

Abstract
The cyclohexane-1,2-diamine-based bisbinaphthyl macrocycles (S)-/(R)-5 and their cyclic and acyclic analogs are synthesized. The interactions of these compounds with various chiral acids are studied. Compounds (S)-/(R)-5 exhibit highly enantioselective fluorescent responses and high fluorescent sensitivity toward α-hydroxycarboxylic acids and N-protected amino acids. Among these interactions, (S)-mandelic acid (10−3 M) led to over 20 fold fluorescence enhancement of (S)-5 (1.0 × 10−5 M in benzene/0.05% DME) at the monomer emission and (S)-hexahydromandelic acid (10−3 M) led to over 80 fold fluorescence enhancement. These results demonstrate that (S)-5 is useful as an enantioselective fluorescent sensor for the recognition of the chiral acids. On the basis of the study of the structures of (S)-5 and the previously reported 1,2-diphenylethylenediamine-based bisbinaphthyl macrocycle (S)-4, the large fluorescence enhancement of (S)-5 with achirality-matched α-hydroxycarboxylic acid is attributed to the formation of a structurally rigidified host-guest complex and the further interaction of this complex with the acid to suppress the photo-induced electron transfer fluorescent quenching caused by the nitrogens in (S)-5.
Introduction
Study of fluorescent sensors has attracted broad research attention in areas such as pH sensing, metal ion detection and biological tags, since fluorescence spectroscopy can provide high sensitivity as well as multiple signaling modes.1–4 Recently, there are also growing interests in developing fluorescent sensors for chiral recognition.5,6 Using these sensors can potentially lead to rapid analysis of the enantiomeric compositions of chiral organic compounds.7,8 In our laboratory, we are particularly interested in developing enantioselective fluorescent sensors for the recognition of α-hydroxycarboxylic acids, because these compounds are the structural units of many organic compounds as well as versatile functional synthons.9 The enantioselective fluorescent recognition of α-hydroxycarboxylic acids may allow the development of high throughput catalyst screening methods for their asymmetric synthesis.
We initially designed compound (S)-1 as a fluorescent sensor to recognize mandelic acid (2), an aromatic α-hydroxycarboxylic acid.10 Interaction of (S)-1 with (S)-mandelic acid could generate complex 3 through three specific hydrogen bonds. The fluorescence of (S)-1 quenched by its nitrogen atom through a photo-induced-electron-transfer process could be turned on when the nitrogen binds with the carboxylic acid proton of mandelic acid. This fluorescence enhancement was enantioselective with the (S)-mandelic acid leading to a greater signal for (S)-1 than (R)-mandelic acid. We also studied the use of the 1,2-diphenylethylenediamine-based bisbinaphthyl macrocycle (S)-4 to recognize the α-hydroxycarboxylic acids.11 This compound gave good enantioselective response at its excimer emission in the presence of (S)-mandelic acid. Both (S)-1 and (S)-4 showed up to 2 – 3 fold fluorescence enhancement in the presence of the chirality matched enantiomer of mandelic acid. In order to further improve these sensors, we incorporated cyclohexane-1,2-diamine into the bisbinaphthyl macrocycle to prepare compound (S)-5. This compound exhibited greatly enhanced sensitivity and enantioselectivity in the recognition of mandelic acid and other chiral acids.12 Herein, the detailed study of (S)-5 and its analogs is reported, and the mechanism of its fluorescent responses is discussed.
Results
1. Synthesis of the Cyclohexane-1,2-diamine-Based Bisbinaphthyl Compounds
Compound (S)-5 was synthesized in a way similar to that reported by Brunner and Schiessling for the synthesis of (S)-4.11,13 Treatment of the methoxymethyl protected (S)-1,1′-bi-2-naphthol (BINOL) with nBuLi followed by the addition of DMF and hydrolysis produced (S)-2,2′-dihydroxy-[1,1′]binaphthalenyl-3,3′-dicarbaldehyde.14 Condensation of this compound with (R,R)-cyclohexane-1,2-diamine followed by reduction resulted in the desired macrocyclic compound (S)-5 in 55% yield over two steps. The enantiomer of (S)-5, (R)-5, was obtained by using a (R)-binaphthyl starting material and (S,S)-cyclohexane-1,2-diamine.
An acyclic analog of (S)-5 was prepared in 78% yield from the reaction of the monoaldehyde binaphthyl compound (S)-615 with (R,R)-cyclohexane-1,2-diamine followed by reduction (Scheme 2). In comparison with the structure of (S)-5, (S)-7 is not only acyclic but also short of a cyclohexane-1,2-diamine unit.
Scheme 2.

Synthesis of the Acyclic Bisbinaphthyl Compound (S)-7.
Compound (S)-12 was prepared as a macrocyclic analog of (S)-7 (Scheme 3). Treatment of the monoprotected BINOL (S)-8 with 1,4-dibromobutane gave (S)-9 in 80% yield. This compound was reacted with nBuLi and DMF to give the dialdehyde (S)-10 in 39% yield. A monoaldehyde side product was obtained from this reaction. Mixing (S)-10 with CF3CO2H in methylene chloride followed by aqueous work-up gave (S)-11 in 91% yield.16 Condensation of (S)-11 with (R,R)-cyclohexane-1,2-diamine followed by reduction produced the macrocycle (S)-12 in 54% yield. This compound contains only half of the functional groups of (S)-5.
Scheme 3.

Synthesis of the Bisbinaphthyl Macrocycle (S)-12.
We also extended the conjugation of (S)-5 by introducing a p-ethoxyphenyl group to the 6-position of each of the naphthalene units to make compound (S)-14 (Scheme 4). Compound (S)-13 was synthesized as previously reported.11b Condensation of (S)-13 with (R,R)-cyclohexane-1,2-diamine followed by reduction gave (S)-14 in 53% yield.
Scheme 4.

Synthesis of Compound (S)-14 with more Extended Conjugation.
2. X-Ray Structure of (R)-5
A single crystal of (R)-5 was obtained from its acetone/hexane solution and its X-ray analysis established the molecular structure of (R)-5. As shown in Figure 1, the central cavity of the molecule is defined by two almost parallel naphthol units separated by 8.13 Å. The cavity is large enough to host an acetone solvent molecule which is sandwiched between the two naphthol rings. The distances between the C atoms of acetone and naphthol range between 3.77 and 4.20 Å. All of the O atoms of the naphthol units are on the same side of the macrocycle. The macrocycle rings are stabilized by intramolecular O-H…N hydrogen bonds with the average O…N donor-acceptor distance of 2.74 Å.
Figure 1.

ORTEP Drawing (30% Ellipsoids) of (R)-5.
3. UV Spectra of Compounds (S)-5, (S)-7, (S)-12 and (S)-14
Figure 2 gives the UV spectra of compounds (S)-5, (S)-7, (S)-12 and (S)-14 in benzene at 1.0 × 10−5 M. As shown, the long wavelength absorption maximum of the macrocycle (S)-5 is shifted to the red of that of the acyclic analog (S)-7, that is, from 336 nm to 340 nm. This indicates a different conjugation in these compounds which could originate from the possibly different binaphthyl dihedral angles of (S)-5 and (S)-7. The macrocyclic structure of (S)-5 might have restricted the rotation of the naphthalene units around the 1,1′-bonds and forced them to achieve a better conjugation than the flexible acyclic compound. The absorption maxima of the two macrocycles (S)-5 and (S)-12 are almost identical. The four p-ethoxyphenyl groups of (S)-14 greatly increased the absorptions in the range of 270 – 320 nm. The new long wavelength absorption of (S)-14 at 352 nm (sh) is due to the more extended conjugation of (S)-14 than (S)-5, (S)-7 and (S)-12.
Figure 2.
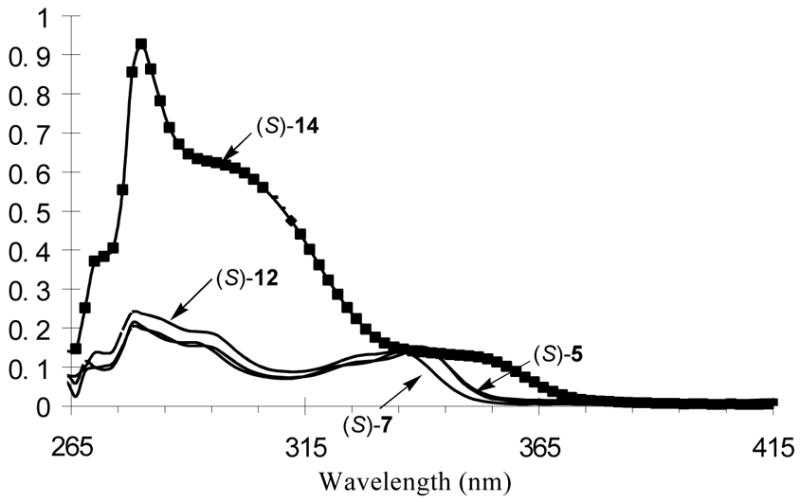
UV Spectra of (S)-5, (S)-7, (S)-12 and (S)-14 (1.0 × 10−5 M in benzene).
4. Fluorescence Spectra of the Bisbinaphthyl Compounds (S)-5, (S)-7, (S)-12 and (S)-14
The fluorescence spectra of the bisbinaphthyl compounds in benzene at 1.0 × 10−5 M while excited at 332 nm are shown in Figure 3. All the three macrocycles (S)-5, (S)-12 and (S)-14 exhibit dual emissions with a short wavelength peak attributed to the monomer emission and the long wavelength one to the excimer emission. The acyclic compound (S)-7, however, shows predominantly the monomer emission. The more flexible structure of (S)-7 may allow it to be better solvated than the more rigid macrocycles and thus produce less excimers. Each of compounds (S)-7 and (S)-12 has two less nitrogen atoms than (S)-5 and (S)-14, and thus there is less fluorescence quenching by the nitrogens through the photo-induced-electron-transfer process. This leads to the significantly greater fluorescence intensity of (S)-7 and (S)-12 than that of (S)-5 and (S)-14 when excited at 332 nm where they all have a similar absorption.
Figure 3.
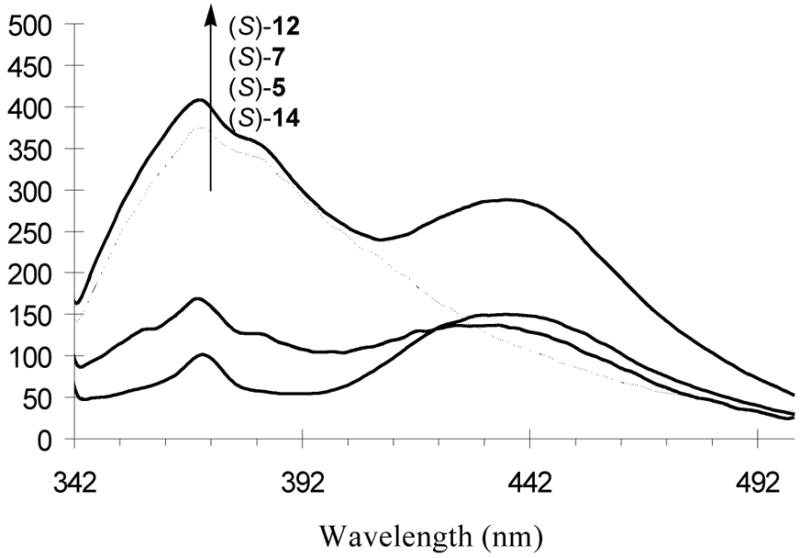
Fluorescent Spectra of (S)-5, (S)-7, (S)-12 and (S)-14 in Benzene at 1.0 × 10−5 M. (λexc = 332 nm, slit = 3.5; 6.5 nm)
The UV spectra of these compounds did not show concentration dependence in terms of peak positions and shapes. However, the emission maxima of the macrocycles (S)-5, (S)-12 and (S)-14 strongly depended on their concentrations. Figure 4 gives the fluorescence spectra of (S)-5 at various concentrations. As the concentration increased from 1.0 × 10−6 M to 1.0 × 10−4 M in benzene, the excimer emission of (S)-5 at the long wavelength (λ = 435 nm) increased significantly. At 1.0 × 10−6 M, (S)-5 showed mainly the monomer emission. The same observations were also made for the fluorescence spectra of (S)-12 (see Figure S16) and (S)-14. The acyclic compound (S)-7 showed much less excimer emission even with the increase of the concentration (see Figure S17).
Figure 4.
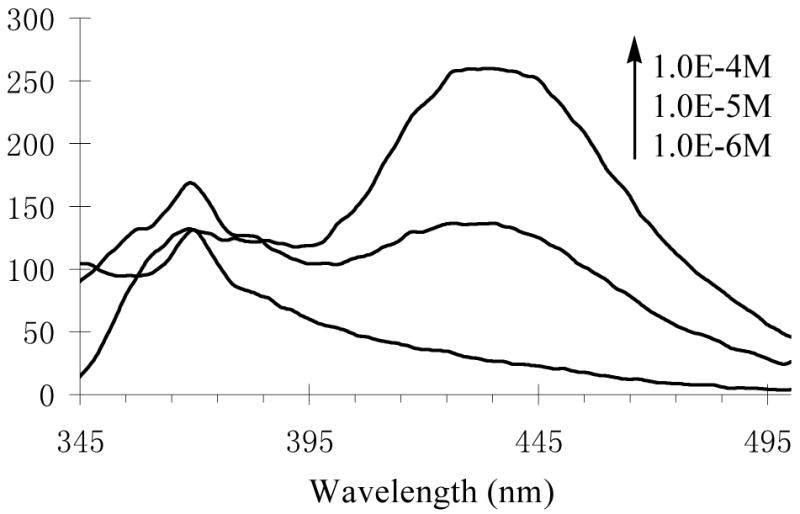
Concentration Effect on the Fluorescence Spectra of (S)-5 in Benzene (λexc = 332 nm, slit = 3.5/6.5 nm).
5. CD Spectra of the Bisbinaphthyl Compound
Figure 5 shows the CD Spectra of compounds (S)-5, (S)-7, (R)-12 [the enantiomer of (S)-12] and (S)-14 in methylene chloride. The CD spectra of the bisbinaphthyl compounds in benzene solution were also obtained, but because of the interference of the solvent, only signals at wavelengths greater than 270 nm could be observed. Most of these compounds exhibit very different CD effects from each other. The macrocycle (S)-5 gives a intense positive Cotton effect at 239 nm, but the acyclic compound (S)-7, though having the same chiral configurations as those of (S)-5 at both the binaphthyl units and the carbon centers, shows an intense negative Cotton effect at 237 nm. These inverted Cotton effects indicate that these two compounds should have very different conformations. In comparison with the CD spectra of the 1,1′-binaphthyl compounds in the literature,17,18 the following conformations of (S)-5 and (S)-7 could be proposed. The binaphthyl units in (S)-5 probably have a transoid conformation, that is, the dihedral angel as shown in Figure 6 is greater than 90°, and the binaphthyl units in (S)-7 probably have a cisoid conformation with the dihedral angle less than 90°. It was previously observed that when the two substituents L are of large sizes in a binaphthyl molecule, the compound prefers the transoid conformation in order to reduce the steric interaction, and when the two L groups are small or capable of intramolecular hydrogen bonding, the compound would prefer a cisoid conformation. The transoid and cisoid conformations of a binaphthyl molecule exhibit the opposite Cotton effects. The intramolecular hydrogen bonds in (S)-7 might generate a cisoid conformation for its binaphthyl units, whereas the rigid macrocyclic structure of (S)-5 probably forces the binaphthyl units to take a transoid conformation. The macrocycle (R)-12 gives a strong negative Cotton effect at 239 nm. This also indicates that the cyclic structure of (R)-12 [or (S)-12] probably forces its binaphthyl units to take a transoid conformation. That is, the binaphthyl dihedral angles of (S)-5 and (R)-12 [or (S)-12] are similar but different from those of the acyclic compound (S)-7. The UV spectra of (S)-5, (S)-7 and (S)-12 suggest that the transoid conformations of (S)-5 and (S)-12 should have a better conjugation than the cisoid conformation of (S)-7.
Figure 5.

CD Spectra of Compounds (S)-5, (S)-7, (R)-12 and (S)-14 (1.0 × 10−5 M in CH2Cl2).
Figure 6.
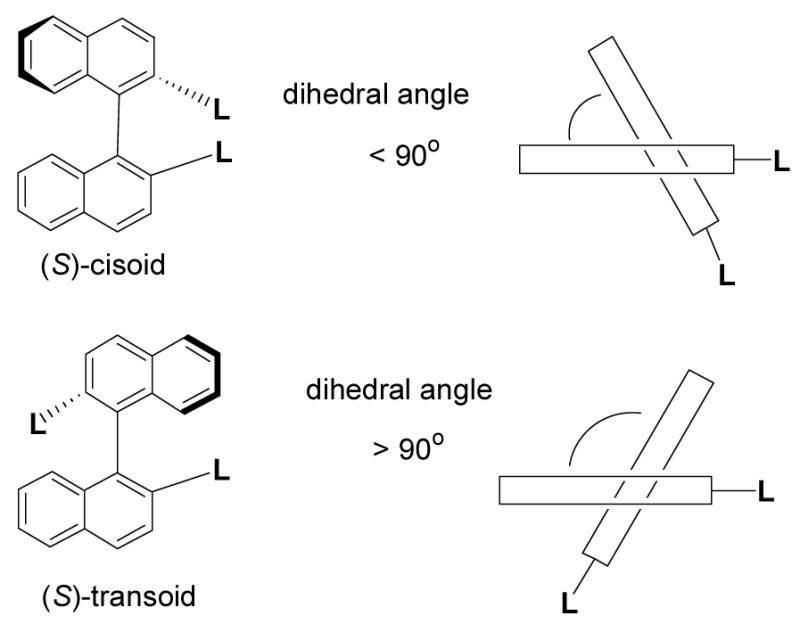
Conformations of 1,1′-Binaphthyl Compounds.
Compound (S)-14 shows a strong exciton coupling signal centered at 269 nm in its CD spectrum which is similar to the previously reported 6,6′-aryl substituted binaphthyl compounds.17,19 Because of the macrocyclic structure, the binaphthyl conformations in (S)-14 are most likely the same as those in (S)-5, that is, transoid.
6. Interaction of (S)- and (R)-5 with Chiral Acids
a. Fluorescent Recognition of Mandelic Acid
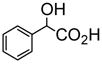
The interaction of the macrocycle (S)-5 with the enantiomers of mandelic acid was studied. The UV spectrum of (S)-5 showed only a slight decrease in the absorption intensity when treated with mandelic acid, but no change in peak shape and position and almost no difference between the effect of (R)- and (S)-mandelic acid were observed. In contrast, a dramatic difference was observed for the fluorescence responses of the macrocycle towards (R)- and (S)-mandelic acid. As shown by Figure 7a, (R)-mandelic acid (5.0 × 10−4 M) had almost no effect on the fluorescence of (S)-5 (1.0 × 10−5 M in benzene/0.05% DME), whereas (S)-mandelic acid increased the fluorescence intensity of (S)-5 by over 20 fold at the monomer emission. In the fluorescent measurement, a small amount of DME was used to improve the solubility of mandelic acid in benzene.
Figure 7.

Fluorescence Spectra of (a) (S)-5 and (b) (R)-5 with/without (R)- and (S)-Mandelic Acid (λexc = 332 nm, slit = 3.5/3.5 nm).
In order to ascertain that the observed large difference in the fluorescence responses of (S)-5 toward (R)- and (S)-mandelic acid is due to an inherent chiral recognition, we studied the interaction of (R)-5, the enantiomer of (S)-5, with (R)- and (S)-mandelic acid. Figure 7b gives the fluorescence spectra of (R)-5 (1.0 × 10−5 M in benzene/0.05% DME) in the presence/absence of (R)- and (S)-mandelic acid (5.0 × 10−4 M). While (S)-mandelic acid caused little change on the fluorescence of (R)-5, (R)-mandelic acid greatly enhanced the fluorescence of (R)-5. Thus, there is a mirror image relationship between the fluorescence responses shown in Figure 7a and Figure 7b. This demonstrates that the fluorescence interaction of the macrocycle with mandelic acid is indeed highly enantioselective.
Figure 8a plots the fluorescence enhancement (I/I0) of (S)-5 versus the concentration of (R)- and (S)-mandelic acid. In the plot, the error bars were obtained by 4 independent measurements. As the concentration of the acid increased, the (S)-enantiomer greatly enhanced the fluorescence of (S)-5, but the (R)-enantiomer did not. When the acid concentration was 5.0 × 10−4 M, the enantiomeric fluorescence difference ratio (ef) [ef = (IS − I0)/(IR − I0)] was as high as 46. Figure 8b plots the fluorescence enhancement of (R)-5 versus the concentration of (R)- and (S)-mandelic acid. The fluorescence responses in Figure 8a are the mirror image of those in Figure 8b.
Figure 8.

Fluorescence Enhancement of (a) (S)-5 and (b) (R)-5 (1.0 × 10−5 M in benzene/0.05% DME) versus Concentration of (R)- and (S)-Mandelic acid (λexc = 332 nm).
Figure 9 plots the fluorescence enhancement of (S)-5 with respect to a broader concentration range (1.0 × 10−4 – 2.0 × 10−3 M) of mandelic acid. It shows that the fluorescence enhancement of (S)-5 reached maximum as the concentration of (S)-mandelic acid increased to around 7.0 × 10−4 M. Further increasing the concentration of (S)-mandelic acid led to a decrease in the fluorescence enhancement.
Figure 9.

Fluorescence Enhancement of (S)-5 (1.0 × 10−5 M) versus Concentration of (R)- and (S)-Mandelic acid (λexc = 332 nm).
The effect of the enantiomeric composition of mandelic acid on the fluorescence of (R)-5 was studied. Curve A in Figure 10 is the fluorescence enhancement of (R)-5 (1.0 × 10−5 M in benzene/0.05%DME) in the presence of mandelic acid (5.0 × 10−4 M) at various compositions of the (R)-and (S)-enantiomers. Curve B in Figure 10 is the fluorescence enhancement of (R)-5 (1.0 × 10−5 M in benzene/0.05%DME) when treated with the optically pure (R)-mandelic acid at concentrations corresponding to those of (R)-mandelic acid in the enantiomeric mixture. Thus, with the same amount of (R)-mandelic acid, the optically pure acid caused a greater fluorescence enhancement than the one containing both the (R)- and (S)-enantiomers.
Figure 10.
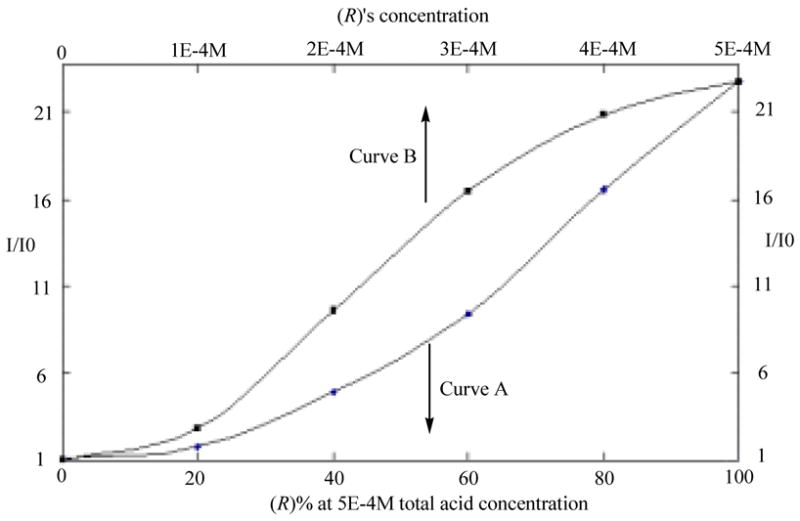
Fluorescence Enhancement of (R)-5 in the Presence of (A) Mandelic Acid at Various Enantiomeric Compositions and (B) the Optically Pure (R)-Mandelic Acid.
Earlier, we demonstrated that at 10−4 M in benzene the 1,2-diphenylethylenediamine-based bisbinaphthyl macrocycle (S)-4 showed 2 – 3 fold fluorescence enhancement at its excimer emission in the presence of (S)-mandelic acid (5.0 × 10−3 – 2.0 × 10−2 M) and no significant change in the presence of (R)-mandelic acid. The fluorescence enhancement of (S)-4 and its enantioselectivity at its monomer emission was much lower. We also examined the fluorescence response of (S)-5 toward mandelic acid at 10−4 M. When (R)-5 (1.0 × 10−4 M in benzene/0.1% DME) was treated with mandelic acid, we found that at lower concentrations of (R)-mandelic acid, (R)-5 showed fluorescence enhancements at both the excimer and monomer emissions. As the amount of (R)-mandelic acid increased, the fluorescence enhancement at the monomer emission became the dominating one. As shown in Figure 11, in the presence of 1.0 × 10−3 M of (R)-mandelic acid, the monomer emission of (R)-5 increased over 41 fold, whereas (S)-mandelic acid caused little change on the fluorescence of (R)-5. This demonstrates that the cyclohexane-1,2-diamine-based compound (S)- or (R)-5 is much more sensitive as well as enantioselective than the corresponding 1,2-diphenylethylenediamine-based compound (S)- or (R)-4.
Figure 11.

Fluorescence Enhancement of (R)-5 (1.0 × 10−4 M in benzene/0.1 % DME) in the Presence of (R)- and (S) -Mandelic Acid (λexc = 332 nm).
The fluorescence quantum yield of (R)-5 was estimated by using a quinine sulfate solution in 1 N H2SO4 (φF = 0.55) as the standard.20 The intergrations of the corrected emission spectra excited at 332 nm for quinine sulfate and the samples were obtained. Following equation was applied to calculate the quantum yield:
Wherein φF,ref, Aref, nD,ref and aref are the reference quantum yield, the reference absorbance, the refraction index of the solvent of the reference, and the integration of the area under the fluorescence signal of the reference, respectively. Compound (R)-5 (1.0 × 10−5 M in benzene) was found to have a very low fluorescence quantum yield of 0.38%. The quantum yields of compounds (S)-7 and (S)-12 were found to be 0.6% and 0.8% respectively which are greater than that of (R)-5 because of their reduced photo-induced electron transfer quenching caused by a smaller number of nitrogens. We measured the effect of mandelic acid on the florescence quantum yield of (R)-5. The quantum yield of (R)-5 (1.0 × 10−5 M) increased 14 fold to 5.3% in the presence of (R)-mandelic acid (5.0 × 10−4 M in benzene/0.05% DME), and only to 0.6% in the presence of (S)-mandelic acid under the same conditions.
b. NMR Study
The interaction of the bisbinaphthyl macrocycle with mandelic acid was studied by using the NMR spectroscopy. We found that a 1:1 mixture of (S)-5 and (S)-mandelic acid in benzene-d6/4% acetone-d6 caused a large up-field shift (Δδ = 1.0 – 1.1 ppm) for the 1H NMR signal of the α-proton of (S)-mandelic acid, that is, from δ5.20 to δ4.1 ~ 4.2. However, under the same conditions, the chirality mismatched mixture of (S)-5 and (R)-mandelic acid only led to a small up-field shift (Δδ = 0.25 ppm) for the α-proton of (R)-mandelic acid. This proton only showed 0.02 ppm up-field shift when mandelic acid was treated with dibenzylamine in the same solvent. These observations suggest that in the macrocycle-mandelic acid complex, (S)-mandelic acid is probably located much deeper inside the chiral cavity of (S)-5 which allows the α-proton of (S)-mandelic acid to be significantly shielded by the aromatic rings of the macrocycle. This could be the origin of the dramatic difference in the fluorescence responses of the (S)-5 toward the two enantiomers of mandelic acid. Unlike (S)-4, the signals of the 3,3′-methylene protons in the 1,1′-binaphthyl units of (S)-5 also changed significantly with the addition of (S)-mandelic acid, which suggests the inclusion of (S)-mandelic acid inside the cavity of (S)-5.
We conducted the NMR titration of (S)-mandelic acid with (S)-5 in benzene-d6/4% acetone-d6. The total concentration of (S)-mandelic acid+(S)-5 was maintained at 4.0 × 10−3 M. Figure 12 plots the up field shift of the signal Δδ [Δδ = δ0 − δ. δ0: the chemical shift for (S)-mandelic acid without (S)-5. δ: the chemical shift for (S)-mandelic acid with (S)-5] for the α-proton of (S)-mandelic acid against the ratio of (S)-5/(S)-mandelic acid. As this ratio approached 1:1, the chemical shift change reached saturation. The large error bars (±0.2 ppm) shown in the saturation region of the plot is because the signal of the α-proton of (S)-mandelic acid merged with those of the 3,3′-methylene protons in the 1,1′-binaphthyl units of (S)-5 at (S)-5/(S)-mandelic acid ≥ 1. This NMR titration experiment demonstrates that (S)-5 forms a 1:1 complex with (S)-mandelic acid.
Figure 12.

The Chemical Shift Change (Δδ) of (S)-Mandelic Acid versus the Ratio of (S)-5/(S)-Mandelic Acid.
On the basis of the NMR study, we calculated the association constant of the complex (S)-5+(S)-mandelic acid by using the following NMR version of the Rose-Drago equation:21
| (1) |
Wherein Δδmax is the difference in chemical shifts between that observed in the guest molecule and that observed in the host-guest complex, Δδ is the measured change in chemical shift (upon addition of host species) referenced to that of the uncomplexed guest. Ka is the association constant, [H0] is the known total concentration of host and [G0] is the known total concentration of guest. The association constant was estimated to be over 2000.
c. Fluorescent Recognition of Other Chiral Acids
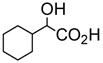
We studied the interaction of (R)-5 with other chiral acids. Figure 13 gives the fluorescence spectra of (R)-5 (1.0 × 10−5 M in benzene/0.4% DME) with/without (R)- and (S)-hexahydromandelic acid (4.0 × 10−3 M). It shows that although (S)-hexahydromandelic acid did not change the fluorescence of (R)-5 significantly, (R)-hexahydromandelic acid increased the monomer emission of (R)-5 by over 80 fold. The ef is 64. Thus, (R)-5 is highly sensitive and enantioselective toward the aliphatic α-hydroxycarboxylic acid. Figure 14 shows the fluorescence change of (R)-5 at three different concentrations (4.0 × 10−4 – 4.0 × 10−3 M) of (R)- and (S)-mandelic acid respectively.
Figure 13.
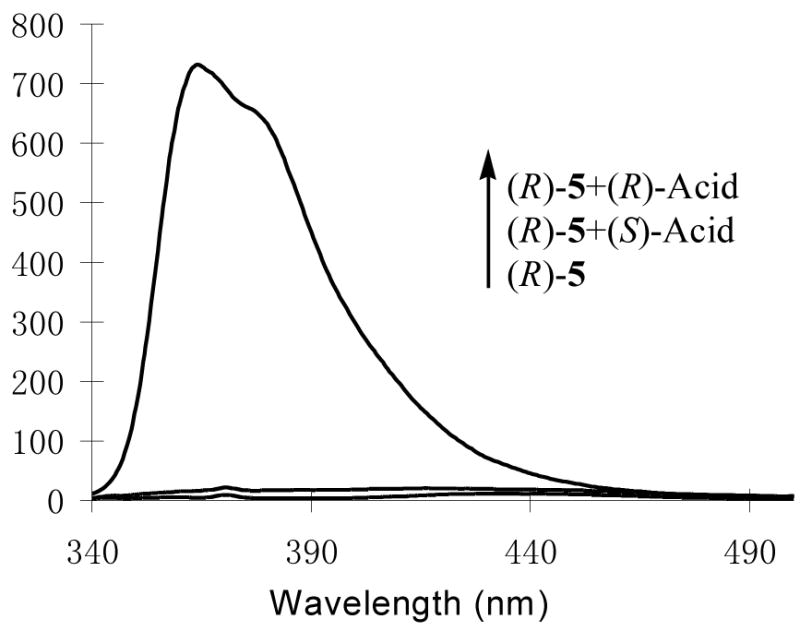
Fluorescence Spectra of (R)-5 (1.0 × 10−5 M in benzene/0.4%DME) with/without (R)- and (S)-Hexahydromandelic Acid (4.0 × 10−3 M) (λexc = 332 nm, slit = 2.5/2.5 nm).
Figure 14.
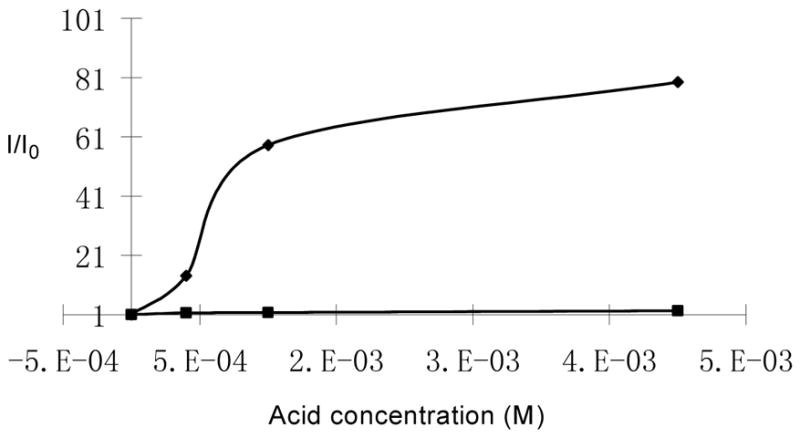
Fluorescence Enhancement of (R)-5 (1.0 × 10−5 M in benzene/0.4%DME) versus the Concentration of (R)- and (S)-Hexahydromandelic Acid (λexc = 332 nm, slit = 2.5/2.5 nm).
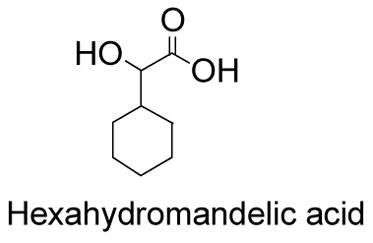
The fluorescence quantum yields of (R)-5 (1.0 × 10−5 M in benzene/0.4% DME) in the presence of (R)- and (S)-hexahydromandelic acid were found to be 12.9% and 0.9% respectively. Thus there was a large increase (34 fold) in the fluorescence quantum yield of (R)-5 when it bound with the chirality matched (R)-hexahydromandelic acid.
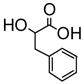
The fluorescence responses of (R)-5 (1.0 × 10−5 M in benzene/1% DME) toward additional chiral acids such as 3-phenyllactic acid, N-benzyloxycarbonylphenylglycien (Phg-boc) and N-benzyloxycarbonylphenylalanine (Phe-boc) were measured. Table 1 summarizes the fluorescence responses of (R)-5 towards various chiral acids. These results demonstrate that (S)- and (R)-5 are useful enantioselective fluorescent sensors for the recognition of both α-hydroxycarboxylic acids and N-protected α-amino acids.
Table 1.
Fluorescence Responses of (R)-5 toward Chiral Acids.
| Acid | Acid Concentration | (R)-5 Concentration | I/I0 | ef [= (IR − I0)/(IS − I0)] |
|---|---|---|---|---|
|
5.0 × 10−4 M | 1.0 × 10−5 M (benzene/0.05% DME) | 20 | 46 |
|
4.0 × 10−3 M | 1.0 × 10−5 M (benzene/0.4% DME) | 80 | 64 |
|
8.0 × 10−3 M | 1.0 × 10−5 M (benzene/1% DME) | 6 | 3 |
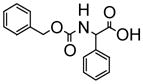
|
8.0 × 10−3 M | 1.0 × 10−5 M (benzene/1% DME) | 5 | 7 |
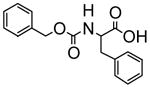
|
8.0 × 10−3 M | 1.0 × 10−5 M (benzene/1% DME) | 5 | 2 |
We also studied the interaction of (S)-5 with O-acetyl mandelic acid. Under the same conditions as the use of mandelic acid, almost no fluorescence enhancement was observed with either (R)- or (S)-O-acetyl mandelic acid. Thus, both the α-hydroxyl group and its chiral configuration are very important for the binding of the acid with the macrocyclic receptor.
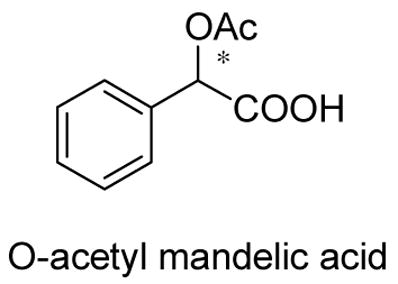
The fluorescence response of (S)-5 toward acetic acid was investigated. It was found that there was only very small fluorescence enhancement in (S)-5 (1.0 × 10−5 M in benzenen/0.05% DME) when the concentration of acetic acid increased from 1.0 × 10−4 M to 1.0 × 10−3 M (see Figure S18).
7. Interaction of (S)-7 with Mandelic Acid
The fluorescence response of the acyclic compound (S)-7 toward mandelic acid was investigated. This acyclic molecule showed very small fluorescence enhancement by either (R)- or (S)-mandelic acid with little enantioselectivity. In the presence of 5.0 × 10−4 M of (S)-mandelic acid, there was only 1.5 fold fluorescence enhancement for (S)-7(1.0 × 10−5 M in benzenen/0.05% DME) (see Figure S19).
8. Interaction of (R)-12 with Mandelic Acid
We synthesized compound (S)- or (R)-12 that contains only half of the hydrogen bond donors (-OH) and hydrogen bond acceptors (-N) of (S)-/(R)-5 but still has a macrocyclic structure. It is a cyclic analog of the acyclic compound (S)-7. This compound is used to further explore the role of each structural component of (S)-/(R)-5 in the enantioselective fluorescent recognition of mandelic acid.
Figure 15a shows the fluorescence spectra of (R)-12 (1.0 × 10−5 M in benzene/0.05% DME) in the presence of mandelic acid (8.0 × 10−5 M). Unlike (S)-5 and (S)-7, the fluorescence enhancement of (R)-12 in the presence of mandelic acid was observed predominantly at its long wavelength excimer emission. The excimer emission maximum of (R)-12 also underwent blue shift by ca 20 nm with the addition of both (R)- and (S)-mandelic acid. (R)-Mandelic acid caused a greater fluorescence enhancement than (S)-mandelic acid (ef = 2.0). Similar fluorescence responses were observed when the concentration of (R)-12 was increased to 1.0 × 10−4 M (Figure 15b) (ef = 1.7). The effect of the concentration of mandelic acid on the excimer emission of (R)-12 was investigated (see Figure S20). We also compared the fluorescence responses of (R)-12 toward mandelic acid with those of (S)-12 and observed the expected mirror image relation. These data demonstrate that the enantioselectivity and sensitivity of the macrocyclic (R)-/(S)-12 is significantly lower than that of (R)-/(S)-5 but greater than that of the acyclic (S)-/(R)-7 in the fluorescent recognition of mandelic acid.
Figure 15.

Fluorescence Spectra of (R)-12 at (a) 1.0 × 10−5 M (slit = 3.5/5.0 nm) (b) 1.0 × 10−4 M (slit = 3.5/3.5 nm) with/without (R)- and (S)-Mandelic Acid (8.0 × 10−5 M in benzene/0.05% DME) (λexc = 332 nm).
The 1H NMR spectrum of (S)-12 in the presence of (S)-mandelic acid in benzene-d6 (2% DME) was studied. It was found that the α-proton signal of (S)-mandelic acid gave a maximum of 0.025 ppm down-field shift when treated with (S)-12. This chemical shift change is not only much smaller than the >1.0 ppm up field shift observed for the a-proton signal of (S)-mandelic acid in the presence of (S)-5 but also in the opposite direction. It suggests a very different type of interaction. On the basis of the NMR study, the Job plot22 of (S)-12 with (S)-mandelic acid was obtained (Figure 16). As shown in Figure 16, there are multiple binding modes between (S)-12 and (S)-mandelic acid including the formation of a 1:2 complex. The 1:2 complex should be produced from the interaction of the two nitrogen atoms in a (S)-12 molecule with the carboxylic acid protons of two (S)-mandelic acid molecules. The multiple binding modes between (S)-12 with (S)-mandelic acid probably contribute to the significantly smaller enantioselectivity of (S)-12 than (S)-5.
Figure 16.

The Job Plot of (S)-12 with (S)-Mandelic Acid [X: mole fraction of (S)-mandelic acid. Δδ: chemical shift change of (S)-mandelic acid].
9. Interaction of (S)-14 with Mandelic Acid
When (S)-14 was treated with mandelic acid, the fluorescence enhancement was observed mainly at the monomer emission with (S)-mandelic acid causing a greater enhancement than (R)-mandelic acid. The fluorescence enhancements of (S)-14 at 10−4, 10−5 and 10−6 M respectively were studied when (S)-14 was treated with (R)- and (S)-mandelic acid (see Figure S21). Among these, the highest sensitivity and enantioselectivity were shown by the sensor at 10−4 M which gave ef up to 2.8. Thus, the introduction of the four p-ethoxyphenyl groups in (S)-14 significantly changed its fluorescent responses toward mandelic acid.
Discussion
Previously we found that the 1,2-diphenylethylenediamine-based bisbinaphthyl macrocycle (S)-4 exhibited up to 2 – 3 fold fluorescence enhancement when treated with (S)-mandelic acid. A similar magnitude of fluorescence enhancement was observed when the acyclic bisbinaphthyl sensor (S)-1 was treated with the chirality-matched mandelic acid. We attributed these fluorescence enhancements to the suppressed photo-induced electron transfer23 quenching when the nitrogen atoms of these sensors bond with the carboxylic acid proton of mandelic acid. However, this explanation cannot account for the extremely large fluorescence enhancement when the cyclohexane-1,2-diamine-based bisbinaphthyl macrocycle (S)-5 was treated with (S)-mandelic acid (over 20 fold) and (S)-hexahydromandelic acid (over 80 fold). In addition, the fluorescence enhancement of (S)-4 toward mandelic acid was mainly observed at its excimer emission, whereas that of (S)-5 were observed predominantly at its monomer emission. The big difference in the fluorescence responses of (S)-5 and (S)-4 toward mandelic acid indicates there should be an additional and important fluorescence enhancement mechanism for the interaction of (S)-5 with (S)-mandelic acid besides the suppressed photo-induced electron transfer process.
In order to understand the difference between the fluorescent recognition properties of the two macrocycles, their structures are compared. Figure 17 gives the space filling models for the X-ray structures of (S)-4 and (R)-5. In both macrocycles, there are two parallel naphthalene rings across the chiral cavity with 7 ~ 8 Å apart. However, there is one major difference between the structures of (R)-5 and (S)-4. The distance between the two oxygen atoms of the two unparallel naphthol rings in (S)-4 is 4.63, much longer than that in (R)-5 (2.80 Å). Thus, the structure of (S)-4 is like a cylinder through which a small molecule could pass. However, the structure of (R)-5 is more like a bucket with the two parallel naphthol moieties and the atoms linking them together forming its wall and the two oxygen atoms of the two unparallel naphthols that are close to each other forming its bottom. Small molecules could be included inside the bucket but could not pass through it. This structural difference between the two macrocycles might have contributed to their very different fluorescence responses to the enantiomers of mandelic acid.
Figure 17.
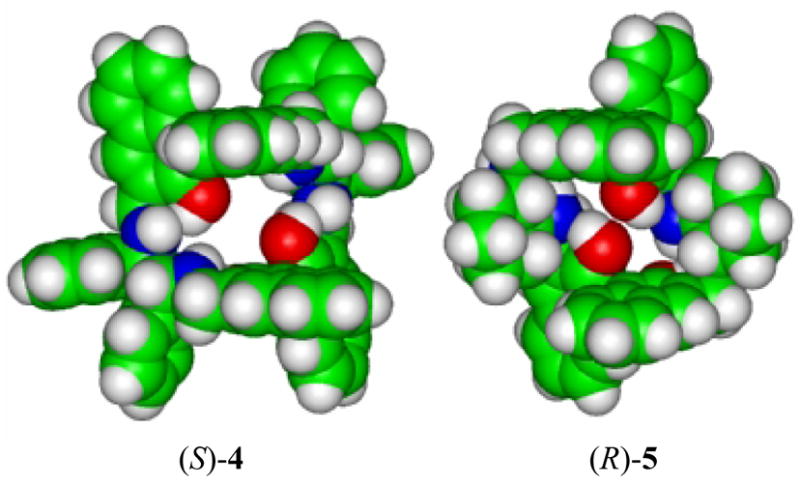
The Space-Filling Models for the X-Ray Structures of (S)-4 and (R)-5.
On the basis of the X-ray structure of (R)-5, we propose that (R)-5 could include a molecule of (R)-mandelic acid inside its cavity. The phenyl ring of (R)-mandelic acid could be sandwiched in between the two parallel naphthalene rings of the macrocycle while the hydroxyl and carboxylic acid groups of (R)-mandelic acid are forming multiple hydrogen bonds with the hydroxyl and amine groups of (R)-5. In this complex, the α-hydrogen of (R)-mandelic acid will be shielded by the aromatic current of one of the naphthalene rings, contributing to its large up-field shift in its 1H NMR signal. Inclusion of (R)-mandelic acid inside the cavity of (R)-5 should greatly rigidify the structure of the macrocycle and contribute to the observed large fluorescence enhancement.
The NMR titration experiment of (S)-mandelic acid with (S)-5 supports the formation of a 1:1 complex (see Figure 12). However, the fluorescence experiment shows that there is continuous enhancement in the fluorescence of (S)-5 as the concentration of (S)-mandelic acid increased to over 60 fold of (S)-5 (see Figures 8, 9 and 11). We propose the following explanation to account for the NMR and fluorescence experiments. After the formation of the 1:1 complex between (S)-mandelic acid and (S)-5 [or (R)-mandelic acid and (R)-5], additional (S)-mandelic acid located outside the cavity of (S)-5 can interact with the complex through multiple hydrogen bonds between the carboxylic acid proton and the nitrogen atoms of the macrocycle. This interaction is weaker and increases with the increase of the acid concentration. It does not cause much shift on the 1H NMR signal of (S)-mandelic acid, but it should suppress the photo-induced electron transfer quenching of the 1:1 complex by the nitrogen atoms and turn on the inherently high fluorescence of this structurally rigid macrocycle. Therefore, both the formation of a structurally rigid 1:1 complex through the host-guest inclusion inside the chiral cavity and the subsequent hydrogen bond interactions outside the cavity are important for the observed large fluorescence enhancement. The chirality mismatched (R)-mandelic acid cannot be included inside the cavity of (S)-5 which explains the very small changes in both the 1H NMR signal of (R)-mandelic acid and the fluorescence of (S)-5.
Compound (S)-4 cannot form a structurally rigid 1:1 complex with (S)-mandelic acid through inclusion and thus exhibits much smaller fluorescence enhancement than the interaction of (S)-5 with (S)-mandelic acid. The very low fluorescence enhancement of (S)-5 in the presence of the small acetic acid molecule also demonstrates that the interaction of the nitrogen atoms of the sensor with the carboxylic acid protons to suppress the photo-induced electron transfer process without formation of a structurally rigid complex cannot enhance the fluorescence significantly.
Figure 10 shows that the fluorescence enhancement of (R)-5 by the optically pure (R)-mandelic acid is greater than that by the enantiomeric mixture containing both (R)- and (S)-mandelic acid even though the mixture contains the same amount of (R)-mandelic acid and an additional amount of (S)-mandelic acid. There are two possible explanations for the effect of the enantiomeric mixture. (a) Mandelic acid could exist in equilibrium between the monomer and the intermolecularly hydrogen bonded dimers in the solution, and the dimers may not be able to enhance the fluorescence of the sensor. If the heterodimer (R-S) were more stable than the homodimer (R-R), the enantiomeric mixture would have a reduced effective concentration of (R)-mandelic acid and give the reduced fluorescence enhancement of (R)-5. (b) Even though (S)-mandelic acid cannot form a rigid 1:1 inclusion complex with (R)-5, it could still be in competition with (R)-mandelic acid in the binding with the nitrogen atoms of (R)-5 and reduce the effective concentration of (R)-mandelic acid. The binding of (S)-mandelic acid with (R)-5 should be much weaker than that of (R)-mandelic acid, and the racemic mixture of mandelic acid still give large fluorescence enhancement. For samples containing < 20% (R)-mandelic acid, the fluorescence enhancement of (R)-5 was small because of the large amount of (S)-mandelic acid (>80%). However, this sample could be analyzed by using the enantiomeric sensor (S)-5 which should show large fluorescence enhancement. Thus, using both enantiomers of the sensor and measuring the difference between their fluorescence responses toward the substrate under the same condition will allow the determination of any enantiomeric composition of the chiral acid.
Previously, we reported the synthesis and study of the 1,2-diphenylethylenediamine-based bisbinaphthyl macrocycle (S)-16.16 The fluorescence responses of (S)-16 toward mandelic acid are very similar to those of (S)-12, which is in sharp contrast to the large differences observed for
(S)-4 versus (S)-5. Both (S)-16 and (S)-12 showed fluorescence enhancement at the excimer emission with about 20 nm blue shift when treated with mandelic acid. Under the same conditions, their enantioselectivities were also similar with ef approaching 2 – 4. These observations suggest that these two macrocycles should bind mandelic acid in a very similar way. The NMR study of (S)-12 indicates that it does not form an 1:1 complex with mandelic acid like (R)-5 with (R)-mandelic acid. Figure 18 gives a molecular modeling structure of (S)-12 obtained by using the PC-Spartan-Pro Semi-Emperical-AM1 program. It shows that all the four naphthyl rings in (S)-12 are pointing toward the opposite directions and are far way from each other. Unlike (R)-5, there are no parallel naphthalene rings in (S)-12 to form a sandwich complex with the benzene ring of mandelic acid. This could explain the much lower enantioselectivity and fluorescence enhancement of (S)-12 than those of (S)-5. The acyclic compound (S)-7 gives even lower enantioselectivity and fluorescence response in the recognition of mandelic acid because of its flexible and undefined binding sites.
Figure 18.
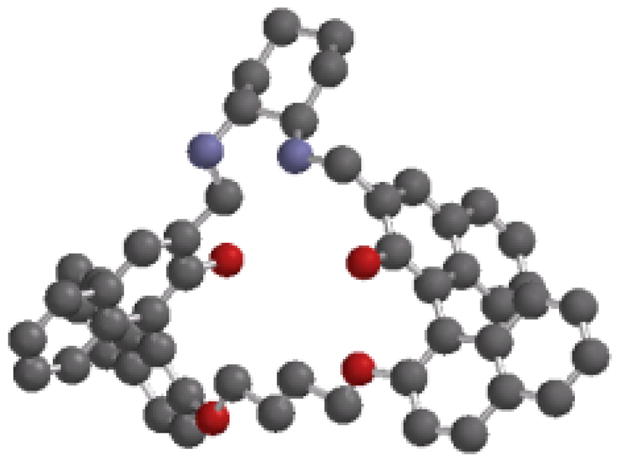
Molecular Modeling Structure of (S)-12 (The hydrogen atoms are omitted for clarity).
The fluorescence intensity of p-ethoxyphenyl substituted macrocycle (S)-14 is similar to that of the unsubstituted macrocycle (S)-5 when excited at 332 nm. However, when (S)-14 is excited at 305 nm, it exhibits greatly increased fluorescence intensity over (S)-5 because of the much greater absorption of (S)-14 at 305 nm (see supporting information). This is similar to what was observed for compound (S)-17.11b Both (S)-14 and (S)-17 show enhancement at their monomer emission with similar enantioselectivity (ef ~ 2) when treated with mandelic acid. The introduction of the four p-ethoxyphenyl group to (S)-5 to make (S)-14 must have significantly disturbed its interaction with mandelic acid and reduced its enantioselectivity in the recognition of mandelic acid.

Summary
We have synthesized the cyclohexane-1,2-diamine-based bisbinaphthyl macrocycles (S)-(R)-5 and their cyclic and acyclic analogs. Highly enantioselective fluorescence responses and high fluorescent sensitivity have been observed when (S)-/(R)-5 are interacted with α-hydroxycarboxylic acids and N-protected amino acids. The mechanism for the enantioselective fluorescent recognition has been explored by comparing (S)-/(R)-5 with several of its analogs. A two stage recognition process is proposed which involves the formation of a structurally rigidified host-guest complex between (S)-5 and a chirality-matched α-hydroxycarboxylic acid and the further interaction of this complex with the acid to suppress the photo-induced electron transfer fluorescence quenching caused by the nitrogens in (S)-5.
Experimental Section
Preparation and Characterization of (S)- and (R)-5
Under nitrogen, (S)-2,2′-dihydroxy-[1,1′]binaphthalenyl-3,3′-dicarbaldehyde (274 mg, 0.80 mmol) and (R,R)-cyclohexane-1,2-diamine [93 mg, 0.80 mmol) were dissolved in dry methylene chloride. The mixture was stirred at room temperature for 2 d. After evaporation of the solvent, the resulting macrocyclic Schiff base was purified by passing through a short alumina column eluted with methylene chloride. This compound was then combined with NaBH4 (88 mg, 2.32 mmol) and ethanol (25 mL). The mixture was degassed by bubbling nitrogen through and then heated at reflux for 4 h. After the reaction was completed, the solvent was removed and the residue was combined with methylene chloride (10 mL) and HCl (aq, 0.4 N, 30 mL). The organic layer was separated from the gel like water layer in a separation funnel and discarded. The water layer was neutralized with sodium bicarbonate and extracted with methylene choride (3 × 20 mL). Removal of the solvent gave pure (S)-5 as a white solidin 55% yield (170 mg). 1H NMR (acetone-d6, 300 MHz) δ0.86–1.16 (m, 8H), 1.48–1.60 (m, 4H), 2.01–2.18 (m, 4H), 2.18–2.32 (m, 4H), 4.27 and 4.43 (AB, 8H, J = 14.4 Hz), 7.08–7.28 (m, 12H), 7.67 (s, 4H), 7.76–7.83 (m, 4H). 13C NMR (acetone-d6, 75 MHz) δ 24.9, 32.3, 51.6, 61.6, 117.4, 122.7, 124.8, 125.5, 126.9, 127.4, 127.9, 128.5, 134.5, 154.8. m.p. >230 °C (dec.). [α]D = −86.3 (c = 0.22, C6H6). HRMS (MALDI) Calcd for C56H57N4O4 (MH+): 849.4374; Found: 849.4382. Anal. Calcd for C56H56N4O4: C, 79.22; H, 6.65; N, 6.60. Found: C, 79.18; H, 6.77; N, 6.45. The enantiomer (R)-5 was obtained in the same way by using the (R)-1,1′-binaphthyl starting material and (S,S)-cyclohexane-1,2-diamine.
Preparation and Characterization of (S)-7
By using a procedure similar to the preparation of (S)-5, a 2:1 mixture of (S)-6 and (R,R)-cyclohexane-1,2-diamine was converted to (S)-7 in 78% yield. Instead of using the acidify-extraction method as described for (S)-5, (S)-7 was purified by column chromatograph on silica gel eluted with 5% acetone in methylene chloride. 1H NMR (CDCl3, 300 MHz) δ1.05–1.30 (m, 4H), 1.65–1.77 (m, 2H), 2.22–2.41 (m, 4H), 4.10 and 4.35 (AB, J = 15.0 Hz, 4H), 5.30 (br, 6H), 6.68–6.71 (m, 2H), 7.04–7.19 (m, 6H), 7.23–7.36 (m, 6H), 7.61–7.80 (m, 8H). 13C NMR (CDCl3, 75 MHz) δ24.7, 31.8, 50.4, 60.5, 113.7, 114.7, 117.8, 123.4, 123.9, 124.8, 125.0, 125.8, 126.5, 127.1, 128.2, 128.4, 128.8, 129.1, 129.4, 129.9, 134.0, 134.2, 151.5, 154.6. m.p. 169–173 °C. [α]D = +85.7 (c = 0.22, C6H6). Mass analysis (FIA_ESI). Calcd. for [C48H43N2O4]+: 711.3. Found: 711.3. Anal. Calcd for C48H42N2O4: C, 81.10; H, 5.96; N, 3.94. Found: C, 81.03; H, 5.87; N, 3.86.
Preparation and Characterization of (S)- and (R)-12
The dialdehyde (S)-11 (100 mg, 0.15 mmol) and (R,R)-cyclohexane-1,2-diamine (0.072 mmol) were dissolved in CH2Cl2 (10 mL) and stirred at room temperature for 2 d. After the solvent was removed, the crude product was purified by passing through a short silica gel column eluted with methylene chloride to give the macrocyclic Schiff base as a yellow solid. The Schiff base was then dissolved in ethanol (25 mL), and combined with NaBH4 (25 mg, 0.67 mmol). The resulting reaction mixture was heated at reflux for 4 h under nitrogen to form a clear colorless solution. After removal of the solvent, methylene chloride (30 mL) and HCl (0.2 N, aq, 30 mL) were added and the solution was stirred for 2 h. NaHCO3 (saturated aq.) was used to adjust the pH to 8. The organic layer was separated and washed with water and brine. The solution was dried over MgSO4 and then passed through a short silica gel column eluted with CH2Cl2/acetone (20:1). After removal of the solvent, (S)-12 was obtained as a white solid in 54% overall yield. For (S)-12: 1H NMR (CDCl3, 300 MHz ) d 7.88-7.84 (m, 4H), 7.68-7.65 (m, 2H), 7.50 (s, 2H), 7.34-7.12 (m, 12H), 7.04-7.01 (m, 2H), 4.25 (d, J = 13.5 Hz, 2H), 4.03 (d, J = 13.5 Hz, 2H), 3.71-3.61 (m, 4H), 2.46-2.32 (m, 2H), 2.20-2.06 (m, 2H), 1.62–1.74 (m, 2H) 1.32–1.16 (m, 8H). 13C NMR (CDCl3, 75 MHz) δ. 154.9, 153.4, 134.3, 134.2, 129.8, 128.4, 128.3, 128.0, 127.5, 126.6, 126.2, 126.1, 125.6, 125.1, 123.9, 123.1, 117.3, 116.7, 70.3, 62.0, 51.5, 31.8, 26.3, 24.9. m.p. 199–202 °C. [α]D = −98.1 (c = 0.50, CH2Cl2). MS. Calcd for C52H49N2O4 (MH+): 765.4. Found: 765.4. The enantiomer (R)-12 was obtained by using (R)-11 and (S,S)-cyclohexane-1,2-diamine.
Preparation and Characterization of (S)-14
By using a procedure similar to the preparation of (S)-5, a 1:1 mixture of (S)-13 and (R,R)-cyclohexane-1,2-diamine was converted to (S)-14 in 53% yield. Instead of using the acidification-extraction method described for (S)-5, (S)-14 was purified by column chromatograph on silica gel eluted with 50% acetone in methylene chloride. 1H NMR (acetone-d6, 300 MHz) d 7.97 (d, J = 1.5 Hz, 4H), 7.70 (s, 4H), 7.63 (d, J = 8.7 Hz, 8H), 7.46 (dd, J1 = 1.5 Hz, J2 = 8.7 Hz, 4H), 7.17 (d, J = 8.7 Hz, 4H), 7.00 (d, J = 8.7 Hz, 8H), 4.42 (d, J = 14.1 Hz, 4H), 4.25 (d, J = 14.1 Hz, 4H), 4.08 (quartet, J = 6.9 Hz, 8H), 2.38-2.26 (m, 4H), 2.26-2.14 (m, 4H), 1.64-1.54 (m, 4H) 1.39 (t, J = 6.9 Hz, 12H), 1.18–0.96 (m, 8H). 13C NMR (acetone-d6, 75 MHz) δ 158.6, 154.7, 134.9, 133.7, 133.2, 128.8, 128.0, 127.7, 127.2, 125.4, 124.9, 124.7, 117.1, 114.9, 63.4, 61.4, 51.5, 32.3, 24.9, 14.9. m.p. >240 °C (dec.). [α]D = −21.9 (c = 0.33, C6H6). MS. Calcd for C88H89N4O8 (MH+): 1329.67. Found: 1329.85.
Preparation of Samples for Fluorescence Measurements
Materials: The sensors were purified by column chromatography and then stored in refrigerator. The enantiomers of mandelic acid were purchased from Aldrich and recrystallized from methanol. All other acids were used directly. All of the solvents were HPLC grade. The benzene stock solutions of the sensors were freshly prepared for each measurement. A 0.001 M stock solution of mandelic acid was freshly prepared using benzene containing 0.1% DME. DME was added to improve the solubility of the acid. For the fluorescence enhancement study, a sensor solution was mixed with the mandelic acid solution at room temperature in a 5 mL volumetric flask and diluted to the desired concentration. The resulting solution was allowed to stand at room temperature for 2 – 4 h before the fluorescence measurement.
Supplementary Material
Details for the NMR experiments and the calculation of the association constant. Additional spectroscopic plots and data. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 1.

Synthesis of the Bisbinaphthyl Macrocycle (S)-5.
Acknowledgments
We are very grateful for the support of this work from the US National Institute of Health (R01GM58454/R01EB002037-05). We also thank Dr. Jeff Ellena at the University of Virginia for assistance on the NMR study.
References
- 1.Czarnik AW, editor. Fluorescent Chemosensors for Ion and Molecular Recognition. ACS Symposium Series 538; American Chemical Society; Washington, DC: 1993. [Google Scholar]
- 2.de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Chem Rev. 1997;97:1515. doi: 10.1021/cr960386p. [DOI] [PubMed] [Google Scholar]
- 3.Fabbrizzi L, Poggi A. Chem Soc Rev. 1995;24:197. [Google Scholar]
- 4.Mason WT, editor. Fluorescent and Luminescent Probes. 2. Academic; San Diego: 1999. [Google Scholar]
- 5.Pu L. Chem Rev. 2004;104:1687–1716. doi: 10.1021/cr030052h. [DOI] [PubMed] [Google Scholar]
- 6.Selected recent examples: Zhao JZ, Davidson MG, Mahon MF, Kociok-Kohn G, James TD. J Am Chem Soc. 2004;126:16179–16186. doi: 10.1021/ja046289s.Xu YF, McCarroll ME. J Am Chem Soc. 2004;108:6929–6932.Mei X, Wolf C. J Am Chem Soc. 2004;126:14736–14737. doi: 10.1021/ja0459781.
- 7.(a) Finn MG. Chirality. 2002;14:534–540. doi: 10.1002/chir.10101. [DOI] [PubMed] [Google Scholar]; (b) Reetz MT. Angew Chem, Int Ed. 2002;41:1335–1338. doi: 10.1002/1521-3773(20020415)41:8<1335::aid-anie1335>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 8.Tsukamoto M, Kagan HB. Adv Synth Catal. 2002;344:453–463. [Google Scholar]
- 9.(a) Coppola GM, Schuster HF. α-Hydroxyl Acids in Enantioselective Synthesis. VCH; Weinheim: 1997. [Google Scholar]; (b) Hanessian S. Total Synthesis of Natural Products: The Chiron Approach. Pergamon; Oxford: 1983. [Google Scholar]
- 10.(a) Lin J, Hu Q-S, Xu MH, Pu L. J Am Chem Soc. 2002;124:2088–2089. doi: 10.1021/ja011971x. [DOI] [PubMed] [Google Scholar]; (b) Xu M-H, Lin J, Hu Q-S, Pu L. J Am Chem Soc. 2002;124:14239–14246. doi: 10.1021/ja020989k. [DOI] [PubMed] [Google Scholar]
- 11.(a) Lin J, Zhang H-C, Pu L. Org Lett. 2002;4:3297–3300. doi: 10.1021/ol026565c. [DOI] [PubMed] [Google Scholar]; (b) Li Z-B, Lin J, Zhang H-C, Sabat M, Hyacinth M, Pu L. J Org Chem. 2004;69:6284–6293. doi: 10.1021/jo049366a. [DOI] [PubMed] [Google Scholar]
- 12.The preliminary study on (S)-5 was communicated. Li Z-B, Lin J, Pu L. Angew Chem Int Ed. 2005;44:1690–1693. doi: 10.1002/anie.200462471.
- 13.(a) Brunner H, Schiessling H. Angew Chem Int Ed Engl. 1994;33:125–126. [Google Scholar]; (b) Brunner H, Schiessling H. Bull Soc Chim Belg. 1994;103:119–126. [Google Scholar]
- 14.Cox PJ, Wang W, Snieckus V. Tetrahedron Lett. 1992;33:2253–2256. [Google Scholar]
- 15.(a) Matsunaga S, Das J, Roels J, Vogl EM, Yamamoto N, Iida T, Yamaguchi K, Shibasaki M. J Am Chem Soc. 2000;122:2252–2260. [Google Scholar]; (b) DiMauro EF, Kozlowski MC. Org Lett. 2001;3:1641–1644. doi: 10.1021/ol0158213. [DOI] [PubMed] [Google Scholar]
- 16.Li Z-B, Pu L. J Mater Chem. 2005;15:2860–2864. [Google Scholar]
- 17.Pu L. Chem Rev. 1998;98:2405–2494. doi: 10.1021/cr970463w. [DOI] [PubMed] [Google Scholar]
- 18.(a) Gottarelli G, Spada GP, Bartsch R, Solladié G, Zimmermann R. J Org Chem. 1986;51:589–592. [Google Scholar]; (b) Mason SF, Seal RH, Roberts DR. Tetrahedron. 1974;30:1671–1682. [Google Scholar]
- 19.Wyatt SR, Hu Q-S, Yan X-L, Bare W, Pu L. Macromolecules. 2001;34:7983–7988. [Google Scholar]
- 20.(a) Demas JN, Crosby J. J Phys Chem. 1971;75:991. [Google Scholar]; (b) Isak SJ, Eyring EM. J Phys Chem. 1998;10:286. [Google Scholar]; (c) Kamat PV, Ford WE. Chem Phys Lett. 1987;135:421. [Google Scholar]
- 21.(a) Fielding L. Tetrahedron. 2000;56:6151–6170. [Google Scholar]; (b) Rose NJ, Drago RS. J Am Chem Soc. 1959;81:6138–6141. [Google Scholar]; (c) Wachter HN, Fried V. J Chem Ed. 1974;51:798–799. [Google Scholar]
- 22.(a) Blanda MT, Horner JH, Newcomb M. J Org Chem. 1989;54:4626–4636. [Google Scholar]; (b) Connors KA. Binding Constants, The Measurement of Molecular Complex Stability. Wiley-Interscience; New York: 1987. pp. 24–28. [Google Scholar]
- 23.(a) Fox MAM, Chanon M, editors. Photoinduced Electron Transfer Parts A–D. Elsevier; Amsterdam: 1988. [Google Scholar]; (b) Bissell RA, de Silva AP, Gunaratna HQN, Lynch PLM, Maguire GEM, McCoy CP, Sandanayake KRAS. Top Current Chem. 1993;168:223–264. [Google Scholar]; (c) Bissell RA, de Silva AP, Gunaratna HQN, Lynch PLM, Maguire GEM, Sandanayake KRAS. Chem Soc Rev. 1992;21:187–195. [Google Scholar]; (d) Czarnik AW. Acc Chem Res. 1994;27:302–308. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details for the NMR experiments and the calculation of the association constant. Additional spectroscopic plots and data. This material is available free of charge via the Internet at http://pubs.acs.org.