

Abstract
A set of cycloisomerizaton methodologies of alkynyl ketones and imines with concurrent acyloxy, phosphatyloxy, or sulfonyloxy group migration, which allow for the efficient synthesis of multisubstituted furans and N-fused heterocycles has been developed. Investigation of the reaction course by way of employing 17O-labelled substrates allowed for elucidation of the mechanisms behind these diverse transformations. It was found that, while the phosphatyloxy migration in conjugated alkynyl imines in their cycloisomerization to N-fused pyrroles proceeded via a [3,3]-sigmatropic rearrangement, the analogous cycloisomerization of skipped alkynyl ketones proceeds through two consecutive 1,2-migrations, resulting in an apparent 1,3-shift, followed by a subsequent 1,2-migration through competitive oxirenium and dioxolenylium pathways. Investigations of the 1,2-acyloxy migration of conjugated alkynyl ketones en route to furans demonstrated the involvement of a dioxolenylium intermediate. The mechanism of cycloisomerization of skipped alkynyl ketones containing an acyloxy group was found to be catalyst dependent; Lewis and Brønsted acid catalysts caused an ionization/SN1' isomerization to the allene, followed by cycloisomerization to the furan, while transition metal-catalysts evoked a Rautenstrauch-type mechanistic pathway. Furthermore, control experiments in the cycloisomerization of skipped alkynyl ketones under transition metal catalysis revealed that, indeed, these reactions were catalyzed by transition metal complexes as opposed to Brønsted acids resulting from hydrolysis of these catalysts with eventual water. Further synthetic utility of the obtained phosphatyloxy-substituted heterocycles was demonstrated through their efficient employment in the Kumada cross-coupling reaction with various Grignard reagents.
Introduction
Furans and pyrroles are frequently occurring heterocyclic units found in numerous naturally occurring and biologically active molecules,1 and find broad application as synthetic intermediates2 and in the material sciences.3 Thus, not surprisingly, numerous methods have been developed for the assembly of the furan and pyrrole ring,4,5 among which transition metal-catalyzed cycloisomerizations,6 perhaps, are the most general and powerful approaches toward these heterocycles.
Recently, we reported the Cu-catalyzed cycloisomerization of alkynyl imines and ketones to afford monocyclic and fused pyrroles and furans.6n,p It was proposed that the alkynyl imine or ketone 1 undergoes a base-assisted prototropic 1,3-H shift, leading to allene 2 which, upon a copper-catalyzed cycloisomerization, transforms into pyrrole or furan 3 (eq 1). This methodology, however, is limited to the synthesis of heterocycles lacking substituents at the C-3 and C-4 positions, as the hydrogen substituent at C-4 of heterocycle 3 arises from the prototropic 1,3-H migration and H at C-3 comes from the cycloisomerization of 2 to 3.

As a partial solution to this problem, we have recently developed a set of methods for the synthesis of pyrroles and furans 6 possessing a thio-6m or halogen6f substituent at C-3 of heterocycle. These novel transition metal-catalyzed cascade transformations involve isomerization of 4 into allene 5, followed by cycloisomerization of the latter into 6, during which, a 1,2-shift of the thio- or halogen group occurs (eq 2). Aiming at expanding the scope of the migrating group (MG), we explored the possibility of

engaging a 1,3-migration of different oxygen-based migrating groups, known to occur in propargyl/allenyl systems via a [3,3]-sigmatropic shift.7 It was found, however, that depending on the substitution pattern in 7 and/or reaction conditions, different regioisomeric products 9 or 11 were formed (eq 3).6a It was reasoned that, in the former case, the expected [3,3]-shift of the migrating group occurs to produce allene 8, followed by its cycloisomerization to 9. In the latter case, a prototropic rearrangement of 7 gives rise to allene 10, which cycloisomerizes with concomitant 1,2-migration into furan 11.

Herein, we describe a more detailed study of the scope and mechanisms of different modes of cycloisomerization8 of alkynyl ketones and cyclic imines proceeding with concurrent migration of acyloxy and phosphatyloxy groups to produce multisubstituted furans and fused pyrroles, together with the synthetic application of the obtained heterocyclic phosphates in Kumada cross-coupling reactions.
Results and Discussion
Synthesis of Trisubstituted Heterocycles via [3,3]-Phosphatyloxy Migration in Conjugated Alkynyl Ketones and Imines
Our initial attempts explored the possibility of engaging a [3,3]-phosphatyloxy migration in alkynyl ketones 12 into 13 as an approach toward furans 14 (Scheme 1). To this end, cycloisomerization of several phosphatyloxy alkynyl ketones9 was examined in the presence of CuCl (5 mol%) in DMA. Since this transformation no longer involves a base-assisted prototropic H shift, we tested this reaction in the absence of amine. We were pleased to find that under these conditions the cycloisomerization proceeded smoothly, furnishing the corresponding furans 14a and 14b in good yield. While CuCl appeared to be a poor catalyst for the cycloisomerization of phenyl-substituted 12c, we found that the employment of AgBF4 in dichloroethane at 80°C gave the desired furan 14c in good yield (Scheme 1).
Scheme 1.

Cascade [3,3]-migration/cycloisomerization of phosphatyloxy alkynyl ketones into furans
Next, we investigated the applicability of this approach for the synthesis of phosphatyloxy-substituted indolizines 17 (Scheme 2). It was found that the cycloisomerization of alkynyl pyridines 15 under the same conditions proceeded uneventfully to furnish the desired indolizines 17a-e in good to excellent yields. Of note, while the standard cycloisomerization of alkynyl imines to give monosubstituted indolizines required 50 mol% Cu salts,6p this novel cascade [3,3]-migration/cycloisomerization protocol proceeded efficiently in the presence of 5 mol% of CuCl only (Scheme 2).
Scheme 2.

Cascade [3,3]-migration/cycloisomerization of phosphatyloxy alkynyl pyridines into indolizines
It deserves mentioning that in both cases (Schemes 2 and 3) we did not observe the intermediate allenes (13 and 16), which supports their formation at the rate limiting event. In an effort to obtain a better understanding of the mechanism behind the propargyl-allenyl isomerization and their subsequent transformation into heterocycles, we investigated the cycloisomerization of phosphoryl oxygen [17O]-enriched alkynyl pyridine 18, the labeled analogue of 15c (Scheme 3), by means of 17O NMR spectroscopy.10 It was reasoned that if the isomerization indeed proceeds via a [3,3]-sigmatropic shift, the oxygen label should be located at the phosphate ester position in the intermediate allene 19, and thus in the indolizine, as shown in 20. Unsurprisingly, the NMR spectrum of the isolated indolizine product displayed a broad signal at 71 ppm corresponding to the bridging phosphate oxygen.11 Only traces of the phosphoryl oxygen signal, resulting from formation of 21 were detected. Thus, the selective formation of 20 strongly supports the mechanistic rationale involving a propargyl-allenyl isomerization proceeding via a sigmatropic [3,3]-phosphatyloxy shift (Schemes 1-3).
Scheme 3.

Cyloisomerization of labeled phosphatyloxy alkynyl pyridine 18
Synthesis of Trisubstituted Furans via 1,2-Migration of Acyloxy Group in Conjugated Alkynyl Ketones
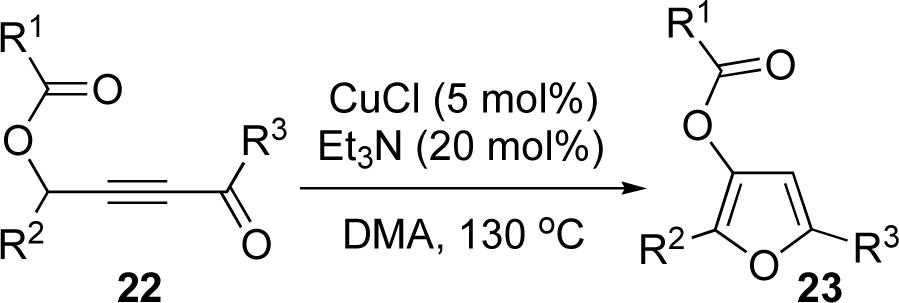
Encouraged by the successful development of conditions for the efficient [3,3]-migration/cycloisomerization of phosphatyloxy-containing propargyl ketones, we next turned our attention to the elaboration of analogous transformations involving acyloxy systems. Our initial attempts at migration/cycloisomerization were disappointing. In the presence of catalytic CuCl, acetate 22a gave a low yield of the expected cycloisomerization product 23a7 and trace amounts of an unexpected regioisomeric product 23b (Scheme 4). The same reaction in the presence of Et3N additive however, produced the other regioisomer, 23b, as a major product. Intrigued by this finding, we decided to investigate the latter transformation further, as it may provide access to heterocycles of a different substitution pattern. Switching to phenyl and tert-butyl alkynyl ketones resulted in increases in both the yields and the regioselectivity of the products (Table 1). Thus, a series of alkynyl ketones 22 with different acyloxy groups under these reaction conditions, underwent smooth cycloisomerization to produce acyloxy furans 23 in good to excellent yields.
Scheme 4.

Table 1.
Cascade 1,2-migration/cycloisomerization of acyloxy alkynyl ketones 22 into furans 23
 | |||||
|---|---|---|---|---|---|
| entry | substrate 22 | time, h | product 23 | yield, %a | |
| 1 | 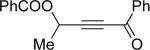 |
c | 22 | 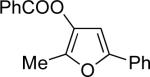 |
82 |
| 2 | 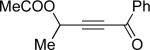 |
d | 1 | 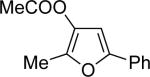 |
81 |
| 3 | 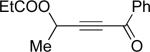 |
e | 9 | 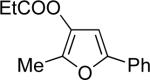 |
69 |
| 4 | 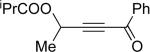 |
f | 2 | 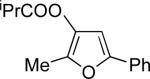 |
90 |
| 5 | 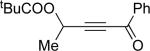 |
g | 17 | 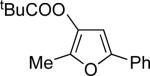 |
86 |
| 6 | 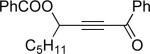 |
h | 23 |  |
80 |
| 7 |  |
i | 32 |  |
80 |
| 8 |  |
j | 46 | 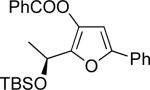 |
83 |
Run on 1.0 mmol scale
We propose the following plausible mechanisms to account for the observed unusual regiochemistry of the cycloisomerization products obtained (Scheme 5). The upper path A depicts the formation of allene 25 from 24 via a base-assisted prototropic shift which, upon metal activation of the carbonyl, forms dioxolenylium species 26. Subsequent transformation of the latter to furan 27 occurs by an intramolecular AdN-E process.12 Alternatively (path B), 24 could undergo a Rautenstrauch-type13 metal-assisted 1,2-acyloxy14 migration15 via dioxolenylium species16 28 to give metal carbenoid 29. Intramolecular attack of the ketone to give 30, followed by tautomerization, gives furan 27. While, on the basis of our current studies, we cannot rule out either mechanistic pathway, the prerequisite of base for the selective formation of the observed regioisomer of furan 23b over 23a (Scheme 4) supports possible involvement of allene intermediate 25.17
Scheme 5.

Proposed mechanisms for cycloisomerization of acyloxy alkynyl ketones into furans via dioxolenylium intermediates
We hypothesized that the acyloxy group migration may also proceed via a three-membered oxirenium species (Scheme 6), analogously to sulfur and halogen group migrations reported earlier.6f,m The involvement of either dioxolenylium (Scheme 5) or oxirenium (Scheme 6) motifs could be established through reaction of a labeled substrate 24. While dioxolenylium-involving pathway A and B both lead to the same isotopomer 27 (Scheme 5), a migration occurring via oxirenium intermediates (paths A' and B') would lead to a different isotopomer 29 (Scheme 6).
Scheme 6.

Alternative mechanisms for cycloisomerization of acyloxy alkynyl ketones into furans via oxirenium intermediates
To test the above hypothesis, we synthesized 17O enriched 339 and subjected it to the cycloisomerization conditions (Scheme 7). The reaction was stopped at about 60% conversion. NMR analysis indicated that the reaction resulted in the formation of a 7:1 mixture of isotopomeric furans 34 and 35. Additionally, it was found that the recovered starting material showed no additional signals in the 17O spectrum, thus indicating no scrambling during the reaction. Furthermore, resubmission of the isolated furans to the reaction conditions did not result in a discernable change in the ratio of 34:35, thus ruling out possible scrambling of the product. These observations, confirm that the major product, furan 34, arises through either of the mechanistic pathways outlined in Scheme 5, with migration occurring via a dioxolenylium species. Formation of minor product 35, however, is consistent with either of the two mechanisms depicted in Scheme 6 or an irreversible ionization/recombination to form the allene (see below, Scheme 11).
Scheme 7.

Cycloisomerization of 17O-labeled acyloxy alkynyl ketone 33
Scheme 11.

Proposed mechanisms for formation of allene 37 from 36
Synthesis of Tetrasubstituted Furans via a Cascade Migration in Skipped Alkynyl Ketones
Encouraged by successful cycloisomerization of various alkynyl ketones to furans (vide supra), we proposed that a propargyl-allenyl isomerization of isomeric propargyl ketone 36 via a [3,3]-migration might provide access to allene 37, the fully substituted analogue of 10 (eq 3), which, upon cycloisomerization with subsequent 1,2-migration, would lead to fully substituted furan 38 (eq 4). Additionally, if successful, formation of allenyl intermediate 37 would provide additional support for our mechanistic proposal above (Scheme 5, path A).

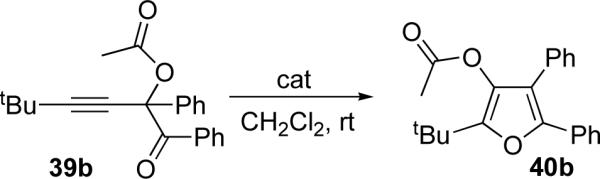
First, we examined the potential migration of acyloxy group in 39 (Y=CR4) en route to tetrasubstituted furan 40. Our optimization studies indicated that several metal salts are able to efficiently catalyze this transformation (Table 2). While the silver salts AgBF4 and AgOTf proved efficient for the cycloisomerization of tert-butyl substituted 39b (entries 5 and 7), they were less desirable for n-butyl substituted 39a (entries 4 and 6). However, it was found that Cu(OTf)2 was effective for the formation of n-butyl-substituted furan 40a (entry 1). AuCl3 worked reasonably well with both tBu- and nBu substituted substrates (entries 10 and 11). Interestingly, Lewis acids18 also were effective in promoting this transformation (entries 13−16). Remarkably, even Brønsted acid (HOTf) efficiently catalyzed this transformation (entry 17).
Table 2.
Optimization of acyloxy migration/cycloisomerization
 | ||||
|---|---|---|---|---|
| entry | R1 | [M] (mol%) | solvent | yield 40, %a |
| 1 | nBu (a) | Cu(OTf)2 (5) | PhMe | 78 |
| 2 | tBu (b) | “ | “ | 66 |
| 3 | nBu (a) | PdCl2(MeCN)2 (5) | “ | 72 |
| 4 | “ | AgBF4 (5) | DCM | 50b |
| 5 | tBu (b) | “ | “ | 99 |
| 6 | nBu (a) | AgOTf (5) | PhMe | 60 |
| 7 | tBu (b) | “ | “ | 88 |
| 8 | nBu (a) | AgSb6 (5) | “ | 50 |
| 9 | “ | PtCl2 (5) | “ | 43 |
| 10 | tBu (b) | AuCl3 (5) | “ | 72 |
| 11 | nBu (a) | AuCl3 (5) | “ | 62 |
| 12 | “ | AuCl(PEt3) (5) + AgPF6 (5) | “ | complex mixture |
| 13 | tBu (b) | TMSOTf (10) | DCM | 63 |
| 14 | “ | Sn(OTf)2 (10) | “ | 72 |
| 15 | “ | Sc(OTf)3 (10) | “ | 65 |
| 16 | “ | Zn(OTf)2 (10) | “ | 56 |
| 17 | “ | HOTf (10) | “ | 80 |
NMR yield.
Run at 0°C.
Remarkably, this cascade method allowed for efficient synthesis of fused furan 40g (entry 7), bicyclic scaffold inaccessible by our previous cycloisomerization techniques.6n
Next, cycloisomerization of several differently-substituted propargyl acetates and pivaloates 39 under the optimized conditions was examined. It was found that the cascade cycloisomerization of 39 proceeded smoothly to produce fully-substituted furan 40 in good to excellent yields (Table 3). Noteworthy, lengthened reaction times were required for the reaction of pivaloates as compared to the corresponding acetate (entries 2 and 9 vs 1 and 8, respectively). As the TMS substituted alkyne function in 39i is incompatible with silver salts,19 employment of AuCl3 gave the desired furan 40i in good yield (entry 10).
Table 3.
Sequential [3,3]/1,2-acyloxy migration/cycloisomerization
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate 39 | cat. | time, min | product 40 | yield, %a | |
| 1 | 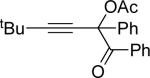 |
b | AgBF4 | 2 | 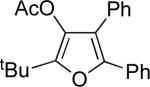 |
>99 |
| 2 | 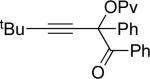 |
c | AgBF4 | 180 | 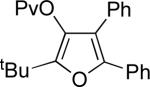 |
73b |
| 3 | TMSOTf | 600 | 70b | |||
| 4 | 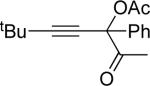 |
d | AgBF4 | 15 | 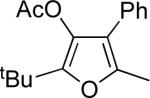 |
73 |
| 5 | 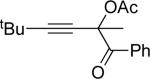 |
e | AgBF4 | 15 | 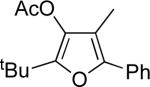 |
84 |
| 6 | 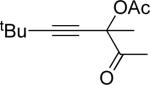 |
f | AgBF4 | 15 | 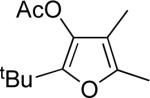 |
90 |
| 7 | 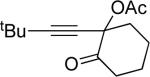 |
g | AgBF4 | 10 | 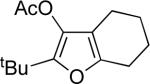 |
86 |
| 8 | 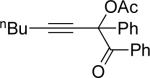 |
a | Cu(OTf)2 | 15 | 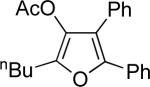 |
60c |
| 9 | 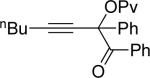 |
h | Cu(OTf)2 | 180 | 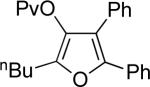 |
81b |
| 10 | 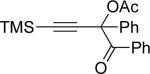 |
i | AuCl3 | 15 | 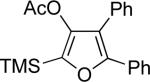 |
78b,c |
| 11 | 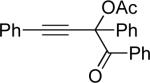 |
j | Cu(OTf)2 | 15 | 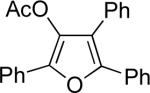 |
91c |
Run on 1.0 mmol scale.
Run on 0.5 mmol scale.
Run in toluene.
Next, we examined possible cascade involving migration of phosphatyloxy group cycloisomerization (Scheme 8). It was found that 41, in the presence of AgBF4 in dichloromethane at room temperature, smoothly converted to allene 42, thus, providing evidence for the formation of allenyl intermediates in the reaction.20 Subsequent treatment of the allene 42 with AgBF4 in dichloroethane at 60°C afforded furan 43 in 77% yield. Additionally, the latter conditions were effective for the formation of furan 43 directly from the propargyl phosphate 41 (Scheme 8).
Scheme 8.

Sequential phosphatyloxy migration/cycloisomerization
Aiming at expanding the scope of this transformation, we examined the analogous transformation of propargyl tosylates under similar reaction conditions. Unexpectedly, our attempts to synthesize 44 led to the direct formation of tosyl allene 45 (Scheme 9). When subjected to AgBF4 in dichloroethane at 60°C, allene 45 was smoothly converted to the tosyl-substituted furan 46 in 82% yield.
Scheme 9.

Sequential tosyloxy migration/cycloisomerization
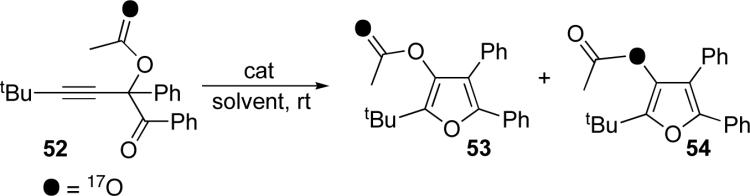
Understandably, we were interested in establishing the mechanism behind the cascade cycloisomerization in skipped systems. From the outset, we postulated two general mechanisms to explain the formation of furans from propargyl substituted ketones. The first operates via an allenyl intermediate 48 (or 37, as depicted in eq 4), whereas the second goes through carbenoid species 50 (Scheme 10). If one of the oxygens in the migrating group is selectively labeled, as in 47, it would be possible to distinguish between the two mechanistic paths. Thus, as depicted in the upper pathway, 47 first undergoes a [3,3]-shift, with “inversion” of the labeled oxygen to form allene 48. A subsequent 1,2-shift via a dioxolenylium species would result in a second “inversion” to produce the Y=O oxygen-labeled furan 49. Alternatively, if 47 undergoes a 1,2-shift to form metal carbenoid 50, the label would be located at the bridged oxygen atom. The intramolecular nucleophilic attack and subsequent cycloisomerization proceeds with no migration, so the label would remain at the bridged oxygen position in the furan product 51.
Scheme 10.

Proposed mechanisms for sequential migration/cycloisomerization of labeled 47
Thus, labeled acetate 529 was tested under several reaction conditions proved efficient in catalyzing this transformation (Table 4). When subjected to the CuCl/Et3N conditions, carbonyl oxygen-labeled 53 was formed as a minor product, with the major product being the ester oxygen-labeled 54 (entry 1). More remarkably, reactions using AgBF4, AuCl3, or Cu(OTf)2 (entries 2, 3, and 4) gave 54 exclusively. Furthermore, the Lewis acids Sn(OTf)2 and TMSOTf (entries 5 and 6) gave mixtures of nearly equal amounts of isotopomers 53 and 54. Employment of the Brønsted acid (TfOH, entry 7) also resulted in the formation of a mixture of isotopomers (Table 4).21
Table 4.
Cycloisomerization of carbonyl labeled 52
 | ||||
|---|---|---|---|---|
| entry | cat (mol%) | solvent | ratio (53:54) | yield, % |
| 1 | CuCl (5) + Et3N (20) | DMA | 38 : 62 | 89a,b |
| 2 | AgBF4 (5) | DCM | 0 : 100 | 69 |
| 3 | AuCl3 (10) | PhMe | 0 : 100 | 64 |
| 4 | Cu(OTf)2 (10) | PhMe | 0 : 100 | 84 |
| 5 | Sn(OTf)2 (10) | DCM | 38 : 62 | 42 |
| 6 | TMSOTf (10) | “ | 42 : 58 | 51a |
| 7 | HOTf (10) | “ | 26 : 74 | 80 |
| 8 | AuCl3 (5) + TTBP (10) | “ | 0:100 | 84 |
NMR of recovered starting material showed no formation of another isotopomer. See Supporting Material for details.
Run at 130°C.
In light of the recent observations that eventual Brønsted acids are the true catalysts in some transition metal-catalyzed transformations,22 we investigated what role, if any, Brønsted acids may play in the described herein cycloisomerization reaction.23 To this end, in the test experiment, we examined the cycloisomerization of 39b in the presence of Brønsted base additive, which expected to quench eventual Brønsted acids which may be present in the reaction media. It was found that hindered base, 2,4,6-tri-tertbutylpyrimidine (TTBP), serves the purpose: addition of 15 mol% of TTBP to reaction mixture containing 10 mol% TfOH completely shuts down the reaction, which is otherwise complete in 6 hours (Table 5, entry 1). With this tool in hand, we then examined cycloisomerization of 39b catalyzed by several transition metal catalysts in the presence and absence of TTBP (entries 2−6). It was found that in all cases, the reaction course was unaffected by the presence of the base: the reactions with TTBP additive proceeded with rates comparable to those lacking the base, thus ruling out possible involvement of Brønsted acids in these transition metal-catalyzed transformations. Moreover, it was found that the cycloisomerization of 52 in the presence of Au catalyst and TTBP (Table 4, entry 8) did not change the isotopomeric distribution over the base-free experiment (entry 3).
Table 5.
Effect of Brønsted base on cycloisomerization with different catalysts
 | |||
|---|---|---|---|
| entry | cat (mol%) | % conversion of 39ba (no base) | % conversion of 39ba (with TTBPb) |
| 1 | TfOH (10) | 100c | <1d |
| 2 | Cu(OTf)2 (5) | 100e | 100e |
| 3 | PdCl2(MeCN)2 (5) | 100e | 100e |
| 4 | AgOTf (5) | 100e | 100e |
| 5 | PtCl2 (5) | 100f | 100f |
| 6 | AuCl3 (5) | 100e | 100e |
based on GC analysis. Yields comparable to that reported in Table 2.
1.5 eq (vs catalyst) 2,4,6-tri-tertbutylpyrimidine (TTBP) added.
reaction complete after 6 hours.
reaction checked after 24 hours.
reaction complete within 30 minutes.
reaction complete within 3.5 hours.
Based on the results of the labeling studies, we believe that the cycloisomerization of acetates follows one of two mechanistic pathways, depending on the catalyst. Transition metal-catalysts such as Ag(I), Au(III), and Cu(II) likely cause an initial 1,2-migration to form a metal carbenoid (Scheme 10, lower path). Subsequent cycloisomerization of this intermediate then provides the corresponding furan (Scheme 16). Alternatively, Lewis/Brønsted acid catalysts, such as TMSOTf, Sn(OTf)2, and HOTf, or under high temperature conditions, a partial ionization of the propargyl acetate 36 to a propargylium/allenylium cation24 occurs during which the 17O label is scrambled: consistent with the observed mixture of isotopomers 53 and 54 (Scheme 10 and 11). To obtain additional support for the ionization mechanism, we tested cycloisomerization of acetate 39b in the presence of an external nucleophile, methallyl TMS (Scheme 12).25 Unsurprisingly, the 1,5-enyne 57 was obtained in high yield, along with small amounts of the allene 58.26 Further studies indicated that enyne 57 does not convert to allene 58 under the reaction conditions. As the allylation of propargylic acetates is known to follow an SN1 mechanism,18 these results support the proposed ionization pathway (Scheme 11).
Scheme 16.

Generalized mechanism for migration/cycloisomerization of 36 to 38
Scheme 12.

Allylation of acetate 39b
Since allenes were cleanly obtained in the cycloisomerization of propargyl phosphates and tosylates, it occurred to us that phosphates and acetates were following distinct mechanistic pathways. Thus, we decided to further investigate the mechanism of this intriguing transformation using labeled phosphates. Initially, we subjected phosphoryl oxygen-labeled 599 to the conditions under which the allene could be obtained (Scheme 13). Surprisingly, the obtained allene 60 maintained the phosphoryl oxygen-labeling pattern, with no traces of the other isotopomer, thus completely ruling out possible involvement of [3,3]-sigmatropic shift in this transformation! Moreover, conversion of the allene 60 to furan resulted in the formation of phosphoryl oxygen-labeled 61 as a major component over the bridged oxygen-labeled 62. Similar results were obtained when alkyne 59 was converted directly to the furan under elevated temperatures. In a control experiment, 59 rapidly (ca in 10 min) isomerized to the allene with 10 mol % triflic acid. Consistent with an ionization pathway, as experienced with acetates, allene 60 was formed accompanied with comparable amounts of its isotopomer.27 When left for over 15 hours, the cycloisomerization of isotopomeric allenes proceeded to give furans 61 and 62 in 83% combined yield.
Scheme 13.

Sequential migration/cycloisomerization of phosphoryl oxygen-labeled 59
Apparently, the mechanism of migration of the phosphatyloxy group presented an interesting challenge. Involvement of the allene was demonstrated, as it was isolated and shown to convert to the furan. Furthermore, the position of the oxygen label ruled out a [3,3]-sigmatropic shift (Scheme 10, upper path) as the product allene 60 from alkyne 59 possessed the oxygen label solely at the phosphoryl oxygen (Scheme 13). Such a migration would have resulted in the other isotopomer and ionization would result in the formation of both. It was clear that both of these proposed mechanisms were in disagreement with the obtained experimental results. To rationalize the selective formation of the observed isotopomer of allene, we propose the following potential mechanisms (Scheme 14). The first path (path A) presumes two consecutive 1,2-migrations via dioxolenylium species (64 and 66) and carbenoid intermediate 65. Net retention of the Y=O label is the result of two “inversions.” Alternative path B occurs via two retentive 1,2-shifts proceeding through oxirenium entities 68 and 70 and carbenoid 69, which is isotopomeric to carbenoid 65 (Scheme 14). While path A is more consistent with previous mechanistic studies, based on our current results, we cannot rule out path B.
Scheme 14.

Mechanisms for formation of phosphatyloxy-substituted allene 67
The formation of two furan isotopomers in the cycloisomerization of isotopomerically pure allene 60 (Scheme 13) can be explained via two competing mechanisms for the 1,2-phosphatyloxy migration (Scheme 15). Apparently, “retentive” 1,2-migration via oxirenium species 71 would provide the major isotopomer, phosphoryl oxygen-labeled furan 72, whereas 1,2 migration via dioxolenylium species 73 produces furan 74, with the bridged oxygen labeled.
Scheme 15.

Proposed mechanisms for conversion of allene 67 to furans 72 and 74
Our mechanistic studies discussed above suggest that the mechanism for the migration/cycloisomerization of propargyl substituted ketones depends on both the nature of the migrating group and catalyst employed. A generalized mechanism for these transformations is depicted in Scheme 16. Our initial postulate that allene 37 could be accessed via a [3,3]-sigmatropic migration is in conflict with our current experimental data (Scheme 16). In the case of acyloxy migration in the presence of transition metal catalysts, the metal carbenoid 75, formed via a 1,2-migration, could be trapped by the carbonyl oxygen of the ketone to give furan 38 directly (Rautenstrauch-type). Alternatively, metal carbenoid 75 could be trapped by the carbonyl of the ester moiety to give the allene 37. A 1,2-migration via a dioxolenylium would result in the formation of furan 38 with inversion of 17O label. As the isotopomeric outcome is the same in either of these two mechanisms, neither mechanistic possibility can be ruled out. Analogous transformation in the presence of Lewis acids proceeds with substantial 17O scrambling in the product, which implies formation of scrambled allene 37 via an SN1' mechanism. In the cycloisomerization of phosphates, the first step is likely also a 1,2-migration via a dioxolenylium species to metal carbenoid 75. However, since the allene 37 was isolated without formation of furan 38, a second 1,2-migration, also through a dioxolenylium intermediate via attack of the phosphoryl oxygen at the carbenoid center, produces the observed allene with exclusive retention of 17O label. The 1,2-migration required to go from allene 37 to furan 38 likely follows two competing mechanistic pathways, among which a retentive 1,2-migration via oxirenium species predominates, producing furan 38 with the 17O label at the Y=O position as a major isotopomer (Scheme 16).
Kumada Cross-Coupling of Hetaryl Phosphates
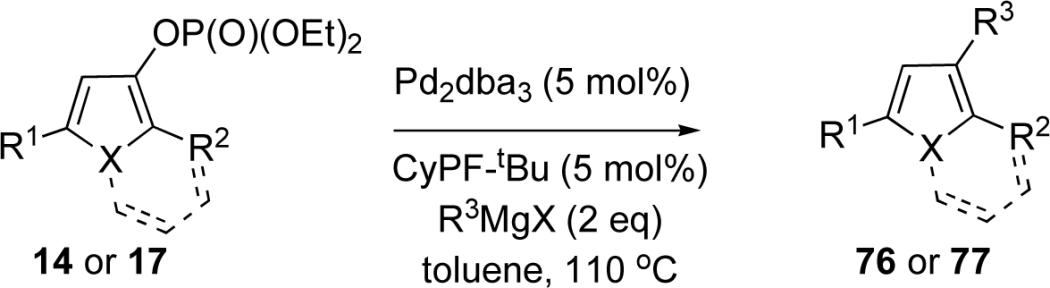
To demonstrate the synthetic utility of the obtained phosphatyloxy-substituted heteroaromatics, we examined their reactions with Grignard reagents under Kumada cross-coupling conditions. It deserves mentioning that, while successful examples of Kumada cross-coupling of Grignard reagents with enol phosphates28 and aryl phosphates29 are well precedented, to the best of our knowledge, no examples of the analogous reaction involving heteroaromatic phosphates have ever been reported. We have found that Kumada cross-coupling of furyl- and indolizinyl phosphates is possible. Substantial optimization indicated that this transformation proceeded most efficiently in the presence of Pd2dba3/CyPF-tBu30 combination. The scope of cross-coupling was examined employing these optimized conditions (Table 6). Kumada cross-coupling of phosphatyloxy-containing furans 14c and indolizines 17b, proceeded smoothly with various Grignard reagents to give C-3 substituted furans 76a-d and C-1 substituted indolizines 77a-d in good to excellent yields (Table 6).
Table 6.
Cross-coupling of phosphatyloxy-substituted heteroaromatics
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate 14 or 17 | R3 | product 76 or 77 | yield, % | ||
| 1 |  |
14c | Me |  |
76a | 94 |
| 2 | “ | “ | Et | 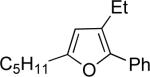 |
76b | 72 |
| 3 | “ | “ | Bn | 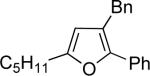 |
76c | 97 |
| 4 | “ | “ | Ph | 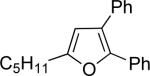 |
76d | 66 |
| 5 | 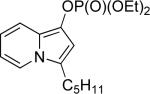 |
17b | Me | 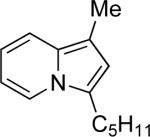 |
77a | 74 |
| 6 | “ | “ | Bn | 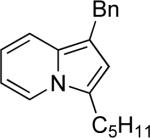 |
77b | 94a |
| 7 | “ | “ | Ph | 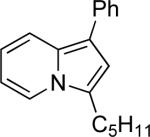 |
77c | 78 |
| 8 | “ | “ | Me3SiCH2 | 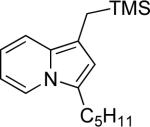 |
77d | 48 |
NMR yield
Conclusion
In summary, we have developed different modes of cascade cycloisomerizations of alkynyl ketones and imines proceeding via various types of migration of acyloxy, phosphatyloxy, and tosyloxy groups to give multisubstituted furans and indolizines in good to high yields. This set of methodologies allows for the efficient synthesis of tri- and tetrasubstituted furans and N-fused heterocycles. Mechanistic studies employing 17O-labeled starting materials indicated that the reaction mechanism depends on the cyclization mode and nature of migrating group. It was shown that 1,3-phosphatyloxy migration in the cycloisomerization of conjugated alkynyl imines proceeds via a [3,3]-shift. The 1,2-migration of the acyloxy group in conjugated alkynyl ketones was found to proceed via a dioxolenylium intermediate. In contrast to acyloxy migration in conjugated alkynyl ketones, the mechanism of acyloxy migration in skipped systems depends on the reaction conditions. While Lewis and Brønsted acids and transition metal complexes catalyze the cycloisomerization of skipped alkynyl ketones, they follow different mechanistic pathways! When an acid (either Lewis or Brønsted) catalyst is used, formation of the postulated allenyl intermediate occurs through an SN1' pathway. However, when transition metal catalysts are used, the cycloisomerization follows either a Rautenstrauch-type 1,2-migration of the acyloxy group followed by the cycloisomerization to the furan directly or, alternatively, two 1,2 migrations of the acyloxy group to form the postulated allenyl intermediate, which cycloisomerizes into the furan via another 1,2-acyloxy shift. Furthermore, control experiments and 17O-labeling studies demonstrated that the transition metal-catalyzed reactions were actually catalyzed by the metal and not by eventual protons. The analogous cycloisomerization of skipped phosphatyloxy alkynyl ketones was also shown to be induced by both Brønsted acid and transition metal catalysts. As in acetates, acids induce an ionization to form the allene via an SN1' mechanistic pathway, whereas under transition metal catalysis, the allene forms via two 1,2-phosphatyloxy migrations. Furthermore, as an extension, we demonstrated the synthetic utility of phosphatyloxy furans and indolizines in the efficient Kumada cross-coupling reaction.
Experimental Section
Concise representative procedures for the preparation of starting materials are provded here. Detailed procedures for the preparation of all starting materials, as well as for the cycloisomerization reactions, can be found in the Supporting Information.
12a
The preparation of 12a is representative. To a microreactor were sequentially added DMAP (11 mg, 0.09 mmol), anhydrous dichloromethane (1 mL), triethylamine (91 μL, 0.65 mmol), 5-hydroxy-3-decyne-2-one (93 μL, 0.50 mmol), and diethylchlorophosphate (94 μL, 0.65 mmol). The reaction was stirred at room temperature for 2.5 hours until judged complete by TLC and GC analysis. The reaction was washed with saturated sodium chloride solution (10 mL), the aqueous layer was extracted twice with dichloromethane (3 mL), and the organic extracts were dried over sodium sulfate and then concentrated. The residue was purified by column chromatography (silica gel, 1:2 ethyl acetate/hexanes) to afford 12a (111 mg, 73%). 1H NMR (500.13 MHz, CDCl3) δ 5.09 (q, J=6.85 Hz, 1H), 4.18−4.10 (m, 4H), 2.35 (s, 3H), 1.87 (dt, J=15.04 Hz, J=7.52 Hz, 2H), 1.51−1.43 (m, 2H), 1.37−1.29 (m, 10H), 0.93−0.85 (m, 3H). 13C NMR (125.77 MHz, CDCl3) δ 184.1, 88.4, 85.5, 67.6, 64.6, 36.2, 36.1, 33.0, 31.5, 24.7, 22.8, 16.6, 16.5, 14.4; LRMS m/z (M++H, 275), 155 (100), 99 (100).
15a
The preparation of 15a is representative. To a solution of 2-ethynyl pyridine (300 μL, 3 mmol) in THF (10 mL) at −78°C was added, dropwise, nBuLi (2.67M, 1.2 mL, 3.3 mmol). The reaction was stirred for a further 5 minutes before being brought to 0°C and stirred for 20 minutes. The reaction was returned to −78°C and butyraldehyde (280 μL, 3.3 mmol) was added slowly. The reaction was brought to and stirred at 0°C for 30 minutes and treated with chlorodiethylphosphate (564 μL, 3.9 mmol). The reaction was brought to room temperature and stirred for an hour, until TLC analysis indicated the reaction was complete. The reaction was diluted with ether (90 mL) and poured into sat. NH4Cl(aq) (75 mL). The organic phase was washed (sat. NH4Cl(aq), 75 mL), dried (MgSO4), filtered, and concentrated. The residue was purified by column chromatography (silica gel, EtOAc) to afford 15a as a viscous oil (880 mg, 2.8 mmol, 94%). 1H NMR (500.13 MHz, CDCl3) δ 8.24 (d, J=4.0 Hz, 1H), 7.36 (tt, J=7.8 Hz, J=1.9 Hz, 1H), 7.11 (d, J=7.9 Hz, 1H), 6.92−6.97 (m, 1H), 4.87−4.93 (m, 1H), 3.78−3.90 (m, 4H), 1.52−1.67 (m, 2H), 1.21−1.30 (sext, J=7.3 Hz, 2H), 0.98−1.04 (m, 6H), 0.65 (t, J=7.4 Hz, 3H); 13C NMR (125.76 MHz, CDCl3) δ 149.3, 141.6, 135.5, 126.5, 122.6, 85.4, 84.9, 67.2 (d, J=5.5 Hz), 63.2 (d, J=5.5 Hz), 63.1 (d, J=5.5 Hz), 37.7 (d, J=5.5 Hz), 17.4, 15.3 (d, J=5.5 Hz), 12.8.
39
To a round bottomed two-necked flask were successively added anhydrous THF (10 mL) and 3,3-dimethyl-1-butyne (0.62 mL, 5.0 mmol). The solution was cooled to −78°C and nBuLi (2.5M, 2.1 mL, 5.2 mmol) in hexanes was added dropwise. The resulting solution was allowed to warm to room temperature. To an additional 50 mL round bottomed two-necked flask the appropriate dione (4.8 mmol) was dissolved in anhydrous THF (10 mL). The dione solution was cooled to −78°C and the acetylide solution from the first flask was added slowly via cannula to the dione solution at −78°C. The resulting solution was allowed to warm to room temperature, then cooled down again to −78°C and acetic anhydride (0.76 mL, 8.0 mmol) was added. The reaction was brought to room temperature and the solution was then poured into a separatory funnel containing saturated NH4Cl(aq) (200 mL) and diethyl ether (50 mL). After extraction, the organic layer was separated, dried (MgSO4), concentrated, and purified (silica gel) to afford pure acetoxy propargyl ketone 39.
41
To a round bottomed, two-necked flask were successively added anhydrous THF (10 mL) and 3,3-dimethyl-1-butyne (0.62 mL, 5.0 mmol). The solution was cooled to −78 °C while stirring and nBuLi (2.5 M, 2.2 mL, 5.5 mmol) was added dropwise. The flask was removed from the cooling bath and allowed to warm to room temperature. Benzil (1.05 g, 5.0 mmol) was dissolved in anhydrous THF (10 mL) in a 50 mL round bottomed, two-necked flask. The solution was cooled to −78°C, and the acetylide solution from the first flask was transferred dropwise via cannula. The resulting purple-black solution was allowed to warm to room temperature while stirring and then cooled to −78 °C again. Diethylchlorophosphate (0.8 mL, 5.5 mmol) and anhydrous triethylamine31 (0.8 mL, 5.5 mmol) were successively added. The solution was allowed to return to room temperature. The resulting amber solution was then poured into a separatory funnel containing 300 mL saturated NH4Cl solution and 50 mL diethyl ether. After thorough extraction, the organic layer was separated, dried (MgSO4), and concentrated. The resulting residue was purified by column chromatography (silica gel, 1:3 ethyl acetate/hexanes) to afford pure 41 (653 mg, 30%) and a second fraction of slightly contaminated 41 (878 mg, 41%). 1H NMR (500.13 MHz, CDCl3) δ 7.89 (d, J=8.4 Hz, 2H), 7.70 (dd, J=7.8 Hz, J=0.9 Hz, 2H), 7.43−7.34 (m, 4H), 7.26 (t, J=7.7 Hz, 2H), 4.24−4.10 (m, 2H), 4.07−3.97 (m, 2H), 1.31 (t, J=7.1 Hz, 3H), 1.24 (t, J=8.1 Hz, 3H), 1.23 (s, 9H); 13C NMR (125.76 MHz, CDCl3) δ 191.6, 137.8, 134.1, 133.0, 131.0, 129.7, 129.1, 128.0, 127.8, 103.8, 82.6, 75.3, 64.1 (d, J=38.9 Hz), 64.1 (d, J=39.0 Hz), 30.6, 28.3, 16.5 (d, J=12.7 Hz), 16.4 (d, J=12.8 Hz); 31P NMR (202.46 MHz) δ −7.30; LRMS m/z 428 (M+, 15), 155 (66), 105 (PhCO, 100).
45
To a 25 mL round-bottomed, two-necked flask were successively added anhydrous THF (10 mL) and 3,3-dimethyl-1-butyne (5.0 mmol, 0.62 mL). The solution was allowed to cool to −78°C while stirring and nBuLi (2.5M, 2.2 mL, 5.5 mmol) in hexanes was added dropwiseThe reaction was allowed to warm to room temperature. Sepearately, a round bottomed, two-necked 50 mL flask was successively loaded with benzil (5.0 mmol, 1.05 g) and anhydrous THF (10 mL). The solution was stirred and cooled to −78°C, and the acetylide solution was then transferred dropwise to the benzil solution via cannula. The resulting solution was allowed to reach room temperature before being returned to −78°C and treated sequentially with p-toluenesulfonyl chloride (1.05 g, 5.5 mmol) in anhydrous THF (1 mL) and anhydrous triethylamine (0.77 mL, 5.5 mmol) were successively added. The flask was then removed from the cooling bath and allowed to warm to room temperature. The solution was then poured into a separatory funnel containing saturated NH4Cl(aq) (300 mL) and diethyl ether (50 mL). After thorough extraction, the organic layer was separated, dried (MgSO4), and concentrated. The residue was purified by column chromatography(silica gel, 1:10 ethyl acetate/hexanes) to afford pure 45 (1.32 g, 60%) as a yellow oil which solidifies upon refrigeration. 1H NMR (500.13 MHz, CDCl3) δ 7.81 (dd, J=8.3 Hz, J=1.2 Hz, 2H), 7.63 (d, J=8.4 Hz, 2H), 7.56 (t, J=7.4 Hz, 1H), 7.41 (t, J=7.8 Hz, 2H), 7.32−7.38 (m, 5H), 7.10 (d, J=7.9 Hz, 2H), 2.35 (s, 3H), 1.01 (s, 9H); 13C NMR (125.76 MHz, CDCl3) δ 196.6, 192.2, 145.1, 137.0, 136.2, 133.3, 132.8, 132.7, 129.6, 129.5, 128.8, 128.6, 128.3, 128.1, 120.5, 36.0, 27.4, 21.6.
Supplementary Material
Acknowledgment
The support of the National Institutes of Health (GM-64444) is gratefully acknowledged.
Footnotes
Supporting Information Available: General methods, experimental procedures for the preparation of starting materials, and analytical and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Bellina F, Rossi R. Tetrahedron. 2006;62:7213. [Google Scholar]; b Hou XL, Yang Z, Wong HNC. In: Progress in Heterocyclic Chemistry. Gribble GW, Gilcrist TL, editors. Vol. 15. Pergamon; Oxford: 2003. p. 167. [Google Scholar]; c Keay BA, Dibble PW. In: Comprehensive Heterocyclic Chemistry II. Katritzky AR, Rees CW, Scriven EFV, editors. Vol. 2. Elsevier; Oxford: 1997. p. 395. [Google Scholar]; d Donnelly D, M. X., Meegan MJ. In: Comprehensive Heterocyclic Chemistry. Katritzky AR, Rees CW, editors. Vol. 4. Pergamon; Oxford: 1984. p. 657. [Google Scholar]; e Dean FM. Naturally Occuring Oxygen Ring Compounds. Butterworths; London: 1963. p. 1. Chapter 1. [Google Scholar]
- 2.a Lee H-K, Chan K-F, Hui C-W, Yim H-K, Wu X-W, Wong HNC. Pure Appl. Chem. 2005;77:139. [Google Scholar]; b Wong HNC, Yu P, Yick C-Y. Pure Appl. Chem. 1999;71:1041. [Google Scholar]; c Lipshutz BH. Chem. Rev. 1986;86:795. [Google Scholar]
- 3.a Ferrero F, Napoli L, Tonin C, Varesano A. J. Appl. Polym. Sci. 2006;102:4121. [Google Scholar]; b Venkatatraman S, Kumar R, Sankar J, Chandrashekar TK, Senhil K, Vijayan C, Kelling A, Senge MO. Chem. Eur. J. 2004;10:1423. doi: 10.1002/chem.200305558. [DOI] [PubMed] [Google Scholar]; c Facchetti A, Abbotto A, Beverina L, van der Boom ME, Dutta P, Evmenenko G, Pagani GA, Marks TJ. Chem. Mater. 2003;15:1064. [Google Scholar]; d Zhang L-Z, Chen C-W, Lee C-F, Wu C-C, Luh T-Y. Chem. Commun. 2002:2336. doi: 10.1039/b207489c. [DOI] [PubMed] [Google Scholar]; e Lee C-F, Yang L-M, Hwu T-Y, Feng A-S, Tseng J-C, Luh T-Y. J. Am. Chem. Soc. 2000;122:4992. [Google Scholar]; f Novak P, Müller K, Santhanum KSV, Haas O. Chem. Rev. 1997;97:207. doi: 10.1021/cr941181o. [DOI] [PubMed] [Google Scholar]
- 4.For recent reviews, see: Patil NT, Yamamoto Y. ARKIVOC. 2007;10:121. Kirsch SF. Org. Biomol. Chem. 2006;4:2076. doi: 10.1039/b602596j. Brown RCD. Angew. Chem., Int. Ed. 2005;44:850. doi: 10.1002/anie.200461668. Jeevanandam A, Ghule A, Ling Y-C. Curr. Org. Chem. 2002;6:841. Keay BA. Chem. Soc. Rev. 1999;28:209. Hou XL, Cheung HY, Hon TY, Kwan PL, Lo TH, Tong SY, Wong HNC. Tetrahedron. 1998;54:1955.
- 5.For recent examples, see: Galliford CV, Scheidt KA. J. Org. Chem. 2007;72:1811. doi: 10.1021/jo0624086. St. Cyr DJ, Martin N, Arndtsen BA. Org. Lett. 2007;9:449. doi: 10.1021/ol062773j. Zhang J, Schmalz H-G. Angew. Chem., Int. Ed. 2006;45:6704. doi: 10.1002/anie.200601252. Gorin DJ, Davis NR, Toste FD. J. Am. Chem. Soc. 2005;127:11260. doi: 10.1021/ja053804t. Kamijo S, Kanazawa C, Yamamoto Y. J. Am. Chem. Soc. 2005;127:9260. doi: 10.1021/ja051875m. Yao T, Zhang X, Larock RC. J. Org. Chem. 2005;70:7679. doi: 10.1021/jo0510585. Larionov OV, de Meijere A. Angew. Chem., Int. Ed. 2005;44:5664. doi: 10.1002/anie.200502140. Duan X.-h., Liu X.-y., Guo L.-n., Liao M.-c., Liu W-M, Liang Y.-m. J. Org. Chem. 2005;70:6980. doi: 10.1021/jo050908d. Lee KY, Lee MJ, Kim JN. Tetrahedron. 2005;61:8705.Jung C-K, Wang J-C, Krische MJ. J. Am. Chem. Soc. 2004;126:4118. doi: 10.1021/ja049377l.Dhawan R, Arndtsen BA. J. Am. Chem. Soc. 2004;126:468. doi: 10.1021/ja039152v.Nishibayashi Y, Yoshikawa M, Inada Y, Milton MD, Hidai M, Uemura S. Angew. Chem., Int. Ed. 2003;42:2681. doi: 10.1002/anie.200351170.Braun RU, Zeitler K, Müller TJJ. Org. Lett. 2001;3:3297. doi: 10.1021/ol0165185.Brown CD, Chong JM, Shen L. Tetrahedron. 1999;55:14233.Shiraishi H, Nishitani T, Sakaguchi S, Ishii Y. J. Org. Chem. 1998;63:6234. doi: 10.1021/jo980435t.
- 6.For recent examples, see: Sniady A, Durham A, Morreale MS, Wheeler KA, Dembinski R. Org. Lett. 2007;9:1175. doi: 10.1021/ol062539t.Smith CR, Bunnelle EM, Rhodes AJ, Sarpong R. Org. Lett. 2007;9:1169. doi: 10.1021/ol0701971.Díaz-Álvarez AE, Crochet P, Zablocka M, Duhayon C, Cadierno V, Gimeno J, Majoral JP. Adv. Synth. Catal. 2006;348:1671.Zhou C-Y, Chan PWH, Che C-M. Org. Lett. 2006;8:325. doi: 10.1021/ol052696c.Kawai H, Oi S, Inoue Y. Heterocycles. 2006;67:101.Sromek AW, Rubina M, Gevorgyan V. J. Am. Chem. Soc. 2005;127:10500. doi: 10.1021/ja053290y.Suhre MH, Reif M, Kirsch SF. Org. Lett. 2005;7:3925. doi: 10.1021/ol0514101.Sromek AW, Kel'in AV, Gevorgyan V. Angew. Chem., Int. Ed. 2004;43:2280. doi: 10.1002/anie.200353535.Hashmi ASK, Sinha P. Adv. Synth. Catal. 2004;346:432.Ma S, Zhang J. J. Am. Chem. Soc. 2003;125:12386. doi: 10.1021/ja036616g.Gabriele B, Salerno G, Fazio A. J. Org. Chem. 2003;68:7853. doi: 10.1021/jo034850j.Ma S, Zhang J. Angew. Chem., Int. Ed. 2003;42:183.Kim JT, Kel'in AV, Gevorgyan V. Angew. Chem., Int. Ed. 2003;42:98. doi: 10.1002/anie.200390064.Kel'in AV, Gevorgyan V. J. Org. Chem. 2002;67:95. doi: 10.1021/jo010832v.Aurrecoechea JM, Pérez E, Solay M. J. Org. Chem. 2001;66:564. doi: 10.1021/jo0014257.Kel'in AV, Sromek AW, Gevorgyan V. J. Am. Chem. Soc. 2001;123:2074. doi: 10.1021/ja0058684.Gabriele B, Salerno G, Fazio A, Bossio MR. Tetrahedron Lett. 2001;42:1339.Hashmi ASK, Schwarz L, Choi J-H, Frost TM. Angew. Chem., Int. Ed. 2000;39:2285. doi: 10.1002/1521-3773(20000703)39:13<2285::aid-anie2285>3.0.co;2-f.Gabriele B, Salerno G, Lauria E. J. Org. Chem. 1999;64:7687. doi: 10.1021/jo994013a.Hashmi ASK, Ruppert TL, Knöfel T, Bats JW. J. Org. Chem. 1997;62:7295. doi: 10.1021/jo970837l.Arcadi A, Marinelli F, Pini E, Rossi E. Tetrahedron Lett. 1996;37:3387.Vieser R, Eberbach W. Tetrahedron Lett. 1995;35:4405.Seiller B, Bruneau C, Dixneuf PH. 1995;51:13089.Fukuda Y, Shiragami H, Utimoto K, Nozaki H. J. Org. Chem. 1991;56:5816.Marshall JA, Robinson ED. J. Org. Chem. 1990;55:3450.
- 7.a Zhao J, Hughes CO, Toste FD. J. Am. Chem. Soc. 2006;128:7436. doi: 10.1021/ja061942s. [DOI] [PubMed] [Google Scholar]; b Zhang L, Wang S. J. Am. Chem. Soc. 2006;128:1442. doi: 10.1021/ja057327q. [DOI] [PubMed] [Google Scholar]; c Wang S, Zhang L. Org. Lett. 2006;8:4585. doi: 10.1021/ol0618151. [DOI] [PubMed] [Google Scholar]; d Wang S, Zhang L. J. Am. Chem. Soc. 2006;128:8414. doi: 10.1021/ja062777j. [DOI] [PubMed] [Google Scholar]; e Buzas A, Gagosz F. J. Am. Chem. Soc. 2006;128:12614. doi: 10.1021/ja064223m. [DOI] [PubMed] [Google Scholar]; f Zhang L. J. Am. Chem. Soc. 2005;127:16804. doi: 10.1021/ja056419c. [DOI] [PubMed] [Google Scholar]; g Schlossarczyk H, Sieber W, Hesse M, Hansen H-J, Schmid H. Helv. Chim. Acta. 1973;56:875. [Google Scholar]
- 8.Preliminary results of this work (Schemes 4, 8, and 9, Table 1 and portions of Table 3) have been previously communicated (see ref. 6h). All other results, described herein (Schemes 5-7, 10-16, Tables 2, 4, 5, and part of Table 3) are new.
- 9.See Supporting Information for synthesis of starting materials.
- 10.For reviews on 17O NMR, see: Boykin DW, editor. 17O NMR Spectroscopy in Organic Chemistry. CRC Press; Boca Raton, FL: 1991. McFarlane W, McFarlane HCE. In: Multinuclear NMR. Mason J, editor. Plenum Press; New York, NY: 1987. p. 403. Chapter 14.Rodger C, Sheppard N, McFarlane C, McFarlane W. In: NMR and the Periodic Table. Harris RK, Mann BE, editors. Academic Press; New York, NY: 1978. p. 383. Chapter 12.Kintzinger JP. In: NMR of Newly Accessible Nuclei. Laszlo P, editor. Vol. 2. Academic Press; New York, NY: 1983. p. 79. Chapter 4.Boykin DW, Baumstark AL. Tetrahedron. 1989;45:3613.
- 11.For discussions on 17O NMR of phosphates in cyclic and biological systems, see:Tsai M-D, Bruzik K. In: Biological Magnetic Resonance. Berliner LJ, Reuben J, editors. Vol. 5. Plenum Press; New York, NY: 1983. p. 129. Chapter 4.Gordillo B, Eliel EL. J. Am. Chem. Soc. 1991;113:2172.Sopchik AE, Cairns SM, Bentrude WG. Tetrahedron Lett. 1989;30:1221.Eliel EL, Chandrasekaran S, Carpenter LE, II, Verkade JG. J. Am. Chem. Soc. 1986;108:6651.Sammons RD, Frey PA, Bruzik K, Tsai M-D. J. Am. Chem. Soc. 1983;105:5455.
- 12.See ref 6m for details.
- 13.For Rautenstrauch-type 1,2-acyloxy migrations, see:Marion N, Nolan SP. Angew. Chem., Int. Ed. 2007;46:2750. doi: 10.1002/anie.200604773.Marion N, Díez-Gonzalález S, de Frémont P, Noble AR, Nolan SP. Angew. Chem., Int. Ed. 2006;45:3647. doi: 10.1002/anie.200600571.Fehr C, Galindo J. Angew. Chem., Int. Ed. 2006;45:2901. doi: 10.1002/anie.200504543.Pujanauski BG, Prasad BAB, Sarpong R. J. Am. Chem. Soc. 2006;128:6786. doi: 10.1021/ja061549m.Marco-Contelles J, Arroyo N, Anjum S, Mainetti E, Marion N, Cariou K, Lemière G, Mouriès V, Festerbank L, Malacria M. Eur. J. Org. Chem. 2006:4618.Faza ON, López CS, Álvarez R, de Lera AR. J. Am. Chem. Soc. 2006;128:2434. doi: 10.1021/ja057127e.Fürstner A, Hannen P. Chem. Eur. J. 2006;12:3006. doi: 10.1002/chem.200501299.Ohe K, Fujita M, Matsumoto H, Tai Y, Miki K. J. Am. Chem. Soc. 2006;128:9270. doi: 10.1021/ja0612955.Cho EJ, Kim M, Lee D. Eur. J. Org. Chem. 2006:3074.Kim M, Miller RL, Lee D. J. Am. Chem. Soc. 2005;127:12818. doi: 10.1021/ja054875v.Johansson MJ, Gorin DJ, Staben ST, Toste FD. J. Am. Chem. Soc. 2005;127:18002. doi: 10.1021/ja0552500.Shi X, Gorin DJ, Toste FD. J. Am. Chem. Soc. 2005;127:5802. doi: 10.1021/ja051689g.Prasad BAB, Yoshimoto FK, Sarpong R. J. Am. Chem. Soc. 2005;127:12468. doi: 10.1021/ja053192c.Mamane V, Gress T, Krause H, Fürstner A. J. Am. Chem. Soc. 2004;126:8654. doi: 10.1021/ja048094q.Miki K, Ohe K, Uemura S. J. Org. Chem. 2003;68:8505. doi: 10.1021/jo034841a.Rautenstrauch V. J. Org. Chem. 1984;49:950.
- 14.For migrations in ß-(acyloxy)alkyl radicals, see:Crich D, Sartillo-Piscil F, Quintero-Cortes L, Wink DJ. J. Org. Chem. 2002;67:3360. doi: 10.1021/jo016388d.Zipse H, Bootz M. J. Chem. Soc., Perkin Trans. 2. 2001:1566.Crich D, Huang X, Beckwith ALJ. J. Org. Chem. 1999;64:1762. doi: 10.1021/jo982385y.Beckwith ALJ, Duggan PJ. J. Am. Chem. Soc. 1996;118:12838.
- 15.For selected examples of metal-catalyzed 1,2-migrations of nonacyloxy groups, see refs 6f,m andPeng L, Zhang X, Zhang S, Wang J. J. Org. Chem. 2007;72:1192. doi: 10.1021/jo0618674.Seregin IV, Gevorgyan V. J. Am. Chem. Soc. 2006;128:12050. doi: 10.1021/ja063278l.Xu F, Shi W, Wang J. J. Org. Chem. 2005;70:4191. doi: 10.1021/jo050109v.Maiereanu C, Kanai A, Weibel J-M, Pale P. J. Carbohydr. Chem. 2005;24:831.
- 16.a Allen AD, Kitamura T, McClelland RA, Stang PJ, Tidwell TT. J. Am. Chem. Soc. 1990;112:8873. [Google Scholar]; b Lorenz W, Maas G. J. Org. Chem. 1987;52:375. [Google Scholar]
- 17.For base-assisted propargyl-allenyl isomerization, see refs 6h,n,p andSonye JP, Koide K. J. Org. Chem. 2006;71:6254. doi: 10.1021/jo060304p.Huang J, Xiong H, Hsung RP, Rameshkumar G, Mulder JA, Grebe TP. Org. Lett. 2002;4:2417. doi: 10.1021/ol020097p.
- 18.For Lewis acid-catalyzed substitution of acyloxy groups in propargyl systems, see:Netz A, Polborn K, Nöth H, Müller TJJ. Eur. J. Org. Chem. 2005:1823.Schwier T, Rubin M, Gevorgyan V. Org. Lett. 2004;6:1999. doi: 10.1021/ol0494055.Müller TJJ. Eur. J. Org. Chem. 2001:2021.
- 19.Orsini A, Viterisi A, Bodlenner A, Weibel J-M, Pale P. Tetrahedron Lett. 2005;46:2259. [Google Scholar]
- 20.This is in contrast to the analogous acyloxy system where attempts at isolation or independent preparation of the allenes failed.
- 21.Triflic acid was tested as a potential catalyst in the base-free reactions depicted in Schemes 1 and 2, but failed to result in product formation.
- 22.There has been a recent discussion on the role of Brønsted acids in homogenous transition metal-catalyzed reactions. For the most relevant references, see:Hashmi ASK. Catal. Today. 2007;122:211.Li Z, Zhang J, Brouwer C, Yang C-G, Reich NW, He C. Org. Lett. 2006;8:4175. doi: 10.1021/ol0610035.Rosenfeld DC, Shekhar S, Takemiya A, Utsunomiya M, Hartwig JF. Org. Lett. 2006;8:4179. doi: 10.1021/ol061174+.Rhee JU, Krische MJ. Org. Lett. 2005;7:2493. doi: 10.1021/ol050838x.
- 23.The authors wish to acknowledge one of the referees, who brought our attention to this possibility.
- 24.For discussions on propargyl/allenyl cations, see:Olah GA, Krishnamurti R, Prakash GKS. J. Org. Chem. 1990;55:6061.Olah GA, Spear RJ, Westerman PW, Denis J-M. J. Am. Chem. Soc. 1974;96:5855.
- 25.For Lewis acid-catalyzed allylation of propargyl acetates with allylsilanes, see: refs 18b,c.
- 26.For mechanistic discussions on regioselectivity of nucleophilic attacks on proparyl cations, see: ref 18c.
- 27.Due to severe signal overlap, quantification of isotopomers was not possible.
- 28.For Kumada cross-coupling of enol phosphates, see:Kobayashi Y, Takeuchi A, Wang Y-G. Org. Lett. 2006;8:2699. doi: 10.1021/ol060692h.Larsen US, Mariny L, Begtrup M. Tetrahedron Lett. 2005;46:4261.Verboom RC, Persson BA, Baeckvall J-E. J. Org. Chem. 2004;69:3102. doi: 10.1021/jo0357667.Hayashi T, Konishi M, Okamoto Y, Kabeta K, Kumada M. J. Org. Chem. 1986;51:3772.Sahlberg C, Quader A, Claesson A. Tetrahedron Lett. 1983;24:5137.Hayashi T, Fujiwa T, Okamoto Y, Katsuro Y, Kumada M. Synthesis. 1981:1001.
- 29.For Kumada cross-coupling of aryl phosphates, see:Huang WG, Jiang YY, Li Q, Li J, Li JY, Lu W, Cai JC. Tetrahedron. 2005;61:1863.Hayashi T, Katsuro Y, Okamoto Y, Kumada M. Tetrahedron Lett. 1981;22:4449.
- 30.(R)-(-)-Di-tert-butyl-{1-[(S)-2-(dicyclohexylphosphanyl)ferrocenyl]ethyl}phosphine [158923−11−6], Strem catalog number 26−0975. For examples on use, see:Fernández-Rodríguez MA, Shen Q, Hartwig JF. Chem. Eur. J. 2006;30:7782. doi: 10.1002/chem.200600949.Shen Q, Shekhar S, Stambuli JP, Hartwig JF. Angew. Chem., Int. Ed. 2005;44:1371. doi: 10.1002/anie.200462629.
- 31.Mikami K, Yoshida A. Tetrahedron. 2001;57:889. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.