

Abstract
Amyloid beta (Aβ), a peptide family produced and deposited in neurons and endothelial cells (EC), is found at subnanomolar concentrations in the plasma of healthy individuals. Simple conformational changes produce a form of Aβ Aβ42, which creates toxic plaque in the brains of Alzheimer’s patients. Oxidative stress induced blood brain barrier degeneration has been proposed as a key factor for Aβ42 toxicity, but cannot account for lack of injury from the same peptide in healthy tissues. We hypothesized that cell state mediates Aβ effect. Thus, we examined the viability of aortic EC, vascular smooth muscle cells (SMC) and epithelial cells (EPI) in different states in the presence of Aβ secreted from transfected Chinese hamster ovary cells (CHO). Aβ was more toxic to all cell types when they were subconfluent. Subconfluent EC sprouted and SMC and EPI were inhibited by Aβ. Confluent EC were virtually resistant to Aβ and suppressed Aβ production by Aβ+CHO. Products of subconfluent EC overcame this resistant state, stimulating the production and toxicity of Aβ42. Confluent EC overgrew ~35% beyond their quiescent state in the presence of Aβ conditioned in media from subconfluent EC. These findings imply that Aβ42 may well be even more cytotoxic to cells in injured or growth states and potentially explain the variable and potent effects of this protein. One may now need to consider tissue and cell state in addition to local concentration of and exposure duration to Aβ. The specific interactions of Aβ and EC in a state-dependent fashion may help understand further the common and divergent forms of vascular and cerebral toxicity of Aβ and the spectrum of AD.
Keywords: Amyloid beta, Endothelium repair, Alzheimer’s, Cerebral amyloid angiopathy
Introduction
The discovery of amyloid deposits in brains of patients with Alzheimer’s disease (AD) provides an attractive mechanistic link between aberrant amyloid beta protein (Aβ) accumulation and local tissue toxicity. Aβ is formed from the cleavage of the amyloid precursor protein into forms of 39 to 42 residues [10]. Two distinct forms of Aβ are produced in neurons; Aβ40 is generated in the trans-Golgi network, and Aβ42 in the endoplasmic reticulum [16]. Aβ polymerizes into fibrils of many different distinct intermediates, but it is the nonfibrillar small soluble oligomers that most toxic [10][19]. Aβ inhibits synaptic AMPA receptors [21] and can induce cerebral amyloid angiopathy (CAA) characterized by Aβ plaque accumulation in the wall of cerebral or leptomeningeal blood vessels [24]. Aβ42 inhibits specific receptors which may play a critical role in hippocampal memory encoding, while no such effects were observed from Aβ40 [21]. Aβ42 is more toxic to rat neuromicrovascular endothelial cells than Aβ40 [11]. Indeed, Aβ40 can even enhance coronary endothelial cell survival in low (nanomolar) concentrations; only becoming toxic in the micromolar range [5]. Yet, it remains unclear at what stage Aβ becomes toxic, whether vascular toxicity predates or follows neurotoxicity, and if Aβ burden correlates with cerebral and vascular disease. The extent of Aβ42 capillary deposits correlate with the presence of neuritic AD [1] but senile plaque abundance does not determine the degree of dementia exhibited by AD patients [25]. Moreover, the oxidative stress-induced blood brain barrier degeneration proposed as a key factor for Aβ42 toxicity cannot account for lack of injury from the same peptide in healthy tissues [15] and therapeutic targeting of amyloid alone may not be sufficient to improve functional deficits over the course of the disease [23].
Previous in vitro studies examined the effects of synthetic peptides as primary cells, both neuronal and non-neuronal, secrete an insufficient amount of Aβ to perform mechanistic experiments. More recently, Chinese hamster ovary cells were stably transfected to overexpress Aβ (Aβ+CHO) such that Aβ oligomers were detectable in the conditioned media (CM) [27]. We hypothesized that tissue state determines amyloid effect, postulating that health tissues may be more resistant to Aβ and diseased tissues more susceptible. In particular, we sought to determine if endothelial integrity affects the vascular consequences of Aβ.
Materials and Methods
Cell culture
Bovine aortic endothelial cells (EC) and Chinese hamster ovary cells were from Promocell (Germany) and Gibco (US), respectively. Rat lung epithelial cells (EPI) were from ATCC (VA) and bovine aortic smooth muscle cells (SMC) were isolated from calf aortas. Amyloid-β (1–42)-secreting Chinese hamster ovary cells (Aβ+CHO) were kindly provided by Prof. Dennis J. Selkoe (Center for Neurologic Diseases, Brigham and Women’s Hospital, Boston, MA). Aβ+CHO secretion of Aβ (Fig 3A) and subsequent Aβ fibril formation (Fig 3B) peak at 48 hours and falls over thereafter. All cells were cultured in DMEM (Gibco, NY) supplemented with 10% fetal bovine serum (HyClone, UT), 200µg/ml of G418 (Cambrex, US), 35 µM streptomycin sulfate, 50 U/ml penicillin and 2 mM glutamine (Gibco) in a 10% CO2 humidified incubator.
Proliferation studies
Proliferation studies were performed using Transwell polystyrene plates (Costar, US) with 0.4µm-pore size polyester membrane. Aβ+CHO were seeded on the membranes at 1.4*E05cells/insert while EC, SMC and EPI were seeded at 10*E04 cells/plate on the bottom. 24h after seeding, inserts were placed inside corresponding plates. In studies done on confluent EC, SMC and EPI, cells were allowed to proliferate and reach confluence for 7 days prior to incorporation of the insert containing Aβ+CHO. Cells were kept in co-culture for 1, 2 and 5 days. At these time points, cell detachment was performed by trypsinization and cell numbers measured using a Beckman Coulter counter.
Collection of conditioned media (CM)
CM was collected for fibril formation and proliferation studies. For the former, media was conditioned with Aβ+CHO and collected at 6, 12, 24, 48 and 120h of culture. For the latter, media was conditioned with Aβ+CHO in co-culture with EC, and with Aβ−CHO in co-culture with EC as control. Additional controls included individual cultures of EC and Aβ−CHO. CM was collected at 24, 48, and 120h of culture, centrifuged and added to a confluent layer of EC. After 5 days, EC were detached and cell number determined. Immediately after collection, CM was filtered using a 30,000 NMWL eppendorf filter unit to separate fibrillar from non-fibrillar fractions. Both initially collected samples (total Aβ42) and filtrate (non-fibrillar Aβ42) were quantified using a commercially available ELISA kit (Biosource, US).
Analysis of fibril formation
Stock solutions of synthetic Aβ42 (Keck Facility, Yale University) were prepared in autoclaved water with addition of 1N NaOH to pH11 and kept on ice. Seed-free Aβ42 was prepared as described by Fezoui et al. [9] for Aβ40. The solution was filtered through a 30,000 NMWL Eppendorf filter unit at 10,000 rpm and 4°C for 10min. Control samples were prepared by diluting the Aβ42 stock solution to 10 µM in Tyrode’s/2mMCa buffer. Fibrils in CM were measured directly after collection or after incubations of 4, 12, 24 and 48h. Thioflavin T (Sigma) was then added to each CM sample for a final concentration of 10µM. 100µl-samples were aliquotted in quadruplicate in black-black 96-well plate and fluorescence measured at Ex440 Em480 in a FLUOstar OPTIMA (BMG Labtechnologies) plate reader.
Results
Aβ42 was cytotoxic to vascular smooth muscle cells (SMC) and lung epithelial cells (EPI) as their expected proliferation 5 days after culturing was suppressed (Figure 1). This reduction in cell number was more profound when cells were subconfluent (33.7±2.6% and 35.1±1.4 for EPI and SMC, respectively p<0.05), a finding all the more remarkable as at growth should have been even greater at these sparse cell densities. The cell density-dependent response to Aβ was especially pronounced in vascular EC. While confluent EC monolayers remained undisturbed and quiescent, indistinct from controls (Figure 1) sub-confluent EC lost their normal control mechanisms and overgrew and sprouted in an aberrant fashion (22.4± 5%, p <0.005, Figure 1).
Figure 1.

Aβ42 was cytotoxic to vascular smooth muscle cells (SMC) and lung epithelial cells (EPI). This reduction in cell number was more profound when cells were subconfluent (33.7±2.6% and 35.1±1.4 for EPI and SMC, respectively p<0.05). While confluent EC monolayers remained undisturbed and quiescent, indistinct from controls, sub-confluent EC lost their normal control mechanisms and overgrew and sprouted in an aberrant fashion (22.4± 5%, p <0.005).
Subconfluent and proliferating cells of all types interact with the Aβ protein, and EC specifically regulate Aβ secretion by CHO cells. Media conditioned from EC at any density had no added effect on EC growth in the absence of Aβ42. Confluent EC remained quiescent when exposed to Aβ+CHO conditioned media (Fig 2) or if Aβ+CHO secreted their products in the presence of confluent EC. In any of these above conditions cell sprouting was not observed and in the latter Aβ secretion by Aβ+CHO was even suppressed (Fig 3A). Subconfluent EC had the reverse effect. Whereas media conditioned in the presence of confluent EC suppressed Aβ, protein secretion was maintained at expected elevated levels in the presence of subconfluent EC (Fig 3A). While Aβ alone had no effects on EC, the very same Aβ concentration incubated with subconfluent EC was cytotoxic. Fibrillar and non-fibrillar Aβ protein levels were identical in media conditioned from Aβ+CHO alone or with subconfluent EC and yet only the latter was toxic to EC. EC and Aβ secreting CHO cells induced EC sprouting (Fig 2A, 33±5 %, p<0.005). The combined media induced marked morphological changes classic for overgrowth and disorganized sprouting in confluent EC after 5-day incubation (Fig 2B). The already significant cytotoxic effects of Aβ42 on SMC were not further enhanced by added effects of EC CM.
Figure 2.
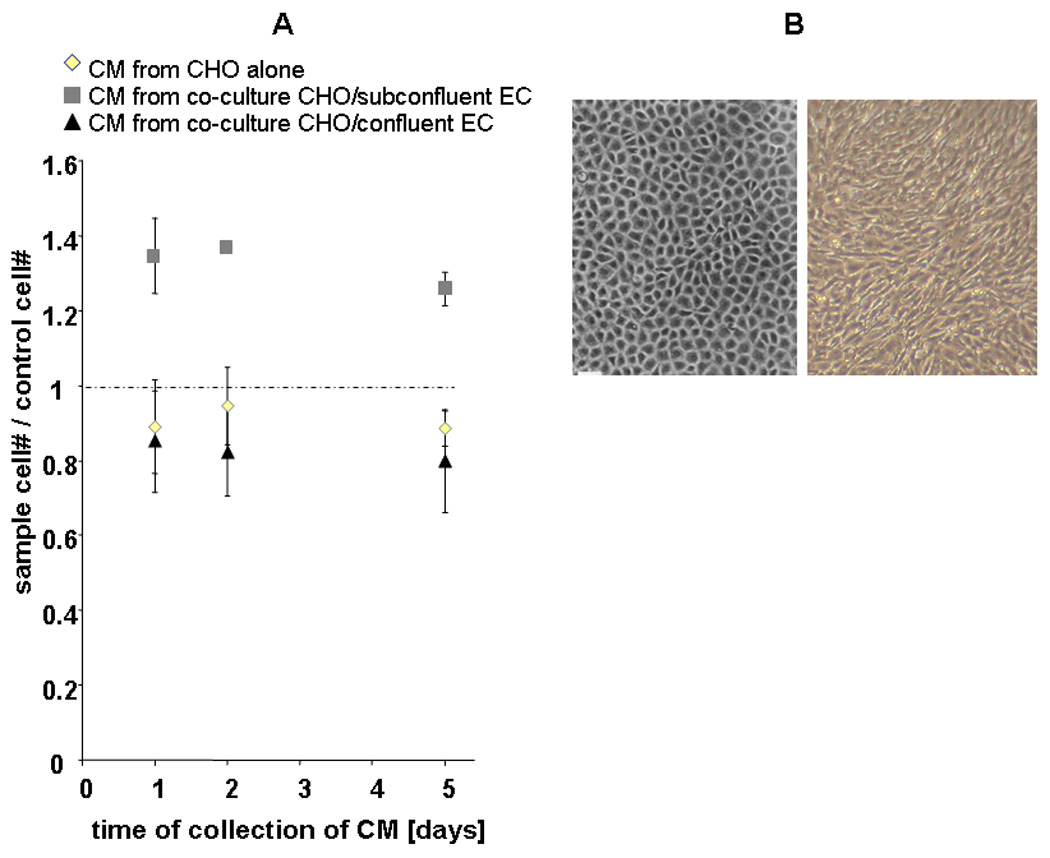
(A) Effect of conditioned media (CM) obtained from Aβ42 secreting cells (Aβ+CHO) alone, or from co-cultures with confluent or sub-confluent EC on the proliferation of confluent EC. Cells were counted after 5 days of incubation with CM. Independent of the collection day, CM from Aβ+CHO/subconfluent EC had a stimulatory effect on the confluent EC (p<0.005 at 24h of CM incubation, p<0.001 at 48h of CM incubation, and p<0.01 at 120h of CM incubation, all compared to Aβ+CHO). No such effect was observed from CM obtained from Aβ+CHO alone, Aβ+CHO /confluent EC, or from Aβ−CHO controls. (B) Morphological changes classic for overgrowth and disorganized sprouting in confluent EC after 5-day incubation (right) and control confluent EC monolayer (left).
Figure 3.

(A) The concentration of Aβ42 in the CM from cultures of Aβ+CHO alone, Aβ+CHO/sub-confluent EC, and Aβ+CHO /confluent EC was determined after 24h, 48h, and 120h of culture. The concentration of Aβ42 was largest in cultures of Aβ+CHO alone and Aβ+CHO/sub-confluent EC at 48h, and much less in the culture of Aβ+CHO/confluent EC at 48h (p<0.005 compared to Aβ+CHO/sub-confluent EC). (B) CM from Aβ42 secreting cells (Aβ+CHO) were collected over 120h, and each sample was further incubated for various times. Fibril formation significantly increased over 48 hours of collection (p<0.001 at 48 hours compared to 24 hours). Prolonged incubation after collection increased fibril formation, especially after 48 hours of collection (p<0.005 compared to no additional incubation time).
Discussion
Oligomers and fibrils of Aβ are the primary putative neurotoxin in Alzheimer’s disease [8][22]. Selective toxicity of Aβ42 over the Aβ40 form is thought to arise from the tendency of the former to polymerize to stable trimeric or tetrameric structures at high concentrations, while the latter remains as a monomer [6]. The toxicity of Aβ may arise from its ability to disrupt the extracellular matrix [17] on the one hand and its interaction with vessels, especially EC on the other. The extracellular matrix effects are well documented and may explain the toxic effects of Aβ-secreting CHO observed on both EPI and SMC. Yet, circulating Aβ protein is not significantly toxic to normal tissues and cells [15]. We now provide added regulatory dimension to these toxic effects wherein Aβ cytotoxicity is density dependent for many cells, and in EC in a manner that is auto-regulatory. Subconfluent EC promote and confluent EC limit Aβ toxicity.
Confluent cells are quiescent and present a phenotype that reflects a functional rather than reparative or proliferative state. Confluent SMC express a greater amount of contractile proteins and in matched array than subconfluent or proliferative SMC [3][12][13]. That Aβ is more toxic to the subconfluent cells may well reflect a susceptible state or even interaction with proteins expressed in the growing and not the quiescent cell. These effects are most profound for the EC. EC state is an index of vascular health. EC dysfunction and injury presages vascular disease and vascular repair directs recovery of endothelial integrity as a means of restoring vascular vigor. The intact endothelial monolayer serves as a powerful bioreactor that secretes factors that inhibit the growth of underlying SMC, compounds like heparan sulfate proteoglycans and TGFβ that bind and inactivate vascular growth factors, maintain vascular tone, control leukocyte adhesion and transmigration and limit local inflammation and immune activation[20]. Vascular damage removes these protective mechanisms and directly exposes underlying SMC to circulating factors without their endothelial filter. Injured SMC, subconfluent EC and activated leukocytes produce an array of chemokines, cytokines and growth factors that are growth promoting, immune activating and vasoconstrictive. Thus, EC state and density define vascular health and modulate vascular repair. Confluent EC promote vascular quiescence and subconfluent EC induce further vascular damage. We now show that these density-dependent effects also regulate Aβ cytoxicity. Disruption of endothelial integrity creates an EC that enhances Aβ toxicity, while the intact endothelium limits not only EC response but Aβ secretion.
A myriad of effects could be envisioned to explain these phenomena and are worthy of future study. Damaged cells, EC in particular, for example, release growth factors like FGF2, and CM from these cells stimulates EC and SMC growth [2]. Aβ40, in low (nanomolar) concentrations, increases mRNA expression and protein production of FGF2 [5], whereas micromolar concentrations of Aβ40 have an opposite effect on FGF2 production. Toxic effects of Aβ can be overcome by higher levels of FGF2. EC that overexpress FGF2 are especially resistant to any Aβ40 injury [7]. Aβ42 may therefore increase FGF2 production in a manner which is density-dependent. In confluent EC, cell machinery may enable FGF2 levels to provide autoprotection but subconfluent EC may not be able to produce such concentrations and only enough added growth factor to promote overgrowth. Growth factor effects may be enhanced by the heparin-binding nature of Aβ. Various domains on the amyloid precursor protein are involved with the binding of heparan sulfate, and fibrillar Aβ may even bind better to heparin-like compounds [18][26]. Modulation of heparan sulfate proteoglycan function is density dependent [14][20] to futher explain cell state control of Aβ toxicity.
The physiologic release of Aβ from transfected cells [27][5] provides the opportunity to examine the pathobiology of AD and related disorders. Continued elucidation of the nature of the synergistic and complimentary effects of Aβ and cell state may help explain the variable but potent effects of Aβ. One may now need to consider tissue and cell state in addition to local concentration of and exposure duration to Aβ. The specific interactions of Aβ and EC in a state dependent fashion may help understand further the common and divergent forms of vascular and cerebral toxicity of Aβ and the spectrum of AD.
Acknowledgement
Dedicated to the memory of Prof. Vernon Ingram. This work was supported in part by NIH GM 49039 to ERE
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Attems J, Lintner F, Jellinger KA. Amyloid beta peptide 1–42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol (Berl) 2004;107:283–291. doi: 10.1007/s00401-004-0822-6. [DOI] [PubMed] [Google Scholar]
- 2.Brooks RA, Burrin JM, Kohner EM. Characterization of relase of basic fibroblast growth factor from bovine retinal endothelial cells in monolayer cultures. Biochem J. 1991;276(Pt 1):113–120. doi: 10.1042/bj2760113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell JH, Kocher O, Skalli O, Gabbiani G, Campbell GR. Arteriosclerosis. 1989 Sep–Oct;9(5):633–643. doi: 10.1161/01.atv.9.5.633. [DOI] [PubMed] [Google Scholar]
- 4.Cantara S, Donnini S, Morbidelli L, Giachetti A, Schulz R, Memo M, Ziche M. Physiological levels of amyloid peptides stimulate the angiogenic response through FGF-2. Faseb J. 2004;18:1943–1945. doi: 10.1096/fj.04-2114fje. [DOI] [PubMed] [Google Scholar]
- 5.Cantara S, Ziche M, Donnini S. Opposite effects of beta amyloid on endothelial cell survival: role of fibroblast growth factor-2 (FGF-2) Pharmacol Rep. 2005;57 Suppl:138–143. [PubMed] [Google Scholar]
- 6.Chen YR, Glabe CG. Distinct early folding and aggregation properties of Alzheimer amyloid-beta peptides Abeta40 and Abeta42: stable trimer or tetramer formation by Abeta42. J Biol Chem. 2006;281:24414–24422. doi: 10.1074/jbc.M602363200. [DOI] [PubMed] [Google Scholar]
- 7.Donnini S, Cantara S, Morbidelli L, Giachetti A, Ziche M. FGF-2 overexpression opposes the beta amyloid toxic injuries to the vascular endothelium. Cell Death Differ. 2006;13:1088–1096. doi: 10.1038/sj.cdd.4401803. [DOI] [PubMed] [Google Scholar]
- 8.Ferreira ST, Vieira MN, De Felice FG. Soluble protein oligomers as emerging toxins in alzheimer's and other amyloid diseases. IUBMB Life. 2007;59:332–345. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- 9.Fezoui Y, Hartley DM, Harper JD, Khurana R, Walsh DM, Condron MM, Selkoe DJ, Lansbury PT, Jr, Fink AL, Teplow DB. An improved method of preparing the amyloid beta-protein for fibrillogenesis and neurotoxicity experiments. Amyloid. 2000;7:166–178. doi: 10.3109/13506120009146831. [DOI] [PubMed] [Google Scholar]
- 10.Finder VH, Glockshuber R. Amyloid-beta aggregation. Neurodegener Dis. 2007;4:13–27. doi: 10.1159/000100355. [DOI] [PubMed] [Google Scholar]
- 11.Folin M, Baiguera S, Tommasini M, Guidolin D, Conconi MT, De Carlo E, Nussdorfer GG, Parnigotto PP. Effects of beta-amyloid on rat neuromicrovascular endothelial cells cultured in vitro. Int J Mol Med. 2005;15:929–935. [PubMed] [Google Scholar]
- 12.Gabbiani G. Control of smooth muscle cell activation. Arterioscler Thromb Vasc Biol. 2004 May;24(5):804–805. doi: 10.1161/01.ATV.0000126677.86124.7e. [DOI] [PubMed] [Google Scholar]
- 13.Glukhova MA, Kabakov AE, Frid MG, Ornatsky OI, Belkin AM, Mukhin DN, Orekhov AN, Koteliansky VE, Smirnov VN. Modulation of human aorta smooth muscle cell phenotype: a study of muscle-specific variants of vinculin, caldesmon, and actin expression. Proc Natl Acad Sci U S A. 1988 December;85(24):9542–9546. doi: 10.1073/pnas.85.24.9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon PB, Choi HU, Conn G, Ahmed A, Ehrmann B, Rosenberg L, Hatcher VB. Extracellular matrix heparan sulfate proteoglycans modulate the mitogenic capacity of acidic fibroblast growth factor. J Cell Physiol. 1989;140:584–592. doi: 10.1002/jcp.1041400325. [DOI] [PubMed] [Google Scholar]
- 15.Hansson O, Zetterberg H, Vanmechelen E, Vanderstichele H, Andreasson U, Londos E, Wallin A, Minthon L, Blennow K. Evaluation of plasma Aβ40 and Aβ42 as predictors of conversion to Alzheimer's disease in patients with mild cognitive impairment. Neurobiol Aging. 2008 May;16 doi: 10.1016/j.neurobiolaging.2008.03.027. (Epub) [DOI] [PubMed] [Google Scholar]
- 16.Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 17.Mok SS, Losic D, Barrow CJ, Turner BJ, Masters CL, Martin LL, Small DH. The beta-amyloid peptide of Alzheimer's disease decreases adhesion of vascular smooth muscle cells to the basement membrane. J Neurochem. 2006;96:53–64. doi: 10.1111/j.1471-4159.2005.03539.x. [DOI] [PubMed] [Google Scholar]
- 18.Mok SS, Sberna G, Heffernan D, Cappai R, Galatis D, Clarris HJ, Sawyer WH, Beyreuther K, Masters CL, Small DH. Expression and analysis of heparin-binding regions of the amyloid precursor protein of Alzheimer's disease. FEBS Lett. 1997;415:303–307. doi: 10.1016/s0014-5793(97)01146-0. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen PH, Li MS, Stock G, Straub JE, Thirumalai D. Monomer adds to preformed structured oligomers of Abeta-peptides by a two-stage dock-lock mechanism. Proc Natl Acad Sci U S A. 2007;104:111–116. doi: 10.1073/pnas.0607440104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nugent MA, Karnovsky MJ, Edelman ER. Vascular cell-derived heparan sulfate shows coupled inhibition of basic fibroblast growth factor binding and mitogenesis in vascular smooth muscle cells. Circ Res. 1993 Dec;73(6):1051–1060. doi: 10.1161/01.res.73.6.1051. [DOI] [PubMed] [Google Scholar]
- 21.Parameshwaran K, Sims C, Kanju P, Vaithianathan T, Shonesy BC, Dhanasekaran M, Bahr BA, Suppiramaniam V. Amyloid beta-peptide Abeta(1–42) but not Abeta(1–40) attenuates synaptic AMPA receptor function. Synapse. 2007;61:367–374. doi: 10.1002/syn.20386. [DOI] [PubMed] [Google Scholar]
- 22.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 23.Seabrook GR, Ray WJ, Shearman M, Hutton M. Beyond amyloid: the next generation of Alzheimer's disease therapeutics. Mol Interv. 2007 Oct;7(5):261–270. doi: 10.1124/mi.7.5.8. [DOI] [PubMed] [Google Scholar]
- 24.Soffer D. Cerebral amyloid angiopathy--a disease or age-related condition. Isr Med Assoc J. 2006;8:803–806. [PubMed] [Google Scholar]
- 25.Watson D, Castaño E, Kokjohn TA, Kuo YM, Lyubchenko Y, Pinsky D, Connolly ES, Jr, Esh C, Luehrs DC, Stine WB, Rowse LM, Emmerling MR, Roher AE. Physicochemical characteristics of soluble oligomeric Abeta and their pathologic role in Alzheimer's disease. Neurol Res. 2005 Dec;27(8):869–881. doi: 10.1179/016164105X49436. [DOI] [PubMed] [Google Scholar]
- 26.Watson DJ, Lander AD, Selkoe DJ. Heparin-binding properties of the amyloidogenic peptides Abeta and amylin. Dependence on aggregation state and inhibition by Congo red. J Biol Chem. 1997;272:31617–31624. doi: 10.1074/jbc.272.50.31617. [DOI] [PubMed] [Google Scholar]
- 27.Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ. Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–7982. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]