

Abstract
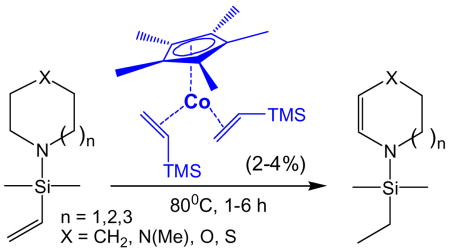
C-H bond activation has been extensively studied with (Cp*)M(L)n (M = Ir, Rh), but cobalt, the third member of this triad, has not previously been shown to activate sp3C-H bonds. Further, practical functionalization of the metal alkyl products of oxidative addition has not been fully explored. Towards these ends, we have developed catalytic dehydrogenation of alkyl amines with a Co(I) catalyst. Amine substrates are protected with vinyl silanes, followed by catalytic transfer hydrogenation to yield a broad range of stable protected enamines and 1,2-diheteroatom substituted alkenes, including several unprecedented heterocycles. (Cp*)Co(VTMS)2 catalyzes transfer hydrogenation under surprisingly mild conditions with high chemo-, regio-, and diastereo- selectivity, while tolerating additional functionality.
Twenty-five years after the seminal reports by Bergman1 and Jones2 on oxidative addition of C-H bonds to (Cp*)M(L) complexes of iridium and rhodium respectively, extensive progress has been made in mechanistic understanding,3 and functionalization for applications in synthesis4. Cobalt, the analogous metal from the first transition series, while known to activate sp2 aromatic and aldehydic C-H bonds5, has been notably absent from reports of sp3 C-H activation. In fact, experimental6 and computational7 studies have indicated that C-H bonds should not oxidatively add to the 16-electron Co(I) center as they do to Ir and Rh. This communication describes for the first time the facile and highly selective activation and functionalization of sp3 C-H bonds by [(Cp*)Co(VTMS)2] (1) (VTMS = vinyltrimethyl silane), allowing synthesis of unique heterocycles.
Direct functionalization of C-H bonds α to nitrogen is a particularly attractive transformation, but catalytic examples are still somewhat rare8. One strategy for functionalization of C-H oxidative addition products is the net dehydrogenation of organic substrates,9 transforming C-H bonds into carbon-carbon bonds. In this area, our group has developed methodology for the synthesis of silyl enol ethers10 and 1,2- diheteroatom alkenes11 via [(Cp*)Rh(VTMS)2] (2) -catalyzed intramolecular transfer dehydrogenation. Enamines12 are versatile reactive intermediates for organic synthesis, so we explored the application of this strategy to their formation (Scheme 1).
Scheme 1.
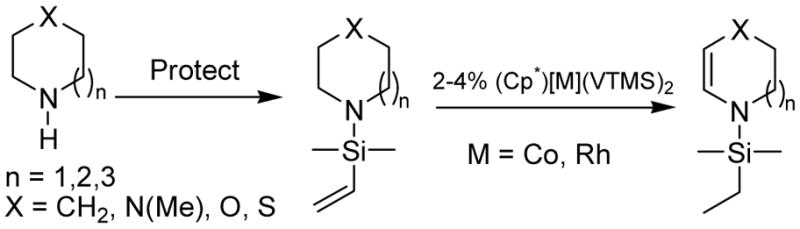
Goldman13 has reported synthesis of enamines from 3° alkyl amines via Ir-catalyzed inter-molecular hydrogen transfer. Our intra-molecular approach to hydrogen transfer is complementary and applicable to (protected) 2° amines. The silicon protecting group serves additionally as hydrogen acceptor and directing group. This strategy affords protected endocyclic enamines which are difficult to access via conventional methodology.
Substrate and catalyst screening was efficiently conducted in screw-cap NMR tubes with C6D12 for convenient monitoring of reaction progress. Preparative scale reactions were then performed in Kontes flasks in pentane solvent (5 mmol substrate, 2% Co catalyst loading, 6h, 80°C) with isolated yields of metal-free products up to 90% (See Supporting Information for details).
Initially we explored conversion of piperidine 3 into protected tetrahydropyridine 4, an attractive target both as a mimic of biological hydrogen transfer agents and as a synthetic intermediate.14 Unprotected endocyclic “enamines” of 2° amines generally react as, and are isolated as, the imine tautomer15, so (protected) enamines should be valuable synthons. Gratifyingly, rhodium catalyst 2 produced the desired enamine 4, albeit requiring 6 hours at 140°C for conversion (Table 1, entry 1). In contrast, cobalt catalyst 1 afforded 4 in < 1h at 80°C (Table 1, entry 2). Encouraged by this rapid conversion under mild conditions, we investigated the scope of this Co-catalyzed transfer dehydrogenation.
Table 1.
Scope of enamine synthesis with 1 and 2
| Substrate | Transfer Product | Cata | t(h) | Yieldb(%) | |
|---|---|---|---|---|---|
| 1 |
3 |
4 |
2c | 6 | >90 |
| 2 |
3 |
4 |
1 | >99 | |
| 3 |
5 |
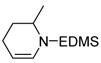
6 |
1 | >99 | |
| 4 |
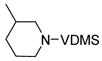
7 |
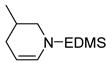
8 |
1 | 2 | >95 |
| 5 |
9 |
10 |
1 | >95 | |
| 6 |
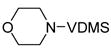
11 |
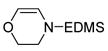
12 |
1 | >99 | |
| 7 |
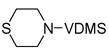
13 |
14 |
1 | 13 | |
| 8 |
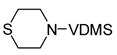
13 |
14 |
2c | 1 | >90 |
| 9 |
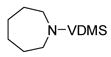
15 |
16 |
1 | 6d | >90 |
| 10 |
17 |
18 |
1 | >95 | |
| 11 |
19 |
20 |
1 | >95 |
Unless noted otherwise, 0.25 mmol substrate, 0.01 mmol 1 (4%), 0.5 mL C6D12, 80°C, 1h.
By 1H NMR spectroscopy
0.25 mmol substrate, 0.01 mmol 2 (4%), 0.5 mL C6D12, 140°C. VDMS = Vinyl(DiMethyl)Silyl. EDMS = Ethyl(DiMethyl)Silyl.
Subjection of 2- and 3-substituted piperidines (Entries 3 and 4) to cobalt-mediated transfer catalysis led to di-substituted olefins 6 and 8 respectively, as the only observable products with no isomerization to the thermodynamically more stable tri-substituted olefins even at long reaction times. Endocyclic enamines have been employed in natural products synthesis16, but typically as the more substituted isomers available by previous methodology17.
One hallmark of late-metal catalysts is their compatability with Lewis basic functionality. Nitrogen- and oxygen-containing functional groups are well tolerated by 1, allowing synthesis of dehydrogenated piperazine and morpholine derivatives (Entries 5 and 6, respectively). Sulfur apparently poisons the active cobalt species; only 13% conversion of 13 was observed with 1, corresponding to a TON of <4. Rhodium catalyst 2 proved effective for this substrate, yielding clean transfer product 14.
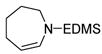
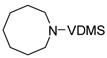
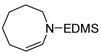
Unsaturated azacycles with ring size greater than 6 are rare18, yet 15 and 17 were transformed into 7- and 8-membered N-protected enamines by this methodology. In the former case (entry 9), starting material was consumed surprisingly rapidly (ca. 10 min), but additional species were observed at early reaction times which slowly isomerized to enamine 16. Intriguingly, these products vary in the position of the double bond (allylic- and homoallylic- amines), indicating oxidative addition of truly “unactivated” C-H bonds to the metal center (See Supporting Information for details).
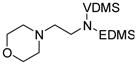
Next, we further probed the regioselectivity of this transformation with catalyst 1. Substrate 19 contains two sites potentially amenable to dehydrogenation: the methylene groups of the diamine or those of the morpholine ring. After 30 minutes at 80°C, only 20 was observed, with no isomerization over several hours. This result establishes that this process is: 1) Applicable to linear as well as cyclic amines. (Primary enamines are notoriously difficult to prepare19, so protected variants are expected to have synthetic utility.) 2) Regioselective; final products observed are consistent with activation of the C-H bond to the protected nitrogen. 3) Diastereoselective. Whereas the Z geometry is thermodynamically favored for 1,2-diheteroatom-substituted olefins, only the E isomer was observed. 4) Consistent with intra-, not inter-molecular hydrogen transfer, as the morpholine moiety was not dehydrogenated.
Cobalt catalyst 1 was ineffective for transfer hydrogenation of the substrate analogous to 19 containing a free N-H (lacking the EDMS protecting group), indicating distinct chemoselectivity compared to Ir catalysts which produce imines from secondary amines20.
These observations are consistent with a mechanism analogous to that for dehydrogenation of protected alcohols with 121. As such, it involves formation of the 16 e− intermediate [(Cp*)Co(mono-olefin)] and oxidative addition of an sp3 C-H bond (generally α to the heteroatom) to the cobalt(I) center. Migratory insertion of the vinyl silane into the cobalt hydride followed by β-hydride elimination (from the 3-position of the amine) and reductive elimination yields the observed enamine products. The more rapid turnover of the Co system relative to the Rh system must stem in part from the lower barrier to dissociation of a vinyl group from the (Cp*)Co(bis-olefin) resting state22.
In conclusion, we have demonstrated a convenient synthetic route to enamines based on cobalt-catalyzed hydrogen transfer of protected amines. This conversion is consistent with cobalt(I) sp3 C-H bond activation, reactivity which was previously held to be the exclusive domain of the heavier group 9 metals. Catalyst 1 exhibits not only high reactivity under milder conditions than the other members of its triad, but also impressive chemo-, regio-, diastereo-, and intra-molecular hydrogen transfer selectivity. Further investigations into the scope of this transformation, mechanistic studies, and applications to synthesis are currently underway in this laboratory.
Supplementary Material
Experimental procedures and characterization of complexes 3–21. This material is available free of charge via the Internet at: http://pubs.acs.org.
Figure 1.
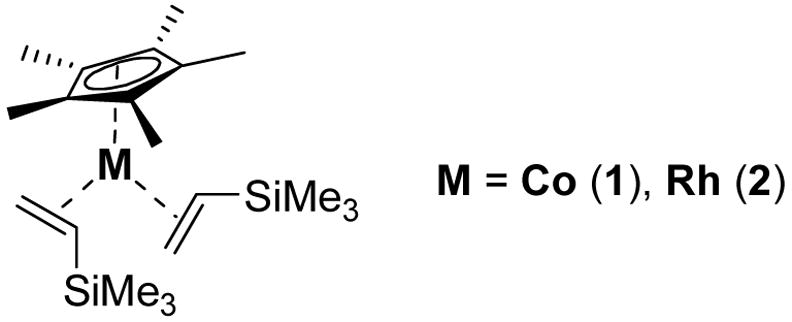
Isostructural Late Metal Catalysts for Transfer Hydrogenation
Acknowledgments
We gratefully acknowledge funding by the National Institutes of Health (Grant No. GM 28939).
References
- 1.Janowicz AH, Bergman RG. J Am Chem Soc. 1982;104:352. [Google Scholar]
- 2.Jones WD, Feher FJ. J Am Chem Soc. 1982;104:4240. [Google Scholar]
- 3.Arndtsen BA, Bergman RG, Mobley TA, Peterson TH. Acc Chem Res. 1995;28:154. [Google Scholar]; (b) Jones WD, Feher FJ. Acc Chem Res. 1989;22:91. [Google Scholar]; (c) Halpern J. Inorg Chim Acta. 1985;100:41. [Google Scholar]
- 4.Wilson RM, Thalji RK, Bergman RG, Ellman JA. Org Lett. 2006;8:1745. doi: 10.1021/ol060485h. and references therein. [DOI] [PubMed] [Google Scholar]; (b) Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. [DOI] [PubMed] [Google Scholar]; (c) Cho J-Y, Tse MK, Holmes D, Maleczka RE, Jr, Smith MR., III Science. 2002;295:305. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]; (d) Chen H, Schlecht S, Semple TC, Hartwig JF. Science. 2000;287:1995. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]
- 5.Lenges CP, Brookhart M, Grant BE. J Organometallic Chem. 1997;528:199. [Google Scholar]; (b) Lenges CP, Brookhart M. J Am Chem Soc. 1997;119:3165. [Google Scholar]; (c) Lenges CP, White PS, Brookhart M. J Am Chem Soc. 1998;120:6965. [Google Scholar]; (d) Wadepohl H, Borchert T, Pritzkow H. J Chem Soc, Chem Comm. 1995:1447. [Google Scholar]
- 6.Bengali AA, Bergman RG, Moore CB. J Am Chem Soc. 1995;117:3879. [Google Scholar]
- 7.Siegbahn PEM. J Am Chem Soc. 1996;118:1487. [Google Scholar]; (b) Poli R, Smith KM. Eur J Inorg Chem. 1999:877. [Google Scholar]
- 8.Campos KR. Chem Soc Rev. 2007;36:1069. doi: 10.1039/b607547a. and references therein. [DOI] [PubMed] [Google Scholar]
- 9.Burk MJ, Crabtree RH. J Am Chem Soc. 1987;109:8025. [Google Scholar]; (b) Liu F, Pak EB, Singh B, Jensen CM, Goldman AS. J Am Chem Soc. 1999;121:4086. [Google Scholar]
- 10.Lenges CP, White PS, Brookhart M. J Am Chem Soc. 1999;121:4835. [Google Scholar]
- 11.Diaz-Requejo MMar, DiSalvo D, Brookhart M. J Am Chem Soc. 2003;125:2038. doi: 10.1021/ja029393n. [DOI] [PubMed] [Google Scholar]
- 12.Rappoport Z, editor. The Chemistry of Enamines (Parts 1 & 2) Wiley; New York: 1994. For a review of metal-mediated syntheses, see: Dehli JR, Legros J, Bolm C. Chem Comm. 2005:973. doi: 10.1039/b415954c.
- 13.Zhang X, Fried A, Knapp S, Goldman AS. Chem Comm. 2003:2060. [PubMed] [Google Scholar]
- 14.Beeken P, Fowler FW. J Org Chem. 1980;45:1336. [Google Scholar]; (b) Wenkert E, Dave KG, Haglid F, Lewis RG, Oishi T, Stevens RV, Terashima M. J Org Chem. 1968;33:747. doi: 10.1021/jo01266a054. [DOI] [PubMed] [Google Scholar]; (c) Cook NC, Lyons JE. J Am Chem Soc. 1966;88:3396. [Google Scholar]
- 15.Cook AG, editor. Enamines: Synthesis, Structure, and Reactions. 2. Marcel Dekker; New York: 1988. p. 460. [Google Scholar]
- 16.Evans DA, Bryan CA, Sims CL. J Am Chem Soc. 1972;94:2891. [Google Scholar]; (b) Evans DA. J Am Chem Soc. 1970;92:7593. [Google Scholar]; (c) Wenkert E. Acc Chem Res. 1968;1:78. [Google Scholar]; (d) Dake GR, Fenster MDB, Hurley PB, Patrick BO. J Org Chem. 2004;69:5668. doi: 10.1021/jo0493572. [DOI] [PubMed] [Google Scholar]
- 17.Takemiya A, Hartwig JF. J Am Chem Soc. 2006;128:6042. doi: 10.1021/ja058299e.For exceptions, see: Larivée A, Charette AB. Org Lett. 2006;8:3955. doi: 10.1021/ol061415d.Hegedus LS, McKearin JM. J Am Chem Soc. 1982;104:2444.
- 18.Cook AG, editor. Enamines: Synthesis, Structure, and Reactions. 2. Marcel Dekker; New York: 1988. p. 442. [Google Scholar]
- 19.Novak BM, Cafmeyer JT. J Am Chem Soc. 2001;123:11083. doi: 10.1021/ja011609i. [DOI] [PubMed] [Google Scholar]
- 20.Gu XQ, Chen W, Morales-Morales D, Jensen CM. J Mol Cat A. 2002;189:119–124. [Google Scholar]
- 21.Diaz-Requejo MMar, DiSalvo D, Brookhart M. J Am Chem Soc. 2003;125:2038. doi: 10.1021/ja029393n. [DOI] [PubMed] [Google Scholar]
- 22.Lenges CP, Brookhart M, Grant BE. J Organometallic Chem. 1997;528:199. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization of complexes 3–21. This material is available free of charge via the Internet at: http://pubs.acs.org.