish the important role for the −66G OPN allele that conveys lower OPN promoter activity in modulating human arterial macrovascular structure and remodeling using a robust non-invasive imaging technique.28
Recently, Brenner reported increased CIMT in stroke patients carrying the C allele of the OPN C-443T SNP.41 Although this variant is very common (allele frequency approaches 50%), unlike the T-66G variant, the C-443T promoter polymorphism has no known functional consequence in transcriptional regulation.28 However, due to its general proximity to the OPN T-66G allele, it is likely that the −443T allele is in significant linkage disequilibrium with the less common, low-expressing −66G allele, and that it is the later that predicts reduced CIMT.28 Given the functional impact of the T-66G genotype on gene transcription, it is reasonable to speculate that the T-66G OPN promoter genotype identifies individuals at greater risk for developing macrovascular disease, and thus, may be used prospectively to identify those who could benefit the most from aggressive therapeutic intervention. Since statins are potent inhibitors of OPN expression by VSMCs and aortic valve myofibroblasts,15,42 future studies could be designed to ascertain potential benefits of statins (in patients without dyslipidemia) based on OPN genotypes. If the OPN −66 TT genotype, which is associated with greater promoter activity and increased CIMT, were to also identify greater risk for cerebrovascular disease, then stroke and vascular dementia risk in these individuals might be ameliorated by aggressive implementation of therapies to limit OPN-dependent increased CIMT. Conversely, those with the favorable OPN −66 G allele may accrue lesser clinical benefit at the same cost for intervention. Given the substantial impact of stroke, unmet needs clearly exist for improving prevention and cost-benefit outcomes.43,44
A significant correlation between circulating OPN levels and the OPN T-66G genotype was not established in this study. Similar results have just been reported by Golledge and colleagues; while they identify that circulating OPN levels are associated with abdominal aortic aneurysm, no connection could be made between OPN genotype and circulating OPN levels or aneurysm risk.45 Circulating OPN is entrained to anabolic stimuli that enhance extracellular matrix turnover and bone formation46; indeed, pulsatile parathyroid hormone treatment promotes bone formation and enhances circulating OPN levels, and concomitantly suppresses aortic OPN expression.46 In the vasculature, OPN plays an important paracrine and matricrine role necessary for vascular remodeling.8,47 Thompson and colleagues have shown that OPN is required for VSMC-dependent recruitment of adventitial fibroblasts into the tunica media during mural injury.48 Elegant studies have identified that a paracrine pyrophosphate-OPN axis inhibits VSMC mineralized matrix deposition.6,49 In an ectopic porcine aortic valve calcification model, Giachelli and co-workers demonstrated that acidification required for calcium egress uses matricrine OPN signals.12 Thus, circulating intact OPN levels do not reflect the paracrine levels of production and exposure experienced by parenchymal cells of the arterial vasculature.8,10,15,16,48,50
Limitations of the study
OPN controls intima-media thickness in preclinical models, upregulated in VSMCs by the very glycemic and mechanical stresses that are critical determinants of CIMT in human.9,29,51 However, the precise levels of carotid VSMC OPN accumulation and MMP activation in the present cohort is unknown. These data would be highly desirable, since our working model postulates that the dysmetabolic state of the metabolic syndrome (MetS) “overrides” the basal, genetically-inherited influence of the OPN T-66G genotype to drive CIMT progression (see Figure 2). The present translational study provides the first direct evidence that OPN T-66G genotype is a significant determinant in Caucasian patients of arterial vascular structure by CIMT, a robust intermediate phenotype of early atherosclerosis that portends increased risk of stroke, and overall cardiovascular mortality. Heritability studies have shown that genetic determinants of IMT may account for 35−66% of the phenotypic variance.52-55 Thus, future studies will evaluate the potential use of this important genotype to “predict, preempt, and personalize” interventions to improve cardiovascular outcomes, including stroke.
Figure 2. Working model for OPN genotype-carotid structure phenotype relationships.

Under basal conditions, vascular OPN expression is determined by the T-66G genotype which has been shown to determine levels of OPN transcription via promoter-Sp1 transcription factor DNA-protein interactions. In the presence of diabetes and/or hypertension, characteristic features of the metabolic syndrome (MetS) that control proximal OPN promoter activity in vascular smooth muscle cells via adjacent upstream stimulatory factor (USF1)/activator protein-1 (AP1) elements, the dysmetabolic milieu drives vascular OPN expression that enhances CIMT.
In conclusion, this study found an association between the T-66G OPN variant and CIMT in humans, consistent with increased arterial OPN expression and signaling and in excellent agreement with genetically altered murine models. The common human OPN promoter −66G allele, which conveys lower promoter activity, was found to be protective in Caucasian patients against increased CIMT, a marker of atherosclerosis. This indicates that, as in mice, the OPN gene contributes to arterial macrovascular structure in humans.
Acknowledgements
The authors would like to acknowledge Melissa Allen, and Arleen P. Loewy, and Sharon L. Heuerman, RN for their contributions to the study.
Funding
Supported in part by grants National Institutes of Health (K12RR023249 to L.d.l.F, HL54473 to C.C.G., R01HL69229 and R01HL81138 to D.A.T., R01HL71782, R01HL58878, K24HL67002, and P50Hl83762 to V.G.D.-R., and M01RR00036 [General Clinical Research Center] to Washington University), the Robert Wood Johnson Foundation (#048875 to L.d.l.F), and the a grant from the Barnes-Jewish Hospital Foundation to the Cardiovascular Imaging and Clinical Research Core Laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest None declared.
References
- 1.Denhardt DT, Noda M, O'Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107:1055–61. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vattikuti R, Towler DA. Osteogenic regulation of vascular calcification: an early perspective. Am J Physiol Endocrinol Metab. 2004;286:E686–96. doi: 10.1152/ajpendo.00552.2003. [DOI] [PubMed] [Google Scholar]
- 3.Giachelli CM, Bae N, Almeida M, Denhardt DT, Alpers CE, Schwartz SM. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J Clin Invest. 1993;92:1686–96. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giachelli CM, Steitz S. Osteopontin: a versatile regulator of inflammation and biomineralization. Matrix Biol. 2000;19:615–22. doi: 10.1016/s0945-053x(00)00108-6. [DOI] [PubMed] [Google Scholar]
- 5.Lai CF, Seshadri V, Huang K, Shao JS, Cai J, Vattikuti R, et al. An osteopontin-NADPH oxidase signaling cascade promotes pro-matrix metalloproteinase 9 activation in aortic mesenchymal cells. Circ Res. 2006;98:1479–89. doi: 10.1161/01.RES.0000227550.00426.60. [DOI] [PubMed] [Google Scholar]
- 6.Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millan JL. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164:1199–209. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Isoda K, Nishikawa K, Kamezawa Y, Yoshida M, Kusuhara M, Moroi M, et al. Osteopontin plays an important role in the development of medial thickening and neointimal formation. Circ Res. 2002;91:77–82. doi: 10.1161/01.res.0000025268.10302.0c. [DOI] [PubMed] [Google Scholar]
- 8.Bruemmer D, Collins AR, Noh G, Wang W, Territo M, Arias-Magallona S, et al. Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112:1318–31. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bidder M, Shao JS, Charlton-Kachigian N, Loewy AP, Semenkovich CF, Towler DA. Osteopontin transcription in aortic vascular smooth muscle cells is controlled by glucose-regulated upstream stimulatory factor and activator protein-1 activities. Journal of Biological Chemistry. 2002;277:44485–96. doi: 10.1074/jbc.M206235200. [DOI] [PubMed] [Google Scholar]
- 10.Takemoto M, Yokote K, Nishimura M, Shigematsu T, Hasegawa T, Kon S, et al. Enhanced expression of osteopontin in human diabetic artery and analysis of its functional role in accelerated atherogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:624–8. doi: 10.1161/01.atv.20.3.624. [DOI] [PubMed] [Google Scholar]
- 11.Ogawa D, Stone JF, Takata Y, Blaschke F, Chu VH, Towler DA, et al. Liver x receptor agonists inhibit cytokine-induced osteopontin expression in macrophages through interference with activator protein-1 signaling pathways. Circ Res. 2005;96:e59–67. doi: 10.1161/01.RES.0000163630.86796.17. [DOI] [PubMed] [Google Scholar]
- 12.Steitz SA, Speer MY, McKee MD, Liaw L, Almeida M, Yang H, et al. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am J Pathol. 2002;161:2035–46. doi: 10.1016/S0002-9440(10)64482-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Speer MY, McKee MD, Guldberg RE, Liaw L, Yang HY, Tung E, et al. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J Exp Med. 2002;196:1047–55. doi: 10.1084/jem.20020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rattazzi M, Bennett BJ, Bea F, Kirk EA, Ricks JL, Speer M, et al. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arterioscler Thromb Vasc Biol. 2005;25:1420–5. doi: 10.1161/01.ATV.0000166600.58468.1b. [DOI] [PubMed] [Google Scholar]
- 15.Rajamannan NM, Subramaniam M, Springett M, Sebo TC, Niekrasz M, McConnell JP, et al. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation. 2002;105:2660–5. doi: 10.1161/01.cir.0000017435.87463.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–4. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohler ER, 3rd, Adam LP, McClelland P, Graham L, Hathaway DR. Detection of osteopontin in calcified human aortic valves. Arterioscler Thromb Vasc Biol. 1997;17:547–52. doi: 10.1161/01.atv.17.3.547. [DOI] [PubMed] [Google Scholar]
- 18.O'Brien KD, Kuusisto J, Reichenbach DD, Ferguson M, Giachelli C, Alpers CE, et al. Osteopontin is expressed in human aortic valvular lesions. Circulation. 1995;92:2163–8. doi: 10.1161/01.cir.92.8.2163. [DOI] [PubMed] [Google Scholar]
- 19.Towler DA, Bidder M, Latifi T, Coleman T, Semenkovich CF. Diet-induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. Journal of Biological Chemistry. 1998;273:30427–34. doi: 10.1074/jbc.273.46.30427. [DOI] [PubMed] [Google Scholar]
- 20.McNeill AM, Rosamond WD, Girman CJ, Heiss G, Golden SH, Duncan BB, et al. Prevalence of coronary heart disease and carotid arterial thickening in patients with the metabolic syndrome (The ARIC Study). Am J Cardiol. 2004;94:1249–54. doi: 10.1016/j.amjcard.2004.07.107. [DOI] [PubMed] [Google Scholar]
- 21.Tzou WS, Douglas PS, Srinivasan SR, Bond MG, Tang R, Chen W, et al. Increased subclinical atherosclerosis in young adults with metabolic syndrome: the Bogalusa Heart Study. J Am Coll Cardiol. 2005;46:457–63. doi: 10.1016/j.jacc.2005.04.046. [DOI] [PubMed] [Google Scholar]
- 22.O'Leary DH, Polak JF, Kronmal RA, Manolio TA, Burke GL, Wolfson SK., Jr. Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N Engl J Med. 1999;340:14–22. doi: 10.1056/NEJM199901073400103. [DOI] [PubMed] [Google Scholar]
- 23.Burke GL, Evans GW, Riley WA, Sharrett AR, Howard G, Barnes RW, et al. Arterial wall thickness is associated with prevalent cardiovascular disease in middle-aged adults. The Atherosclerosis Risk in Communities (ARIC) Study. Stroke. 1995;26:386–91. doi: 10.1161/01.str.26.3.386. [DOI] [PubMed] [Google Scholar]
- 24.Salonen JT, Salonen R. Ultrasonographically assessed carotid morphology and the risk of coronary heart disease. Arterioscler Thromb. 1991;11:1245–9. doi: 10.1161/01.atv.11.5.1245. [DOI] [PubMed] [Google Scholar]
- 25.Bots ML, Hoes AW, Koudstaal PJ, Hofman A, Grobbee DE. Common carotid intima-media thickness and risk of stroke and myocardial infarction: the Rotterdam Study. Circulation. 1997;96:1432–7. doi: 10.1161/01.cir.96.5.1432. [DOI] [PubMed] [Google Scholar]
- 26.Chambless LE, Heiss G, Folsom AR, Rosamond W, Szklo M, Sharrett AR, et al. Association of coronary heart disease incidence with carotid arterial wall thickness and major risk factors: the Atherosclerosis Risk in Communities (ARIC) Study, 1987-1993. Am J Epidemiol. 1997;146:483–94. doi: 10.1093/oxfordjournals.aje.a009302. [DOI] [PubMed] [Google Scholar]
- 27.Hodis HN, Mack WJ, LaBree L, Selzer RH, Liu CR, Liu CH, et al. The role of carotid arterial intima-media thickness in predicting clinical coronary events. Ann Intern Med. 1998;128:262–9. doi: 10.7326/0003-4819-128-4-199802150-00002. [DOI] [PubMed] [Google Scholar]
- 28.Giacopelli F, Marciano R, Pistorio A, Catarsi P, Canini S, Karsenty G, et al. Polymorphisms in the osteopontin promoter affect its transcriptional activity. Physiol Genomics. 2004;20:87–96. doi: 10.1152/physiolgenomics.00138.2004. [DOI] [PubMed] [Google Scholar]
- 29.Iizuka K, Murakami T, Kawaguchi H. Pure atmospheric pressure promotes an expression of osteopontin in human aortic smooth muscle cells. Biochem Biophys Res Commun. 2001;283:493–8. doi: 10.1006/bbrc.2001.4796. [DOI] [PubMed] [Google Scholar]
- 30.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute scientific statement: Executive summary. Circulation. 2005;112:e285–90. [Google Scholar]
- 31.Hanley AJ, Williams K, Stern MP, Haffner SM. Homeostasis model assessment of insulin resistance in relation to the incidence of cardiovascular disease: the San Antonio Heart Study. Diabetes Care. 2002;25:1177–84. doi: 10.2337/diacare.25.7.1177. [DOI] [PubMed] [Google Scholar]
- 32.Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. [PubMed] [Google Scholar]
- 33.ASE Task Force Statement on the Measurement of CIMT J Am Soc Echocardiog. 2008 (in press) - to be updated with final citation at time of proof.
- 34.Newberry EP, Latifi T, Towler DA. The RRM domain of MINT, a novel Msx2 binding protein, recognizes and regulates the rat osteocalcin promoter. Biochemistry. 1999;38:10678–90. doi: 10.1021/bi990967j. [DOI] [PubMed] [Google Scholar]
- 35.Lehto S, Niskanen L, Suhonen M, Ronnemaa T, Laakso M. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol. 1996;16:978–83. doi: 10.1161/01.atv.16.8.978. [DOI] [PubMed] [Google Scholar]
- 36.Sangiorgi G, Rumberger JA, Severson A, Edwards WD, Gregoire J, Fitzpatrick LA, et al. Arterial calcification and not lumen stenosis is highly correlated with atherosclerotic plaque burden in humans: a histologic study of 723 coronary artery segments using nondecalcifying methodology. J Am Coll Cardiol. 1998;31:126–33. doi: 10.1016/s0735-1097(97)00443-9. [DOI] [PubMed] [Google Scholar]
- 37.Duprez DA. Role of the renin-angiotensin-aldosterone system in vascular remodeling and inflammation: a clinical review. J Hypertens. 2006;24:983–91. doi: 10.1097/01.hjh.0000226182.60321.69. [DOI] [PubMed] [Google Scholar]
- 38.Shao JS, Cai J, Towler DA. Molecular mechanisms of vascular calcification: lessons learned from the aorta. Arterioscler Thromb Vasc Biol. 2006;26:1423–30. doi: 10.1161/01.ATV.0000220441.42041.20. [DOI] [PubMed] [Google Scholar]
- 39.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90:251–62. [PubMed] [Google Scholar]
- 40.Qin X, Corriere MA, Matrisian LM, Guzman RJ. Matrix metalloproteinase inhibition attenuates aortic calcification. Arterioscler Thromb Vasc Biol. 2006;26:1510–6. doi: 10.1161/01.ATV.0000225807.76419.a7. [DOI] [PubMed] [Google Scholar]
- 41.Brenner D, Labreuche J, Touboul PJ, Schmidt-Petersen K, Poirier O, Perret C, et al. Cytokine polymorphisms associated with carotid intima-media thickness in stroke patients. Stroke. 2006;37:1691–6. doi: 10.1161/01.STR.0000226565.76113.6c. [DOI] [PubMed] [Google Scholar]
- 42.Kawamura H, Yokote K, Asaumi S, Kobayashi K, Fujimoto M, Maezawa Y, et al. High glucose-induced upregulation of osteopontin is mediated via Rho/Rho kinase pathway in cultured rat aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:276–81. doi: 10.1161/01.ATV.0000112012.33770.2a. [DOI] [PubMed] [Google Scholar]
- 43.Kolominsky-Rabas PL, Heuschmann PU, Marschall D, Emmert M, Baltzer N, Neundorfer B, et al. Lifetime cost of ischemic stroke in Germany: results and national projections from a population-based stroke registry: the Erlangen Stroke Project. Stroke. 2006;37:1179–83. doi: 10.1161/01.STR.0000217450.21310.90. [DOI] [PubMed] [Google Scholar]
- 44.Taylor TN, Davis PH, Torner JC, Holmes J, Meyer JW, Jacobson MF. Lifetime cost of stroke in the United States. Stroke. 1996;27:1459–66. doi: 10.1161/01.str.27.9.1459. [DOI] [PubMed] [Google Scholar]
- 45.Golledge J, Muller J, Shephard N, Clancy P, Smallwood L, Moran C, et al. Association Between Osteopontin and Human Abdominal Aortic Aneurysm. Arterioscler Thromb Vasc Biol. 2006 doi: 10.1161/01.ATV.0000255560.49503.4e. [DOI] [PubMed] [Google Scholar]
- 46.Shao JS, Cheng SL, Charlton-Kachigian N, Loewy AP, Towler DA. Teriparatide (human parathyroid hormone (1−34)) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor-deficient mice. Journal of Biological Chemistry. 2003;278:50195–202. doi: 10.1074/jbc.M308825200. [DOI] [PubMed] [Google Scholar]
- 47.Long MW. Blood cell cytoadhesion molecules. Exp Hematol. 1992;20:288–301. [PubMed] [Google Scholar]
- 48.Li G, Chen YF, Kelpke SS, Oparil S, Thompson JA. Estrogen attenuates integrin-beta(3)-dependent adventitial fibroblast migration after inhibition of osteopontin production in vascular smooth muscle cells. Circulation. 2000;101:2949–55. doi: 10.1161/01.cir.101.25.2949. [DOI] [PubMed] [Google Scholar]
- 49.Towler DA. Inorganic pyrophosphate: a paracrine regulator of vascular calcification and smooth muscle phenotype. Arterioscler Thromb Vasc Biol. 2005;25:651–4. doi: 10.1161/01.ATV.0000158943.79580.9d. [DOI] [PubMed] [Google Scholar]
- 50.Shao JS, Cheng SL, Charlton-Kachigian N, Loewy AP, Towler DA. Teriparatide (human parathyroid hormone (1−34)) inhibits osteogenic vascular calcification in diabetic low density lipoprotein receptor-deficient mice. J Biol Chem. 2003;278:50195–202. doi: 10.1074/jbc.M308825200. [DOI] [PubMed] [Google Scholar]
- 51.de las Fuentes L, Brown AL, Mathews SJ, Waggoner AD, Soto PF, Gropler RJ, et al. Metabolic Syndrome Is Associated With Abnormal Left Ventricular Diastolic Function Independent Of Lv Mass. European Heart Journal. 2007 doi: 10.1093/eurheartj/ehl526. in press. [DOI] [PubMed] [Google Scholar]
- 52.Fox CS, Polak JF, Chazaro I, Cupples A, Wolf PA, D'Agostino RA, et al. Genetic and environmental contributions to atherosclerosis phenotypes in men and women: heritability of carotid intima-media thickness in the Framingham Heart Study. Stroke. 2003;34:397–401. doi: 10.1161/01.str.0000048214.56981.6f. [DOI] [PubMed] [Google Scholar]
- 53.Juo SH, Lin HF, Rundek T, Sabala EA, Boden-Albala B, Park N, et al. Genetic and environmental contributions to carotid intima-media thickness and obesity phenotypes in the Northern Manhattan Family Study. Stroke. 2004;35:2243–7. doi: 10.1161/01.STR.0000142132.20442.d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moskau S, Golla A, Grothe C, Boes M, Pohl C, Klockgether T. Heritability of carotid artery atherosclerotic lesions: an ultrasound study in 154 families. Stroke. 2005;36:5–8. doi: 10.1161/01.STR.0000149936.33498.83. [DOI] [PubMed] [Google Scholar]
- 55.Zannad F, Benetos A. Genetics of intima-media thickness. Current Opinion in Lipidology. 2003;14:191–200. doi: 10.1097/00041433-200304000-00011. [DOI] [PubMed] [Google Scholar]