

Abstract
Protective antigen (PA) is the cell surface recognition moiety of the Bacillus anthracis A-B toxin system, and the active immunogenic component in the currently licensed human anthrax vaccine (BioThrax™, or AVA). The serum antibody response to the PA protein is polyclonal and complex both in terms of the antibody combining sites utilized to bind PA and the PA-associated epitopes recognized. We have cloned, sequenced, and expressed a large panel of PA-specific human monoclonal antibodies from 7 AVA-immunized donors. Dot blots, Western blots, and radio-labeled antigen capture assays employing both proteolytic fragments of PA and engineered PA sub-domain fusion proteins were used to determine the region (domain) of the PA monomer to which each of the cloned human antibodies bound. The domain specificity of the isolated monoclonals was highly biased towards the amino-terminal 20kd fragment of PA (PA20), with the majority (62%) of independently arising antibody clones reacting with determinants located on this PA fragment. A similar bias in domain specificity was also demonstrated in the serum response of AVA-vaccinated donors. Since PA20 is cleaved from the remainder of the monomer rapidly following cell surface binding and has no known role in the intoxication process, the immunodominance of PA20-associated epitopes may directly affect the efficacy of PA-based anthrax vaccines.
Introduction
The Bacillus anthracis binary toxins are major determinants of virulence in anthrax infection. The cell surface recognition element of this toxin system is an 83kd protein known as protective antigen (PA). PA83 is the primary immunogenic component of the currently licensed anthrax vaccine (BioThrax™, or AVA), and recent attempts to develop a “second generation” anthrax vaccine more contemporary in design and formulation have also been based on a recombinant form of PA83. The importance of PA as a vaccine target has driven a significant amount of research into both the biology and immunobiology of this protein toxin.
The role played by PA in toxin function is complex. PA83 recognizes and binds to the cell surface receptors Tumor Endothelial Marker 8 (TEM8) and the capillary morphogenesis gene 2 product (CMG2) [1, 2]. After binding, PA is cleaved by cell associated furin proteases to release the 20kd amino-terminal portion of the molecule (PA20), which has no further role in intoxication. Cell-bound PA63 then self-associates to form a heptameric pre-pore structure that can bind several molecules of the catalytic toxin components lethal factor (LF) and edema factor (EF). Following receptor-mediated endocytosis, the toxin complex inserts into the membrane of the endocytic vacuole and LF/EF is actively translocated into the cytoplasm of the cell. The structure of PA, both as a monomer and heptamer, has recently been determined [3, 4], and the regions of the molecule (domains) involved in the various functions described above have been identified [3–7].
The molecular basis of the immune response to PA in vaccinated humans has only recently been explored in detail. As a large protein antigen, PA would be expected to elicit a polyclonal antibody response, and initial studies indicate this to be the case [8]. Most individual (monoclonal) PA-specific antibodies are not capable of neutralizing toxin function in vitro, suggesting that antibody binding alone is insufficient, and that a particular function of PA must be blocked for toxin neutralization to occur [9]. The intricate role played by PA during intoxication suggests several points at which individual antibodies might inhibit toxin function. These include blocking receptor binding, preventing LF and or EF association, interfering with heptamer formation, or blocking the proteolytic cleavage of PA20. Several murine hybridomas that neutralize toxin have been demonstrated to function by one or another of these modalities[9–11].
We have isolated and characterized a large panel of human PA-specific monoclonal antibodies from multiple AVA immunized donors. In this report, we examine the epitope specificity of the individual antibody binding domains (paratopes).We find that a large and disproportionate number of paratopes are specific for determinants associated with the PA20 region of the PA monomer. We determine this domain bias to be present in the polyclonal serum antibodies of vaccinated donors as well. Since PA20 is rapidly cleaved from the remainder of the molecule following cell surface binding and has no known role in intoxication, this epitope bias may be of consequence in terms of the function and efficacy of PA-based anthrax vaccines. Understanding the mechanism underlying this biased antibody response would facilitate the design and formulation of more effective "next generation" vaccines to prevent anthrax.
Materials and Methods
Subjects
The donors analyzed in this report were recruited from individuals taking part in a larger study of the response to AVA being conducted at Baylor College of Medicine. Human subject protocols were reviewed and approved by the Institutional Review Boards at both Children’s Hospital Oakland and Baylor College of Medicine.
Construction of Fab expression libraries
Fab expression libraries were constructed from mononuclear cells (MNCs) enriched for PA-specific B cells in a manner similar to that previously described for PA and polysaccharide-specific antibody expression libraries [8, 12–15]. PA83, PA20, and PA63 were purchased from List Biological Laboratories, Campbell, CA. PA-specific Fabs were identified using a sensitive 125I-labeled PA capture assay and lysates of individual E. coli expression cultures. Positive isolates were re-cloned, heavy (H) and light (L) chain gene sequence determined, and PA-specific binding confirmed by ELISA. Initial sequence analysis utilized the NCBI IgBlast server (http://www.ncbi.nlm.nih.gov/igblast/) to identify candidate germline gene [16]. Subsequent analysis, alignments and translations were performed using MacVector (Accelrys Inc, Princeton, NJ). H chain V region gene nomenclature is as described in the IMGT database [17, 18]. Complementarity determining regions (CDRs) are as defined in [19]. Selected Fab clones were converted to full chain IgG1 antibodies and expressed in Chinese Hamster Ovary (CHO) cells using an in house PCI (Promega, Madison, WI)-derived bicistronic eukaryotic expression vector and the Flp-in system from Invitrogen (Carlsbad, CA). Antibody was concentrated from the cell culture supernatant for use in binding assays.
Construction of PA20- and D4-GFP fusion proteins
The amino-terminal (residues 1–191) and the domain 4 carboxy-terminal (residues 587 – 735) portion of the PA monomer were cloned using PCR and expressed fused to intact green fluorescent protein (GFP). Cloning primers for the amino-terminal fragment were ATATGAATTCTATGGAAGTTAAACAGGAGAACCG (5’) and ATATGGATCCTCCTTCTACCTCTAATGAATC (3’). Cloning primers for the domain 4 region were GCATTAGAATTCGCATCACCATCACCATCACATGAATATTTTAATAAGAGATAAACG (5’) and CGTATATCTAGAAGGATCCCCTATCTCATAGCCTTTTTTAGAAAAGAT (3’). Fusion proteins were expressed in E. coli and purified by nickel-chelate chromatography.
Domain specificity of PA-specific antibodies
The domain specificity of individual PA-specific antibodies was determined using capture assays, dot blots, western blots of proteolytic fragments of PA, and western blots of PA20- and D4-GFP fusion proteins. In capture assays, 96-well plates coated with light chain-specific antibody were used to capture individual PA-specific monoclonal antibodies. Plates were then washed and incubated with radiolabeled PA83, PA63, PA20, or D4-GFP. Binding was detected using PhosphorImager detection plates (Molecular Dynamics, Sunnyvale, CA). For Western blots of proteolytic PA fragments, 1 µg each of PA83, PA63, and PA20 were electrophoresed using 4–12% Bis-Tris polyacrylamide gels (NuPAGE, Carlsbad, CA), electrically transferred to nitrocellulose membranes, and probed with individual PA-specific antibodies. Binding was visualized by means of an alkaline-phosphatase conjugated goat antibody specific for human kappa or lambda light chains followed by BCIP/NBT color development. Western blots of PA20- and D4-GFP fusion proteins were processed in a similar fashion. For dot blots, the above described PA and PA-derived proteins were spotted onto nitrocellulose membranes using a 96-well manifold. The resulting membrane was then cut into strips such that each strip contained one spot for each protein, and the individual strips probed with PA-specific antibodies as described above.
Antigen binding and Fab concentration assays
Fab concentration was determined by a capture ELISA in which goat anti-human Fd (The Binding Site, Birmingham, UK) or goat anti-IgA (Sigma, St. Louis, MO) immobilized on a microtiter plate captures Fab which is then detected by alkaline-phosphatase labeled goat anti-human L chain (Biosource International, Camarillo, CA). This assay is standardized with a purified Fab standard whose concentration was calculated from UV absorbance at 280 nm. Binding in ELISA was determined for both Fabs and full-chain IgG1 antibodies on 96-well plates coated with 5 µg/ml PA83 and developed with alkaline-phosphatase conjugated goat antibody specific for human kappa or lambda light chains.
Serum inhibition studies
The ability of PA83, PA63, PA20-GFP, and PA Domain 4 (D4-GFP) to inhibit post-vaccination serum antibody binding to PA was determined in an antigen specific ELISA assay. PA83 and PA63 were purchased from List Biological Laboratories, Campbell, CA. PA20 and D4 were GFP-fusion proteins as described above. Serum diluted to achieve approximately 50% maximum binding was incubated with inhibitors overnight at 4°C prior to addition to the ELISA binding plate. All inhibitors were present in equimolar concentrations, and in at least 150 fold excess to PA-specific antibody present in the diluted sample.
Results
Construction and analysis of antibody expression libraries
Fourteen individual recombinant antibody expressions libraries (kappa and lambda) were constructed in E. coli using MNCs isolated from 7 AVA vaccinated donors that had received at least 4 injections. Approximately 48,000 individual clones were screened using a radiolabeled PA capture assay, and approximately 250 PA-specific human monoclonal antibody Fab fragments were isolated. All isolates were re-cloned, re-screened, and soluble Fab protein produced for PA binding and specificity assays. Antibody-coding sequences from selected clones were transferred into an eukaryotic expression vector and expressed as full IgG1 antibodies in CHO cells. The ability of the isolated paratopes to bind PA was verified in ELISA, 125I-PA capture assays, dot blots, and Western blots (see below) using Fabs expressed in E. coli and full chain IgG1 antibodies expressed in CHO cells when available. Examples of PA-specific binding by individual monoclonal antibodies in an ELISA assay are shown in Figure 1. The gene sequence of the antibody variable (V) region genes were determined and the most likely germline V gene of origin assigned using the NCBI IgBlast server [16]. Within each donor, unique heavy (H) chain V region rearrangements were identified based on V gene usage and the sequence of the third complementarity-determining region (CDR3). Sequence analysis identified 121 sequence-unique antibody Fabs representing 64 unique rearrangements (families) and their somatically mutated progeny. Although extensive, our screening was not exhaustive, and additional sampling would be expected to reveal additional PA-specific binding domains.
Figure 1.
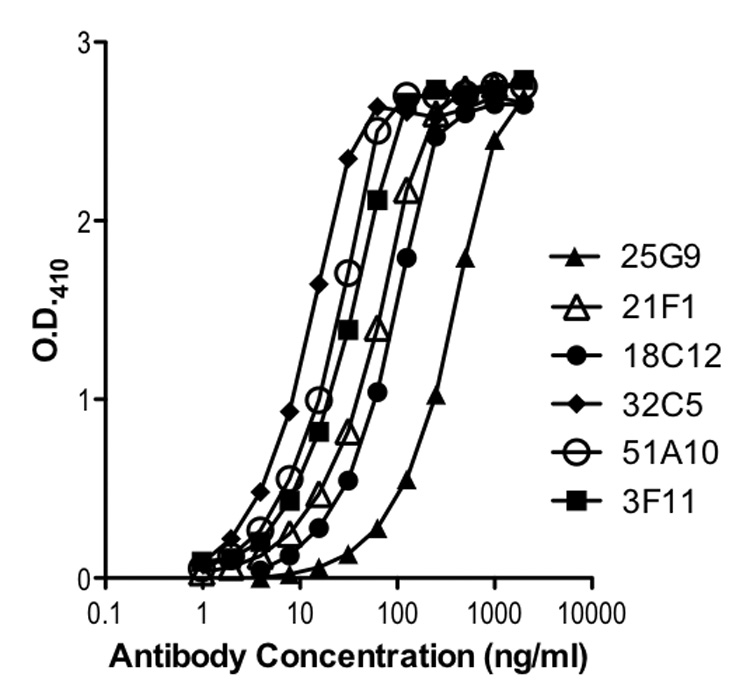
Protective Antigen (PA) binding by representative CHO cell-derived IgG1 PA-specific human monoclonal antibodies.
Domain specificity of individual PA-specific human antibodies
Radiolabeled antigen capture assays, dot blots, and Western blots were utilized to determine the region (domain) of PA to which the individual antibodies bound. In these assays, native PA, its proteolytic fragments PA20 and PA63, and engineered and expressed PA-fragment fusion proteins were used as antigens. The two fusion proteins consisted of the amino-terminal domain (residues 1–191) of PA fused to GFP (PA20-GFP), and the carboxy-terminal domain 4 (residues 587 –735) of the PA monomer fused to GFP (D4-GFP). In the antigen capture assay, immobilized Fabs were allowed to capture 125I-labeled antigen, and binding was detected using a PhosphorImager (see materials and methods). For the dot blot assay, 1 µg each of the 5 antigens described above were spotted onto a nitrocellulose membrane using a 96-well format vacuum manifold and the membrane probed with individual antibodies. Western blots were carried out using the 5 antigens described above and standard methodologies. In the dot blot and Western assays, membranes were probed with both Fabs and, when available, full chain IgG1 antibodies (58 of the 64 antibody families were available as CHO-expressed IgG1 antibodies). Examples of the dot blot results are shown in Figure 2. When individual antibodies were compared in the three assays, none were found to be contradictory in terms of the region of the PA monomer bound. A few paratopes were not internally consistent across all assays. A clone that bound the PA20 proteolytic fragment and PA20-GFP fusion protein in the Western blot might bind PA20 poorly or not at all in the capture assay, for example. We believe these cases are indicative of an alteration of epitope structure in the different methods of fragment preparation and presentation, and highlight the necessity of utilizing multiple methodologies when assaying epitope specificity. There were also a few clones that appeared to be specific for PA63-associated epitopes but reacted weakly with the PA20 proteolytic fragment in the dot blot. These did not bind the PA20-GFP fusion protein in the dot blot, and resolved to PA63 binders in the Western blot. This is most likely due to trace contamination of the PA20 preparation with either PA63 or intact PA. In those families where multiple somatically mutated members were isolated, all members mapped to the same PA fragment. The VH3-15 family of antibodies isolated from donor 5, for example, had 12 sequence-distinct members. All members of this family mapped to PA20. Clones 16A1, 24A10, and 29G6 (donor 3) and clones 18G7 and 40E2 (donor 7) were specific for native PA in the ELISA assay, the capture assay, dot blots, and the Western assay, but failed to react with any of the PA fragments. This might occur if the required epitope spanned the junction between the individual fragments, or if the epitope was not preserved during the manipulations required for the three assays. The domain specificity of these 5 (7.8%) clones remains undetermined (ND). Overall, 40 (62.5%) of the 64 independent isolates were determined to be specific for epitopes present in the PA20 portion of the molecule, 14 (21.9%) were specific for determinants in PA63 (excluding D4), and 5 (7.8%) bound to epitopes present in the carboxy-terminal D4 region. The domain specificity of the 64 antibody families is shown in Table 1a and 1b, and is summarized in Table 2.
Figure 2.
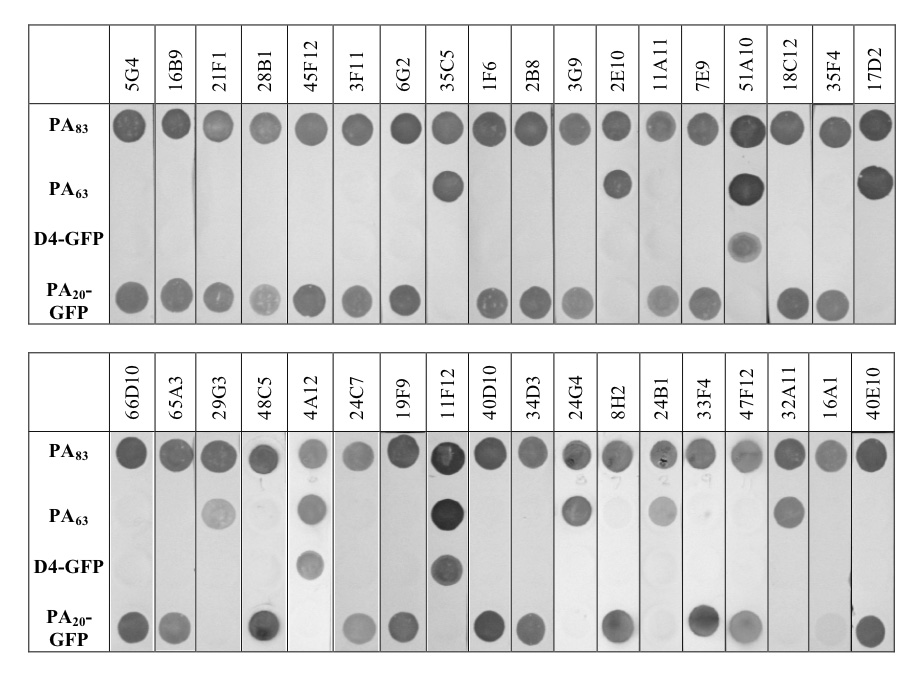
Dot blot assay of representative CHO cell-derived IgG1 PA-specific human monoclonal antibodies. PA83 and PA63 were purchased from List Biological Laboratories. D4-GFP and PA20-GFP were expressed and purified from E. coli as described in the materials and methods section. All antigens were spotted at 1 µg per spot using a 96 well manifold. Blots were cut into strips and probed with individual PA-specific monoclonal antibodies.
Table 1.
Table 1a Domain specificity of the human PA-specific monoclonal antibodies reported in this study. Table 1b Domain specificity of the human PA-specific monoclonal antibodies reported in this study.
| Table 1a | |||||
|---|---|---|---|---|---|
| Clone | VHa | VH CDR3b | Nc | Domain Specificity | |
| Donor 1 | 25G9 | VH1-18 | DIGTIFGVVIDFFDY | 1 | PA20 |
| 45F12 | VH1-2 | SGRPVEY | 1 | PA20 | |
| 47H5 | VH3-11 | DLDVGGYSSSAY | 1 | D4 | |
| 14E2 | VH3-15 | DGPMMQVLTDPRRSDYSYYTLDV | 1 | PA63 | |
| 28B1 | VH3-30 | DRIIVPGRDDYQYYGMDV | 3 | PA20 | |
| 5G4 | VH3-30 | NRVIIPRMAVPGPAAFDL | 1 | PA20 | |
| 51A10 | VH3-30 | PRAVFGVVIGYYFDF | 1 | D4 | |
| 16B9 | VH3-33 | DRVIVPADTSLSGDAFDL | 1 | PA20 | |
| 21F1 | VH3-33 | ERVIIPRTAVAGQAAFDI | 2 | PA20 | |
| 39C2 | VH3-33 | EGVIVPAGSYHYYYYMDV | 1 | PA20 | |
| 48C5 | VH4-39 | HDGKVQGVIFPGGQHMDV | 1 | PA20 | |
| Donor 2 | 3F11 | VH3-30 | ARVIVPAGSNYNQYGMDV | 1 | PA20 |
| Donor 3 | 41C2 | VH1-2 | RGAPVDY | 1 | PA20 |
| 41F6 | VH3-15 | EILGG | 1 | PA20 | |
| 16A1 | VH3-21 | EKDYYDGRGYSSWFDP | 1 | N.D. | |
| 40E10 | VH3-23 | DPESFLESLPTYYFDY | 1 | PA20 | |
| 50A4 | VH3-23 | GPGPPNQSRRVTMIVLPPPRWFDP | 1 | PA63 | |
| 55E3 | VH3-30 | DPYGADEGYYGMDV | 1 | PA20 | |
| 33G8 | VH3-30 | DRIIVPNPYSYYGLDV | 1 | PA20 | |
| 19F9 | VH3-30 | DRVIIPGTMLYYYYGMDV | 2 | PA20 | |
| 34D3 | VH3-33 | DSVIVPAVTSPRGFGMDV | 2 | PA20 | |
| 13D12 | VH3-33 | SSGSYQQPIEY | 1 | PA20 | |
| 6G2 | VH3-33 | VDGSYHQNADY | 2 | PA20 | |
| 24A10 | VH3-48 | ESGLYCGGECYSPSFDP | 2 | N.D. | |
| 29G6 | VH3-74 | GYRFGWDV | 1 | N.D. | |
| 47F9 | VH3-9 | DVGAYYYDSSGYRSAFDI | 1 | PA20 | |
| 13C10 | VH4-31 | GVGSGYYYGARNAFDI | 1 | PA20 | |
| 39B8 | VH5-51 | LGLRAIIPFDS | 1 | PA20 | |
| Table 1b | |||||
|---|---|---|---|---|---|
| Clone | VHa | VH CDR3b | Nc | Domain Specificity | |
| Donor 4 | 18C12 | VH1-2 | LAAVAGDY | 1 | PA20 |
| 33F3 | VH1-2 | TEDIVLGVAAKPHAHFDY | 1 | PA63 | |
| 24C7 | VH3-21 | LDGGDYAFDN | 1 | PA20 | |
| 59G4 | VH3-30 | ARVIVPAGSNYNQYGMDV | 1 | PA20 | |
| 3G9 | VH3-30 | DRVIVPAANRNYYYYGMDV | 6 | PA20 | |
| 2B8 | VH3-30 | DRVIVPGAHGYAYYGVDV | 4 | PA20 | |
| 1F6 | VH3-30 | EPAIVPARNSQHFFGMDV | 4 | PA20 | |
| 13C7 | VH3-30 | ERWTGILDY | 1 | PA63 | |
| 7E9 | VH3-74 | VEDIEMAELDY | 1 | PA20 | |
| 24B1 | VH3-9 | DMYGGGGYFFAK | 1 | PA63 | |
| 32C5 | VH4-34 | VTSAIAVTSTRWYIDL | 1 | PA63 | |
| 66D10 | VH4-39 | DNGGHLTFVVVDS | 1 | PA20 | |
| 32E12 | VH4-4 | DKDYFISGSYYNWFDP | 1 | PA63 | |
| Donor 5 | 11A11 | VH1-18 | DRGSRWFGEFPDEYYFDY | 1 | PA20 |
| 4A12 | VH1-46 | VNWAYGDYDFDY | 1 | D4 | |
| 40D10 | VH3-15 | DVLGIVIIVGAAH | 12 | PA20 | |
| 11F12 | VH3-23 | VVGADLRFDY | 1 | D4 | |
| 17D2 | VH3-30 | WDYVWESYRGKAFDI | 1 | PA63 | |
| 65A3 | VH3-30 | LISYDGNTKYYADSVKG | 1 | PA20 | |
| 47F12 | VH3-30 | ARVIVPAGSNYNQYGMDV | 1 | PA20 | |
| 8H2 | VH3-33 | WGYYYGSGSPPEY | 1 | PA20 | |
| 41F12 | VH3-33 | EDGSYHQGPFDY | 1 | PA20 | |
| 20C7 | VH3-43 | GPNRRDSFGLHYYGLDV | 1 | D4 | |
| 24G4 | VH3-53 | APQYDLWTGPLYGMDV | 1 | PA63 | |
| 51C5 | VH3-74 | EGRPMGLGTSVGMN | 1 | PA20 | |
| 1A5 | VH4-59 | GDMVTGDPGDY | 4 | PA63 | |
| 2E10 | VH5-51 | QSSNWEDYFQH | 1 | PA63 | |
| Donor 6 | 2E7 | VH3-23 | DQNYIDYAPSRRGSHYFYALDV | 1 | PA20 |
| Donor 7 | 18G7 | VH3-21 | EGEHSGSGSRYGMDV | 1 | N.D. |
| 8C11 | VH3-30 | EGVIVPAASNKKNYYFDL | 2 | PA20 | |
| 35F4 | VH3-30 | DRVIIPRTSAYYYYGMDV | 22 | PA20 | |
| 32A11 | VH3-30 | ERLTGILDY | 1 | PA63 | |
| 17A9 | VH3-30 | GGGSGSSDY | 1 | PA63 | |
| 29G3 | VH3-30 | ADYAGGRRFDL | 1 | PA63 | |
| 28E3 | VH3-49 | ADYNNRNYASDV | 4 | PA20 | |
| 40E2 | VH4-59 | HGKYGGFSSGWFDP | 1 | N.D. | |
Germline H chain variable region gene used by the individual monoclonal antibodies.
Heavy chain CDR3 region used to define independent B-cell rearrangements (families).
Number of sequence unique, somatically-derived members of each rearrangement (family) isolated.
Domain specificity of individual antibodies. Specificity was list as N.D. if no conclusive assignment could be made (see text). Listing continued in Table 1b.
Germline H chain variable region gene used by the individual monoclonal antibodies.
Heavy chain CDR3 region used to define independent B-cell rearrangements (families).
Number of sequence unique, somatically-derived members of each rearrangement (family) isolated.
Domain specificity of individual antibodies. Specificity was list as N.D. if no conclusive assignment could be made (see text).
Table 2.
Summary of domain specificity for all isolated antibodies (detailed in Tables 1a and 1b).
| Donor | Familiesa | Nb | PA20 | PA63 | D4 | NDc |
|---|---|---|---|---|---|---|
| 1 | 11 | 14 | 8 | 1 | 2 | 0 |
| 2 | 1 | 1 | 1 | 0 | 0 | 0 |
| 3 | 16 | 20 | 12 | 1 | 0 | 3 |
| 4 | 13 | 24 | 8 | 5 | 0 | 0 |
| 5 | 14 | 28 | 7 | 4 | 3 | 0 |
| 6 | 1 | 1 | 1 | 0 | 0 | 0 |
| 7 | 8 | 33 | 3 | 3 | 0 | 2 |
| Totals | 64 | 121 | 40 | 14 | 5 | 5 |
Number of paratope families (as defined by unique B cell rearrangements) isolated for each donor.
Number of sequence unique, somatically-derived members of each family isolated.
Domain specificity could not be assigned.
Domain specificity of post vaccination serum antibodies
The finding of a significant bias towards PA20 associated epitopes in the panel of human PA-specific monoclonal antibodies was unexpected and prompted us to determine if such a bias was present in the polyclonal serum antibody response following vaccination. Eleven such sera were tested to determine the degree to which antibody binding to native PA could be inhibited by the PA fragments used in the assays described above. PA20-GFP and D4-GFP were employed to remove the possibility of contamination with native PA or unintended fragments. No engineered PA63 fragment was available, necessitating the use of the proteolytically derived PA63 (any trace contamination of PA63 with PA20 or native PA would lead to an overestimation of PA63-specific antibody). The amount of PA-specific antibody present in each sera was determined by comparison to the standard reference serum AVR801 (generously supplied by Conrad P. Quinn, Centers for Disease Control and Prevention, Atlanta, GA [20]). A serum dilution was selected that resulted in approximately 50% maximum binding, and diluted serum and inhibitors were combined and incubated overnight prior to their addition to the ELISA assay. Inhibitors were in at least 150-fold molar excess to serum antibody at the serum dilution utilized. All inhibitors were present at equimolar concentrations. The values for percent inhibition with PA63, D4, and PA20 were calculated as a percentage of the total amount of antibody binding inhibitable with native PA (PA83), and are shown in Figure 3. These results indicate that the serum antibody response following vaccination is also biased toward determinants associated with the PA20 portion of the molecule.
Figure 3.
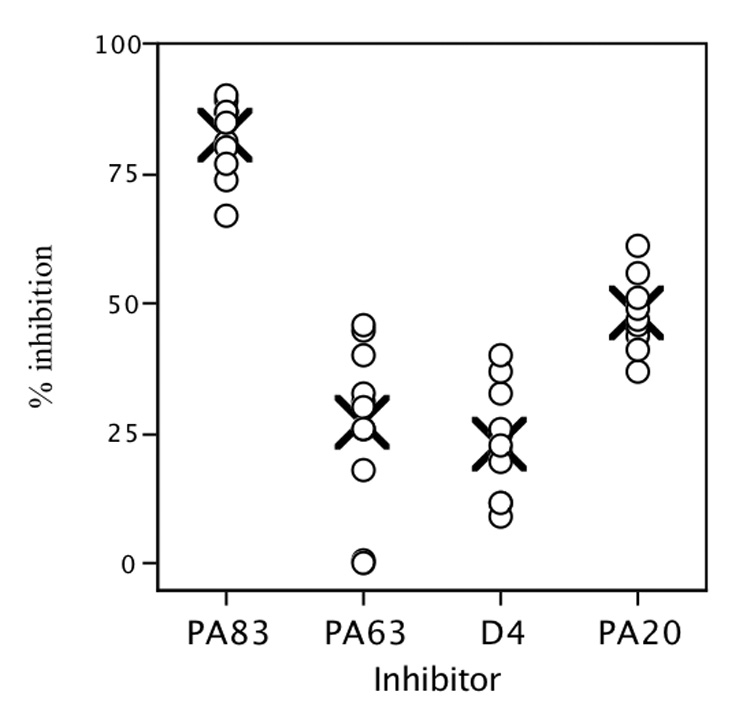
Inhibition of post AVA vaccination serum binding to PA in an antigen-specific ELISA. PA63 is a proteolytic fragment of PA83. Domain 4 (D4) and PA20 fragments were expressed as GFP fusion proteins. All inhibitors were present at approximately 150 fold excess of antibody, and in equal molar concentrations. Values for PA63, PA20, and D4 are expressed as the percentage of PA83-inhibitable binding. For each inhibitor, X indicates the mean values for the 11 donors.
Discussion
Anthrax Vaccine Absorbed (AVA or BioThrax™), manufactured by Emergent BioSolutions (Rockville, MD; formerly BioPort) is the vaccine approved for human use in the United States. It is prepared from a sterile culture filtrate of an avirulent, non-encapsulated derivative of the Sterne strain (V770-NP1-R) that is grown in a defined, protein free medium. The filtrate is absorbed with aluminum hydroxide and contains benzethonium chloride and formaldehyde as preservatives [21]. Although its composition is not well defined, it is widely accepted that the primary protective immunogen in the filtrate-based vaccines is the toxin component PA [22]. Several lines of evidence support this assertion. Strains that have lost the pXO1 plasmid and concomitant PA production fail to produce protective immunity [23]. Multiple animal studies have shown that vaccination with PA alone, produced either from B. anthracis or through recombinant DNA technology, is sufficient to produce immunity to infection. [24, 25]. A “DNA vaccine” encoding PA has been shown to provide protection against infection [26, 27]. It is interesting that although protection from infection is believed to be mediated by antibodies that block PA function, the protection afforded by vaccination with PA against different B. anthracis isolates varies, and it is often difficult to directly correlate anti-PA binding titers with in vitro toxin neutralization or immunity [28–30]. In addition to their ability to directly inhibit intoxication, there is evidence that PA-specific antibodies are also active against spores [31]. No mechanism for this activity has been proposed.
Over 1.4 million people have been vaccinated with AVA, and studies have demonstrated the vaccine to be safe [32–34]. In spite of these facts, there remains significant resistance to AVA among military personnel and the public at large. This resistance, the undefined nature of the vaccine itself, and the extended dosage schedule has prompted calls for the creation of a new vaccine, more in line with contemporary standards of composition and performance. To this end, the development of a vaccine based on a recombinant form of PA (rPA) was initiated. The recombinant vaccine (rPA102) went into phase I clinical trials to determine immunogenicity and safety [35]. Initial results indicated the product to be safe, but immunogenicity did not appear significantly improved over AVA [35, 36].
Although serum from PA-vaccinated animals and humans can readily be demonstrated to neutralize anthrax toxin in vitro, the majority of monoclonal antibodies isolated from either vaccinated mice [9] or humans [37] fail to neutralize PA-mediated cytotoxicity. This is in spite of their ability to bind toxin with high avidity in standard assays of antigen binding. Such findings demonstrate that PA binding alone is insufficient for antibody-mediated neutralization, and suggests that to be effective, an antibody must bind to PA in a manner that interferes with one of the obligatory PA functions. For example, an antibody that recognized an epitope located in the region of the PA monomer involved with cell surface receptor recognition might neutralize toxin by preventing receptor binding. Many of the residues involved in the various functions ascribed to PA during intoxication have been determined, and their location within the various domains of the PA monomer established. Defining the domain specificity of the individual antibody paratopes that comprise the combined serum response to vaccination therefore provides an insight to how they may function to block toxin function during infection. The analytical techniques we employ provide access to the individual antibody paratopes that comprise the overall response, and thus allow us to investigate the functional contribution made by each to the serum antibody pool.
In this report we have demonstrated that the majority of individual antibody paratopes arising following vaccination are specific for determinants that reside in the amino-terminal PA20 region of the toxin monomer. Sixty-two percent of the human monoclonal antibodies we isolated were specific for determinants associated with this fragment, which comprises only 25% of the overall molecular mass of the toxin. Since the majority of functions required for PA-mediated toxicity, such as receptor binding, heptamer formation, LF/EF binding, membrane insertion, and toxin translocation all map to regions of the monomer located within PA63 [3, 6, 7, 38], it is possible that antibodies recognizing determinants in the PA20 region of PA may be deficient in their ability to effectively neutralize toxin. Current studies are underway to determine if PA20-specific monoclonal antibodies differ from those directed towards the more functionally relevant regions of PA in their ability to neutralize PA-mediated cytotoxicity.
It is difficult to determine with certainty that the procedures we employ to generate and identify PA-specific Fabs do not in some way bias our results towards paratopes that bind a particular sub-domain of the molecule, but we believe this to be unlikely. Gel analysis and silver staining of the PA preparation used for biotinylation, cell selection, radio-iodination, and screening indicated a high degree of purity free of any significant contamination with smaller proteolytic fragments. Reactive residues utilized for biotinylation and radio-iodination are evenly distributed throughout the molecule. The use of a soluble antigen capture assay during the initial screening of individual colonies and identification of PA-binding Fabs maximizes the epitope integrity of the antigen. The complexity of the antibody V gene repertoire we isolated in the PA-specific response makes it unlikely that an epitope bias was introduced during the initial PCR reaction based on primer design. Together, these factors suggest that the epitope distribution we observe for our isolated Fabs reflects that present in the ongoing immune response in vaccinated individuals. Significant support for this conclusion is provided by the data presented in figure 3. Measurement of the ability of PA-derived fragments to inhibit the binding of post-vaccination serum antibody to PA in the ELISA is free of any artifactual epitope bias possibly introduced by the paratope cloning methodology we employed to isolate and identify the PA-specific Fabs. Results obtained using this inhibition assay also demonstrate a significant bias in the overall antibody response towards epitopes present in the amino-terminal PA20 portion of the PA monomer. The finding of a similar epitope bias using two assays so disparate in their methodologies strongly supports our conclusion that the human antibody response following vaccination with AVA is biased towards PA20-associated epitopes.
The mechanisms that bias the distribution of responding antibody clones towards PA20 are unknown. The rapid internalization of PA63 once PA83 has bound the cell surface and been cleaved may sequester it within the target cells and render it less available for B cell interaction. Since PA20 remains in the extracellular space and available to the immune system, the altered molar ratio of PA20 to PA63 might bias the response towards epitopes associated with PA20. It has also been shown that PA83 binds directly to B cells by means of the PA receptor. In the presence of LF, this binding leads to an impairment of B cell function [39]. The effects on B cell function of PA binding alone, or PA along with trace amounts of LF possibly present in the anthrax vaccine are unknown. It is also possible that differences in antigen processing between cell-associated PA63 and unbound PA20 may contribute to the observed bias in epitope recognition. During antigen processing, foreign proteins are degraded in the endosomes of dendritic and other cells of the immune systems [40]. Peptide fragments of these proteins are associated with MHC class II molecules and transported to and displayed on the cell surface. Naïve T cells encounter these displayed peptides, and those with receptors specific for MHC-bound peptide determinants proliferate and become activated. These peptide-specific helper T cells provide the signals and cytokines that responding B cells require in order to proliferate and differentiate into antibody secreting and memory B cells. Following vaccination or infection, PA20 is cleaved from cell-bound PA83 rapidly and therefore would be available to enter the “normal” antigen-processing pathway. PA63 on the other hand, actively directs its own entry into the cell. The presence of the PA63 heptamer modifies both the endosomal membrane and the endosomal environment, and may disrupt the complex vacuole trafficking or other steps required for efficient antigen processing and peptide presentation. The result would be that peptides derived from the PA63 region of PA are less efficiently processed and presented as compared to peptides from PA20. This would in turn lead to a PA-specific helper T cell population that is biased towards epitopes present on PA20. In addition, little is known about the effects adjuvant and/or preservatives present in AVA have on antigen presentation, and it is possible that these alter antigen presentation in a manner that biases the response towards PA20-associated determinants.
The functional consequences, if any, of such an epitope bias are also unknown. The majority of molecular interactions required for PA-mediated endocytosis of LF and EF map to determinants located in the PA63 region of the molecule, and antibodies that interact with this part of the molecule are more likely to inhibit toxin function. Antibodies specific for PA20-asociated epitopes may be capable of blocking the required proteolytic cleavage of PA83 at the cell surface, thereby neutralizing PA function. It is also possible that PA20-specific antibodies that do neutralize toxin (by blocking furin cleavage) might be rendered less effective by free PA20 that is generated during intoxication by protease cleavage. It has recently been shown in a murine model that PA is cleaved in vivo independent of cell surface binding, and that both PA20 and PA63 proteolytic fragments circulate in the bloodstream [41]. If this also occurs in humans, circulating PA20 would compete with and possibly render neutralizing PA20-specific antibodies ineffective.
Our findings of a biased epitope distribution in the human immune response to AVA suggest that factors intrinsic to native PA and its proteolytic processing may diminish its effectiveness when used as an immunogen to induce toxin-neutralizing antibodies. These factors would likely influence the antibody response to “second generation” vaccines based on rPA as well. Although the mechanisms responsible for this bias are unknown, it may be possible, through a minimal alteration of the PA primary sequence, to shift the epitope bias of the response towards the more functionally relevant PA63 portion of the molecule. A vaccine for anthrax, rationally designed and mechanism based, may prove more effective than either the first- or second-generation vaccines currently being evaluated. Studies are currently underway in our laboratory utilizing a murine model and sequence-altered forms of the PA molecule to determine if this is the case.
Acknowledgments
The authors gratefully acknowledge Nanette Bond, PA-C for assistance with sample collection, and Betty M. Ho for critically reading the manuscript. This work was supported by Public Health Service Grants AI57932 and AI066508 from the National Institute of Allergy and Infectious Diseases. This research was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR-16226 from the National Center for Research Resources, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature
- 1.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414(6860):225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 2.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci U S A. 2003;100(9):5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385(6619):833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 4.Santelli E, Bankston LA, Leppla SH, Liddington RC. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature. 2004;430(7002):905–908. doi: 10.1038/nature02763. [DOI] [PubMed] [Google Scholar]
- 5.Rosovitz MJ, Schuck P, Varughese M, Chopra AP, Mehra V, Singh Y, et al. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for binding to the cellular receptor and to a neutralizing monoclonal antibody. J Biol Chem. 2003;278(33):30936–30944. doi: 10.1074/jbc.M301154200. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham K, Lacy DB, Mogridge J, Collier RJ. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc Natl Acad Sci U S A. 2002;99(10):7049–7053. doi: 10.1073/pnas.062160399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahuja N, Kumar P, Bhatnagar R. Hydrophobic residues Phe552, Phe554, Ile562, Leu566, and Ile574 are required for oligomerization of anthrax protective antigen. Biochem Biophys Res Commun. 2001;287(2):542–549. doi: 10.1006/bbrc.2001.5613. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J, Ullal A, Liberato J, Sun J, Keitel W, Reason DC. Paratope diversity in the human antibody response to Bacillus anthracis protective antigen. Mol Immunol. 2008;45(2):338–347. doi: 10.1016/j.molimm.2007.06.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Little SF, Leppla SH, Cora E. Production and characterization of monoclonal antibodies to the protective antigen component of Bacillus anthracis toxin. Infect Immun. 1988;56(7):1807–1813. doi: 10.1128/iai.56.7.1807-1813.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Little SF, Novak JM, Lowe JR, Leppla SH, Singh Y, Klimpel KR, et al. Characterization of lethal factor binding and cell receptor binding domains of protective antigen of Bacillus anthracis using monoclonal antibodies. Microbiology. 1996;142(Pt 3):707–715. doi: 10.1099/13500872-142-3-707. [DOI] [PubMed] [Google Scholar]
- 11.Gubbins MJ, Berry JD, Corbett CR, Mogridge J, Yuan XY, Schmidt L, et al. Production and characterization of neutralizing monoclonal antibodies that recognize an epitope in domain 2 of Bacillus anthracis protective antigen. FEMS Immunol Med Microbiol. 2006;47(3):436–443. doi: 10.1111/j.1574-695X.2006.00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reason DC, Wagner TC, Lucas AH. Human Fab fragments specific for the Haemophilus influenzae b polysaccharide isolated from a bacteriophage combinatorial library use variable region gene combinations and express an idiotype that mirrors in vivo expression. Infect Immun. 1997;65(1):261–266. doi: 10.1128/iai.65.1.261-266.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reason DC, Zhou J. Codon insertion and deletion functions as a somatic diversification mechanism in human antibody repertoires. Biol Direct. 2006;1:24. doi: 10.1186/1745-6150-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou J, Lottenbach KR, Barenkamp SJ, Lucas AH, Reason DC. Recurrent variable region gene usage and somatic mutation in the human antibody response to the capsular polysaccharide of Streptococcus pneumoniae type 23F. Infect Immun. 2002;70(8):4083–4091. doi: 10.1128/IAI.70.8.4083-4091.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou J, Lottenbach KR, Barenkamp SJ, Reason DC. Somatic hypermutation and diverse immunoglobulin gene usage in the human antibody response to the capsular polysaccharide of Streptococcus pneumoniae Type 6B. Infect Immun. 2004;72(6):3505–3514. doi: 10.1128/IAI.72.6.3505-3514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lefranc MP. IMGT-ONTOLOGY and IMGT databases, tools and Web resources for immunogenetics and immunoinformatics. Mol Immunol. 2004;40(10):647–660. doi: 10.1016/j.molimm.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Matsuda F, Ishii K, Bourvagnet P, Kuma K, Hayashida H, Miyata T, et al. The complete nucleotide sequence of the human immunoglobulin heavy chain variable region locus. J Exp Med. 1998;188(11):2151–2162. doi: 10.1084/jem.188.11.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest. 5 ed. Bethesday: U.S. Department of Health and Human Services; 1991. [Google Scholar]
- 20.Semenova VA, Steward-Clark E, Stamey KL, Taylor TH, Jr, Schmidt DS, Martin SK, et al. Mass value assignment of total and subclass immunoglobulin G in a human standard anthrax reference serum. Clin Diagn Lab Immunol. 2004;11(5):919–923. doi: 10.1128/CDLI.11.5.919-923.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anthrax Vaccine Adsorbed (Biothrax™) Package Insert. 2002. [Google Scholar]
- 22.Leppla SH, Robbins JB, Schneerson R, Shiloach J. Development of an improved vaccine for anthrax. J Clin Invest. 2002;110(2):141–144. doi: 10.1172/JCI16204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pezard C, Weber M, Sirard JC, Berche P, Mock M. Protective immunity induced by Bacillus anthracis toxin-deficient strains. Infect Immun. 1995;63(4):1369–1372. doi: 10.1128/iai.63.4.1369-1372.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivins BE, Welkos SL, Little SF, Crumrine MH, Nelson GO. Immunization against anthrax with Bacillus anthracis protective antigen combined with adjuvants. Infect Immun. 1992;60(2):662–668. doi: 10.1128/iai.60.2.662-668.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McBride BW, Mogg A, Telfer JL, Lever MS, Miller J, Turnbull PC, et al. Protective efficacy of a recombinant protective antigen against Bacillus anthracis challenge and assessment of immunological markers. Vaccine. 1998;16(8):810–817. doi: 10.1016/s0264-410x(97)00268-5. [DOI] [PubMed] [Google Scholar]
- 26.Gu ML, Leppla SH, Klinman DM. Protection against anthrax toxin by vaccination with a DNA plasmid encoding anthrax protective antigen. Vaccine. 1999;17(4):340–344. doi: 10.1016/s0264-410x(98)00210-2. [DOI] [PubMed] [Google Scholar]
- 27.Williamson ED, Beedham RJ, Bennett AM, Perkins SD, Miller J, Baillie LW. Presentation of protective antigen to the mouse immune system: immune sequelae. J Appl Microbiol. 1999;87(2):315–317. doi: 10.1046/j.1365-2672.1999.00901.x. [DOI] [PubMed] [Google Scholar]
- 28.Ivins BE, Pitt ML, Fellows PF, Farchaus JW, Benner GE, Waag DM, et al. Comparative efficacy of experimental anthrax vaccine candidates against inhalation anthrax in rhesus macaques. Vaccine. 1998;16(11–12):1141–1148. doi: 10.1016/s0264-410x(98)80112-6. [DOI] [PubMed] [Google Scholar]
- 29.Turnbull PC, Broster MG, Carman JA, Manchee RJ, Melling J. Development of antibodies to protective antigen and lethal factor components of anthrax toxin in humans and guinea pigs and their relevance to protective immunity. Infect Immun. 1986;52(2):356–363. doi: 10.1128/iai.52.2.356-363.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fellows PF, Linscott MK, Ivins BE, Pitt ML, Rossi CA, Gibbs PH, et al. Efficacy of a human anthrax vaccine in guinea pigs, rabbits, and rhesus macaques against challenge by Bacillus anthracis isolates of diverse geographical origin. Vaccine. 2001;19(23–24):3241–3247. doi: 10.1016/s0264-410x(01)00021-4. [DOI] [PubMed] [Google Scholar]
- 31.Welkos S, Little S, Friedlander A, Fritz D, Fellows P. The role of antibodies to Bacillus anthracis and anthrax toxin components in inhibiting the early stages of infection by anthrax spores. Microbiology. 2001;147(Pt 6):1677–1685. doi: 10.1099/00221287-147-6-1677. [DOI] [PubMed] [Google Scholar]
- 32.Sever JL, Brenner AI, Gale AD, Lyle JM, Moulton LH, West DJ. Safety of anthrax vaccine: a review by the Anthrax Vaccine Expert Committee (AVEC) of adverse events reported to the Vaccine Adverse Event Reporting System (VAERS) Pharmacoepidemiol Drug Saf. 2002;11(3):189–202. doi: 10.1002/pds.712. [DOI] [PubMed] [Google Scholar]
- 33.Martin SW, Tierney BC, Aranas A, Rosenstein NE, Franzke LH, Apicella L, et al. An overview of adverse events reported by participants in CDC's anthrax vaccine and antimicrobial availability program. Pharmacoepidemiol Drug Saf. 2005;14(6):393–401. doi: 10.1002/pds.1085. [DOI] [PubMed] [Google Scholar]
- 34.Sever JL, Brenner AI, Gale AD, Lyle JM, Moulton LH, Ward BJ, et al. Safety of anthrax vaccine: an expanded review and evaluation of adverse events reported to the Vaccine Adverse Event Reporting System (VAERS) Pharmacoepidemiol Drug Saf. 2004;13(12):825–840. doi: 10.1002/pds.936. [DOI] [PubMed] [Google Scholar]
- 35.Gorse GJ, Keitel W, Keyserling H, Taylor DN, Lock M, Alves K, et al. Immunogenicity and tolerance of ascending doses of a recombinant protective antigen (rPA102) anthrax vaccine: a randomized, double-blinded, controlled, multicenter trial. Vaccine. 2006;24(33–34):5950–5959. doi: 10.1016/j.vaccine.2006.05.044. [DOI] [PubMed] [Google Scholar]
- 36.Keitel WA. Recombinant protective antigen 102 (rPA102): profile of a second-generation anthrax vaccine. Expert Rev Vaccines. 2006;5(4):417–430. doi: 10.1586/14760584.5.4.417. [DOI] [PubMed] [Google Scholar]
- 37.Zhou J, Ullal A, Liberato J, Sun J, Keitel W, Reason DC. Frequency and distribution of neutralizing paratopes in the human immune response to the protective antigen of Bacillus anthracis. Manuscript in preparation. 2008 [Google Scholar]
- 38.Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem. 1994;269(32):20607–20612. [PubMed] [Google Scholar]
- 39.Fang H, Xu L, Chen TY, Cyr JM, Frucht DM. Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J Immunol. 2006;176(10):6155–6161. doi: 10.4049/jimmunol.176.10.6155. [DOI] [PubMed] [Google Scholar]
- 40.Brodsky FM, Guagliardi LE. The cell biology of antigen processing and presentation. Annu Rev Immunol. 1991;9:707–744. doi: 10.1146/annurev.iy.09.040191.003423. [DOI] [PubMed] [Google Scholar]
- 41.Moayeri M, Wiggins JF, Leppla SH. Anthrax protective antigen cleavage and clearance from the blood of mice and rats. Infect Immun. 2007;75(11):5175–5184. doi: 10.1128/IAI.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]