

Abstract
Isopenicillin N synthase (IPNS) is a unique mononuclear non-heme Fe enzyme that catalyzes the four electron oxidative double ring closure of its substrate ACV. A combination of spectroscopic techniques including EPR, absorbance, circular dichroism (CD), magnetic CD, and variable-temperature, variable-field MCD (VTVH-MCD) were used to evaluate the geometric and electronic structure of the {FeNO}7 complex of IPNS coordinated with the ACV thiolate ligand. Density Function Theory (DFT) calculations correlated to the spectroscopic data were used to generate an experimentally calibrated bonding description of the Fe-IPNS-ACV-NO complex. New spectroscopic features introduced by the binding of the ACV thiolate at 13,100 and 19,800 cm−1 are assigned as the NO π*(ip) → Fe dx2−y2 and S π → Fe dx2−y2 charge transfer (CT) transitions, respectively. Configuration interaction mixes S CT character into the NO π*(ip) → Fe dx2−y2 CT transition, which is observed experimentally from the VTVH-MCD data from this transition. Calculations on the hypothetical {FeO2}8 complex of Fe-IPNS-ACV reveal that the configuration interaction present in the {FeNO}7 complex results in an unoccupied frontier molecular orbital (FMO) with correct orientation and distal O character for H-atom abstraction from the ACV substrate. The energetics of NO/O2 binding to Fe-IPNS-ACV were evaluated and demonstrate that charge donation from the ACV thiolate ligand renders the formation of the FeIII-superoxide complex energetically favorable, driving the reaction at the Fe center. This single center reaction allows IPNS to avoid the O2 bridged binding generally invoked in other non-heme Fe enzymes that leads to oxygen insertion (i.e. oxygenase function) and determines the oxidase activity of IPNS.
1. Introduction
Mononuclear non-heme iron enzymes catalyze a wide variety of medically, pharmaceutically and biologically important reactions.1,2 These enzymes can be divided into two groups, one which binds an organic substrate to an FeIII center and activates it for reaction with triplet O2 and the other which binds oxygen to an FeII center and activates it for reaction with an organic substrate.3 The oxygen activating enzymes include the α-ketogulatare (αKG)-dependent dioxygenases, the pterin-dependent hydroxylases, the extradiol dioxygenases and the Rieske dioxygenases. These enzymes share a common active site Fe-binding motif of 2 histidines and one carboxylate, known as the facial triad.4 The majority of these enzymes also exhibit a common reactivity, acting as oxygenases that insert either one or two oxygen atoms from O2 into the substrate or co-factor. However, two FeII/O2 activating enzymes, Isopenicillin N-synthase (IPNS) and 1-aminocyclopropane-1-carboxylic acid oxidase (ACCO), have a high sequence homology to the αKG-dependent dioxygenases but exhibit very different reactivity from the oxygenases. Both IPNS and ACCO are oxidases that utilize O2 for substrate oxidation, which is proposed to occur through an initial H-atom abstraction.5–10
Isopenicillin N-synthase is an enzyme found in fungi and bacteria that catalyzes the formation of isopenicillin N, a bicyclic precursor to the β lactam antibiotics including the penicillins and cephalosporins.5–6 IPNS binds a tripeptide substrate δ-(L-α-aminoadipoyl)-L-cysteinyl-D-valine (ACV) and performs a four electron oxidative ring closure, fully reducing one equivalent of O2 to H2O and closing the β lactam and thiazolidine rings of isopenicillin N (Scheme 1).11–14 The crystal structure of the resting form of the enzyme complexed with Mn shows a six-coordinate metal site with two histidines, one aspartate, two waters and a glutamine.15 Upon substrate binding, the crystal structure of the FeII-IPNS-ACV complex shows that the glutamine is displaced by the cysteinyl S of ACV and one of the water molecules is lost, freeing a coordination position for O2 binding.16 From these crystal structures and ACV analogue studies, a reaction mechanism has been proposed for IPNS in which O2 binds end-on to FeII and abstracts a methylene H from the cysteinyl β C in the first step.5,7,17
Scheme 1.
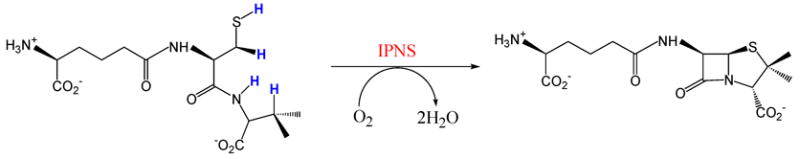
IPNS catalyzes a four electron oxidative double ring closure of ACV to form isopenicillin N.
H-atom abstraction by metal-superoxide complexes has been invoked in the mechanisms of several metalloenzymes, including the noncoupled binuclear Cu enzyme peptidylglycine α-hydroxylating monooxygenase (PHM)18–20 and the binuclear Fe enzyme myo-inositol oxygenase (MIOX).21 Experimentally, a FeIII/FeIII-superoxide intermediate of MIOX has been isolated and characterized by EPR. While the proposed IPNS-ACV reaction mechanism calls for H-atom abstraction by an FeO2 complex, there is little evidence to support the formation of FeIII-superoxide in the reactions of mononuclear non-heme Fe enzymes.
While mononuclear non-heme Fe enzymes are relatively inert to reaction with O2 in the absence of substrate or co-factor, in most cases they react readily with NO to form stable {FeNO}7 complexes both with and without substrates and co-factors.3 In addition, the binding of NO creates an EPR active center, which is also chromophoric, allowing for the application of a variety of spectroscopic techniques.22,23 {FeNO}7 complexes of many mononuclear non-heme Fe enzymes have been prepared including those of phenylalanine hydroxylase24, the Rieske dioxygenases25 and IPNS11,17. The crystal structure of Fe-IPNS-ACV-NO shows a six-coordinate Fe center with NO occupying the coordination position opened by ACV binding to Fe.17 Previous EPR and Mössbauer studies of Fe-IPNS-ACV-NO demonstrate that the complex has an (S=3/2) ground state, which is mostly axial but becomes slightly rhombic upon ACV addition.11 From absorbance studies, the addition of ACV to Fe-IPNS-NO introduces new features at 508 and 720nm.11 A DFT study of Fe-IPNS-ACV-NO calculates a shortened Fe-NO bond distance and larger Fe-N-O bond angle relative to the crystal structure.26
While many studies of {FeNO}7 complexes of mononuclear non-heme enzymes provided valuable information on the structures of the active sites, the electronic structure of the {FeNO}7 unit was not fully understood. Spectroscopic and DFT studies of {FeNO}7 model complexes have elucidated the oxidation and spin states of both the Fe and NO moieties and their bonding interactions within the {FeNO}7 unit.27,28 The electronic structure of the {FeNO}7 unit was shown to be best described as a high-spin FeIII (S=5/2) antiferromagnetically coupled to NO− (S=1) to give a total spin of S=3/2. Five key transitions in the absorbance/CD/MCD spectra of the model complexes were identified as two Fe d → d Ligand Field (LF) transitions and three NO 2π* → Fe d Charge Transfer (CT) transitions. Different combinations of basis sets and functionals were used to optimize the {FeNO}7 model complexes and were calibrated using experimental geometric and spectroscopic parameters for the complexes. Using this calibrated method, NO was replaced by O2 in the model complex and the electronic structure of the hypothetical {FeO2}8 complex was described. The energetics of NO and O2 binding to FeII model complexes were also calculated and found to be exergonic for NO and endergonic for O2, the result of FeIII-NO− forming a stronger bond than FeIII-O2−.
In this study, we apply the spectroscopic and DFT methodology developed to study the {FeNO}7 model complexes to the Fe-IPNS-ACV-NO enzyme complex. We use a combination of EPR, absorbance, CD, MCD and VTVH-MCD spectroscopy to identify new spectral features introduced by the binding of the ACV thiolate to the IPNS {FeNO}7 complex. From these new spectral features, we use DFT calculations to determine the effects of the ACV thiolate on the FeIII-NO− bond. Extending our DFT study to the Fe-IPNS-ACV-O2 complex, we evaluate how the features of the FeIII-O2− bond lead to the unique oxidase activity of IPNS.
2. Experimental Procedures
2.1 Materials
ACV was purchased from Bachem. The IPNS expression vector pIT353 was a gift from Victor Chen (Eli Lilly).29 Cellufine A-800 anion exchange resin was purchased from Millipore and is now available from Chisso America Incorporated.
2.2 Sample Preparation
Isopenicillin N-synthase was overexpressed in E. Coli K12 JM109 and purified from inclusion bodies in the apo form according to previously published procedures.30 A small amount (~ 1 mg) of DNAse I was added to the buffer used to resuspend the cell pellet prior to sonication. Dissolution of the inclusion bodies in 7M urea containing 20 mM DTT, 30mM glycine, pH 10.0 required vigorous stirring for over 2 hours at room temperature. All other steps were performed as described.30 All samples were prepared under inert atmosphere inside a N2-purged wet box. apoIPNS was exchanged into deuterated MOPS buffer (100mM, pD=7.4) and concentrated to 3–4mM using a 10kDa centricon tube. The sample was transferred to a sealed conical vial and made anaerobic by purging under an Ar atmosphere on a Schlenk line at 0°C for approximately one hour. Buffer was sealed in a conical vial and degassed by bubbling Ar through the solution on a Schlenk line. The FeII-IPNS complex was generated by μL addition of an FeII stock solution prepared by dissolving Fe(NH4)2(SO4)2 in degassed MOPS buffer (100mM) to the apoIPNS. The FeII-IPNS-ACV complex was prepared through addition of a stock solution of ACV to the FeII-IPNS complex. ACV in degassed MOPS buffer (1mM) was slowly added to the FeII-IPNS in μL quantities with vigorous stirring between aliquots. NO was added to the Fe-IPNS-ACV complex by stirring the Fe-IPNS-ACV solution under an NO atmosphere for approximately 2–3 minutes. Glycerol-d3 (60% (v/v), degassed by freeze/pump/thaw/heat) was added to the MCD and Abs samples as a glassing agent. CD spectra were collected with and without glycerol to ensure that the site was not affected by the addition of the glassing agent. EPR samples were prepared by diluting the Fe-IPNS-ACV-NO solution with degassed buffer (100mM) to a concentration of approximately 100–150 μM.
2.3 Spectroscopic Methods
Near-IR (600–2000 nm) CD and MCD data were recorded on a Jasco J-200D spectropolarimeter with a liquid N2-cooled InSb detector coupled to an Oxford Instruments SM4000-7 Tesla (T) superconducting magnet. UV/Vis (300–900 nm) CD and MCD data were recorded on a Jasco J810 spectropolarimeter with an extended S-20 photomultiplier tube and adapter to accept an Oxford Instruments SM4000-7T superconducting magnet. MCD spectra were corrected for the natural CD and zero-field baseline effects by subtracting the 0T spectra at each temperature. VTVH-MCD data were collected using a calibrated Cernox resistor (Lakeshore Cryogenics, calibrated 1.5–300 K) inserted into the sample cell to accurately measure the sample temperature. VTVH data were normalized to the maximum observed intensity over all isotherms for a given wavelength and the ground-state parameters were extracted by fitting utilizing published procedures. X-band EPR spectra were collected on a Bruker EMX spectrometer with Bruker ER 041XG/ER microwave bridges and ER 4102ST/ER 5106QT cavitities. Spectra were collected at temperatures between 8 and 50K using an Oxford ITC503 temperature controller with an ESR 900 continuous flow cryostat.
2.4 Computational Methods
The starting geometry for the Fe-IPNS-ACV-NO complex was taken from the crystal structure of Fe-IPNS-ACV-NO from Aspergillus nidulans.17 Protein-derived ligands were truncated with imidizoles modeling histidines and propionate modeling aspartate. The ACV substrate was truncated to remove the 6 carbon aminoadipoyl chain but was otherwise left intact. The alpha-carbons of the protein ligands were frozen relative to each other to impose the constraints of the protein backbone. The cysteine nitrogen and the two valine carboxylate oxygens of ACV were frozen relative to each other and the alpha-carbons of the protein ligands to mimic hydrogen bonding to the substrate in the protein pocket. For the Fe-IPNS-ACV and Fe-IPNS-ACV-O2 complexes, the starting geometry was taken from the optimized Fe-IPNS-ACV-NO complex with the NO moiety either removed or replaced with O2 and optimized under the same conditions.
All complexes were geometry optimized using the Gaussian 03 software package31, with the spin unrestricted BP86 functional32,33 with 10% Hartree-Fock Exchange under tight convergence criteria. This functional was previously calibrated for {FeNO}7 complexes.28 Geometry optimizations were carried out using the Pople 6–311G* basis set on Fe, S, and the NO/O2 unit with the 6–31G* basis set on the remaining atoms. Single point calculations were performed on the optimized structures to generate molecular orbitals using the functional and basis set above. Molecular orbital compositions were calculated using AOMix34,35 and optimized structures and molecular orbitals were visualized using Molden version 4.1.36 Frequencies and thermodynamic parameters were calculated using the split 6–311G*/6–31G* basis set and all frequencies were found to be real. In order determine the effects of the ACV thiolate on the energetics of NO and O2 binding to Fe-IPNS-ACV, single point energies were calculated for the Fe-IPNS-ACV 5C FeII, NO and O2 complexes using the 6–311+G(2d,p) basis set. Solvation effects to the energy of the optimized structure were included using the Polarized Continuum Model (PCM)37 with a dielectric constant ε = 4.0 to model the protein environment.
In order to correlate the calculated electronic structure of Fe-IPNS-ACV-NO to the spectroscopy, ΔSCF excitation energies were calculated with the Amsterdam Density Functional (ADF) package.38–40 (The Gaussian package does not allow for ΔSCF calculations.) Using the Gaussian 03 optimized geometry, exited states were calculated for select charge transfer transitions using the BP86 functional with the uncontracted triple-ξ basis set (TZP) with a single polarization function without any frozen core approximation. As ADF does not allow for the mixing of HF exchange for open-shell systems, the Z of Fe was set to 25.6 (single point calculation) as was previously calibrated for similar calculations on {FeNO}7 model complexes.28 All excitation energies were calculated by exciting a full electron to a virtual orbital except for the NO π*(in-plane) →FeIIIdxz and S pseudo-σ →FeIIIdz2 transitions, which were calculated from Slater transitions because of convergence issues with the full electron excitation. Slater transitions were also evaluated for the transitions that converged in ΔSCF and gave the same results.
3. Results and Analysis
3.1. Spectroscopic Results
A. Temperature-Dependent EPR
Previous EPR studies of Fe-IPNS-ACV-NO reported a signature S=3/2 spectrum with g = 4.22, 3.81 and 1.99 described by the spin Hamiltonian,
| (1) |
where g0 = 2.0 and D and E are the axial and rhombic Zero Field Splitting parameters.11 The E/D value was 0.035, indicative of a close to axial complex, with an increase in rhombicity upon binding of the substrate. (The Fe-IPNS-NO complex had an E/D = 0.015). In order to obtain the value and sign of D, temperature-dependent EPR spectra of Fe-IPNS-ACV-NO were collected from 8K to 50K under non-saturating conditions and the normalized intensity of the g = 4.22 signal was plotted versus temperature (Figure 1, insert). A Boltzmann fit to the Curie law dependence is given by Equation 2
Figure 1.

Temperature-dependent EPR Spectra of Fe-IPNS-ACV-NO and Boltzmann fit to the Curie law of relative EPR intensities to give an axial ZFS value of D = 12.5 ± 0.5cm−1 (inset).
| (2) |
where C is the Curie constant, T is temperature in Kelvin and k is the Boltzmann constant. The best fit of the EPR intensity gives a value of D = +12.5 ± 0.5cm−1. This is in good agreement to the magnitude of D previously obtained from Mössbauer spectroscopy of D = 14 ± 1cm−1.12
B. Absorbance/CD/MCD Spectroscopy
Low temperature absorbance, CD and MCD spectra were collected for Fe-IPNS-ACV-NO complex and simultaneous Gaussian fitting of the spectra requires the presence of at least 7 bands (Figure 2). Previous spectroscopic studies of {FeNO}7 model complexes report a signature pattern of five transitions in the Abs/MCD spectra of these complexes with two weak, low energy features from Fe d→d ligand field (LF) transitions in the 11,000–15,000cm−1 energy region, followed by three NO 2π* →Fe d charge transfer (CT) transitions in the 17,000–22,000cm−1 energy region (Insert, Figure 2 Bottom).27 Comparison of the Fe-IPNS-ACV-NO bands to those of the model complexes reveals two new features introduced by the addition of the ACV thiolate at approximately 13,100cm−1 and 19,800cm−1 (Bands 1 and 5, indicated by arrows in MCD). The lower energy transition is the weaker of the two in intensity in all three spectra, but is more intense than the Fe d→d LF transitions found in the same region of the spectra of the model complexes. There is a moderately intense absorbance feature coincident with Band 1 seen in the room temperature spectrum of Fe-IPNS-ACV-NO.11 The transition at 19,800cm−1 is very intense in low temperature absorbance and CD, dominating both spectra, but is weak and negative in the MCD spectrum (Band 5). The low intensity of this negative MCD band is likely due at least in part to overlap with strong positive bands at lower and higher energy.
Figure 2.

Absorbance (10K, top), CD (5K, middle) and MCD (5K,7T, bottom) spectra collected for Fe-IPNS-ACV-NO with Gaussian best fits. Comparison to the MCD spectrum of Fe-EDTA-NO (inset) reveals two new spectral features seen in the spectra upon addition of ACV to Fe-IPNS-NO (arrows).
Variable temperature-variable field MCD (VTVH) data were collected on the Fe-IPNS-ACV-NO complex to determine the polarization of the bands at 13, 100cm−1 (Band 1) and 22,100cm−1 (Band 6). Collection of VTVH data for Band 5 was unsuccessful because of its low intensity and overlap with the bands to lower and higher energy. VTVH data collected at 13,260 and 22,730 cm−1 exhibit very different nesting behaviors and are consistent with these transitions having different polarizations (Figure 3, insert).41
Figure 3.

Fe-IPNS-ACV-NO VTVH MCD data collected at 13,260cm−1 (left) and 22,730cm−1 (right) demonstrate different nesting behaviors. (Arrows indicate energy where the isotherms were collected.)
MCD intensity is directly proportional to the spin-expectation values of the ground state sublevels, which can be generated from the spin-Hamiltonian. VTVH MCD data collected on a randomly oriented sample with S ≥ ½ can be modeled with Equation 3
| (3) |
where x, y and z refer to the principal axes of the ZFS tensor; Ni is the temperature-dependent Boltzmann population, lx, ly and lz are the direction cosines for the magnetic field relative to the molecular coordinate system; 〈Sx.y.z〉i are the spin expectation values for the ith ground state sublevel; are the products of the polarizations of the electronic transitions and γ is a collection of constants. Using the spin Hamiltonian parameters D, E/D and g0 obtained from the EPR data, the effective transition moments can be obtained from fitting the data (Figure 3, insert) using Equation 3.42 These transition moments can then be used to calculate the percent polarization along the ZFS tensor axes using Equation 4 with cyclic permutations of the indices for the other polarizations.
| (4) |
From the fit of the VTVH isotherms at 22,730cm−1, the 22,100cm−1 transition is 96.9% z polarized. As LMCT bands are polarized along the metal-ligand bond, a strongly z polarized transition is consistent with a transition along the dominant Fe-NNO bond and this band can be assigned as NO π* → Fe d CT transition. (The z axis lies along the short Fe-NNO bond, with the x and y axes designated as lying between the equatorial ligand-Fe bonds with x in the Fe-N-O plane (Fe-NO ∠= 150° vide infra) between the Fe-S and Fe-NHis bonds.) The fit of the 13,260cm−1 isotherms shows the band to be 49.7% z polarized and 50.3% x-y polarized (x and y cannot be distinguished in this close to axial ZFS system), indicating a transition with both NO− →FeIII (z) and RS− → FeIII (x,y) CT character.
3.2. Computational Results
A. Geometry Optimization
DFT calculations were performed on Fe-IPNS-ACV-NO to determine the effects of the Fe-ACV thiolate bond on the geometric and electronic structure of the IPNS {FeNO}7 unit. The optimized Fe-IPNS-ACV-NO complex is a distorted octahedron with an Fe-S bond distance of 2.35Å, an Fe-NNO bond distance of 1.74Å and an Fe-N-O bond angle of 150.3° (Figure 4). This Fe-NNO geometry is markedly different than that reported in the IPNS/Fe/ACV/NO crystal structure (Table 1)17, which has a Fe-NNO bond distance of 2.13Å with a Fe-N-O bond angle of 119.7°. However, the optimized Fe-NNO geometry is in good agreement with the experimental Fe-NNO bond distance determined by EXAFS which have an accuracy of ±0.02Å,13 and with the Fe-NNO bond distance and Fe-N-O bond angle previously determined for the {FeNO}7 model complex Fe-[Me3TACN]-(N3)2-NO.28 The Fe-S bond distance in the optimized Fe-IPNS-ACV-NO is consistent with an FeIII – thiolate S bond. From the comparison to the model complex without thiolate ligation, the presence of the ACV thiolate does not significantly affect the geometry of the {FeNO}7 unit in the Fe-IPNS-ACV-NO complex.
Figure 4.
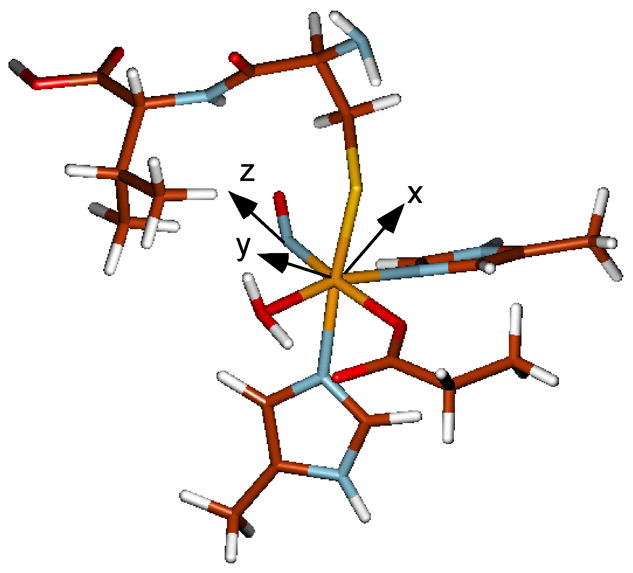
UBP86+10%HF/6–311G*/6–31G* Optimized Fe-IPNS-ACV-NO complex.
Table 1.
Geometric parameters for {FeNO}7 unit of Fe-IPNS-ACV-NO and related model complex
| Fe-IPNS-ACV-NO Crystal Structure | Fe-IPNS-ACV-NO EXAFS | Fe-IPNS-ACV-NO Optimized Structurea | Fe-[Me3TACN]−(N3)2-NO Optimized Structurea | |
|---|---|---|---|---|
| Fe-NNO | 2.13Å | 1.71Å | 1.74Å | 1.72Å |
| N-O | 1.15 Å | N/A | 1.19Å | 1.17Å |
| Fe-N-O | 119.7° | N/A | 151° | 148° |
UBP86+10%HF/6–311G*/6–31G* optimized structures.
B. Electronic Structure
Experimental and computational studies of the {FeNO}7 model complexes Fe-EDTA-NO and Fe-[Me3TACN]-(N3)2-NO have determined the electronic structure of the {FeNO}7 unit to be a high-spin FeIII (S=5/2) antiferromagnetically coupled to NO− (S=1).27,28 The ground state wave function of this complex is best described by five unoccupied β Fe d molecular orbitals (MOs) and two unoccupied α NO 2π* MOs, with one NO π* MO located in the Fe-N-O plane (in-plane) and the other perpendicular to the Fe-N-O plane (out-of-plane). These unoccupied molecular orbitals are used to describe the bonding as they reflect the uncompensated occupied counterparts which give the major contribution to bonding. The MOs and energy level diagram for the optimized Fe-IPNS-ACV-NO complex are given in Figures 5 and 6, respectively. As seen in the model complexes, the two lowest unoccupied α MOs of Fe-IPNS-ACV-NO have mostly NO 2π* in-plane (ip) and out-of-plane (op) character, respectively, with small contributions from the Fe d orbitals and the ACV thiolate. In the β manifold, the five lowest unoccupied MOs are comprised mostly of Fe d character, with some contribution from the NO π* and ACV thiolate orbitals. (The corresponding occupied α Fe d MOs are stabilized by approximately 6eV by spin polarization.) In the optimized Fe-IPNS-ACV-NO complex the spin densities on the Fe and NO are 3.44 and -0.79, respectively, which is consistent with the values calculated for the Fe-[Me3TACN]-(N3)2-NO model complex.28
Figure 5.
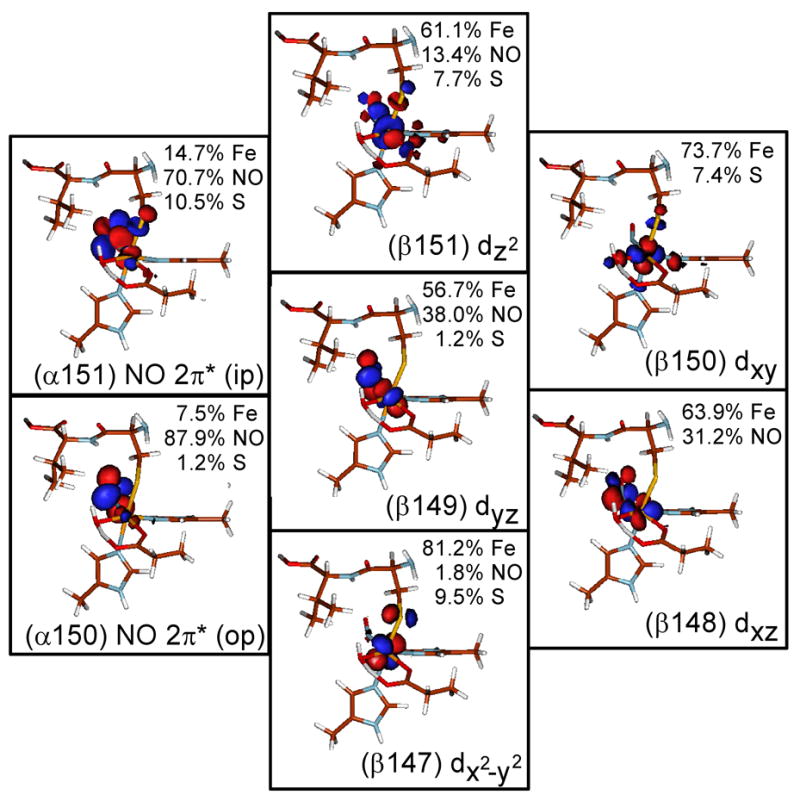
Contours of the lowest unoccupied α and β molecular orbitals from the ground state wavefunction of Fe-IPNS-ACV-NO.
Figure 6.

Molecular orbital diagram of Fe-IPNS-ACV-NO. The 5 lowest unoccupied β orbitals are comprised of mostly Fe d character, while the 2 lowest unoccupied α orbitals are comprised of mostly NOπ* character, consistent with an electronic structure description of a high-spin FeIII antiferromagnetically coupled to NO−.
A previous density functional study of Fe-IPNS-ACV-NO described the electronic structure of the complex as high-spin FeII (S=2) antiferromagnetically coupled to NO. (S=1/2) based upon calculated spin densities and Mössbauer parameters.26 While the calculated Fe-NNO bond length used to calibrate the optimized structure by Zhang et al. (1.83Å) is shorter than that reported in the crystal structure, it is longer than the experimental value obtained by EXAFS13 and the calculated values of the {FeNO}7 model complexes28 and the geometry optimized structure obtained here. A combination of MCD, resonance Raman and XAS has shown the model is a high-spin FeIII (S=5/2) antiferromagnetically coupled to NO− (S=1/2).27 As the {FeNO}7 unit contains a very covalent Fe-NO bond, the Mössbauer parameters for {FeNO}7 complexes generally lie between those reported for high spin octahedral FeII and FeIII; however, Mössbauer studies of a series of {FeNO}6–8 model complexes also concluded that the Fe center in {FeNO}7 is best described as a high-spin FeIII.43 Based on the better agreement with experimental geometric parameters, consistency with model complexes, and the occupancy of the MO orbitals, the electron structure of the Fe-IPNS-ACV-NO complex is best described as a high-spin FeIII (S=5/2) antiferromagnetically coupled to NO− (S=1).
In addition to the Fe-NO bonding description, the MO and energy diagrams for Fe-IPNS-ACV-NO describe the interactions of the FeIII and the ACV thiolate. The Fe-bound thiolate has two 3p orbitals (the third is tied up in the C-S bond) that can interact with the FeIII d orbitals in either a pseudo-σ or π orientation.44 From the Fe-IPNS-ACV-NO ground state wave function, the ACV thiolate lone pairs have a π interaction with dx2−y2 orbital (Figure 5, Contour β147, Axes defined in Figure 4) and a pseudo-σ interaction with the dz2 and dxy orbitals (Figure 5, Contours β151 and β150). These same interactions are also reflected in the α manifold, with the thiolate pseudo-σ interacting with the dz2 orbital, which also interacts with the unoccupied NO π*(ip) orbital (Figure 5, Contour α151). This MO has 10.5% S character, compared to 1.2% S character in the NO π*(op) orbital (Figure 5, Contour α150), demonstrating a significant configuration interaction between the thiolate pseudo-σ and NO π*(ip) MOs mediated by the Fe dz2 orbital. This configuration interaction raises the NO π*(ip) MO in energy (Figure 6) and reverses the order of the NO π* orbitals relative to that of the model complexes.
3.3. Spectral Assignment
A. MCD Analysis
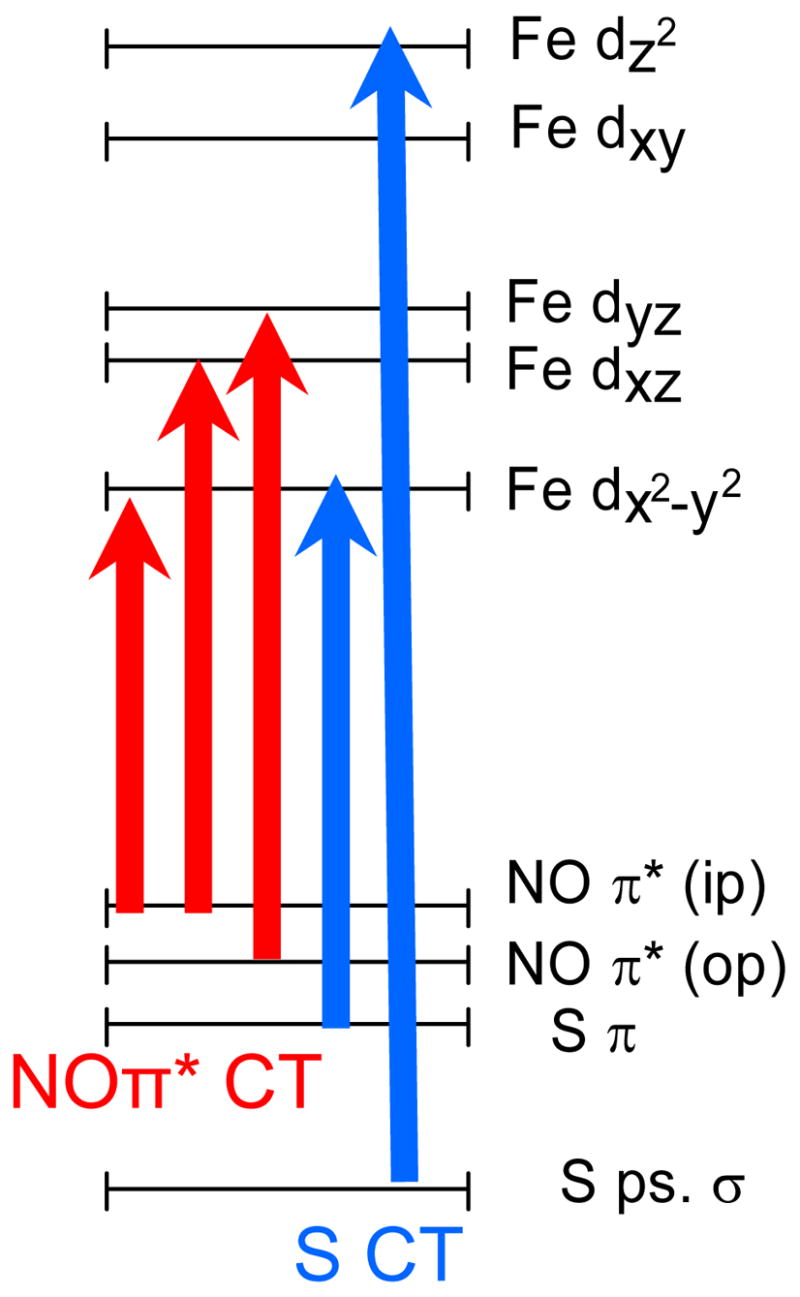
{FeNO}7 complexes demonstrate signature Absorbance/CD/MCD spectra with a common pattern of five transitions.27 The first two are d→d ligand field (LF) transitions, which are spin forbidden but gain intensity through exchange coupling to the FeIII, followed by three charge transfer (CT) transitions: NOπ*(in-plane) →FeIIIdx2−y2, NO π*(in-plane)→FeIIIdxz, and NO π*(out-of-plane)→FeIIIdyz with NO π*(out-of-plane)→FeIIIdyz being the most intense due to increased mixing of the NO π*(out-of-plane) and FeIIIdyz orbitals. Comparison of the Fe-IPNS-ACV-NO MCD spectrum to that of the Fe-EDTA-NO model complex (Figure 2, insert) shows that Bands 4 and 6 in the Fe-IPNS-ACV-NO spectrum correspond both in energy and relative intensity to Bands 4 and 5 in the Fe-EDTA-NO spectrum, which are assigned as the NO π*(in-plane)→FeIIIdxz, and NO π*(out-of-plane)→FeIIIdyz transitions in the Fe-EDTA-NO complex. Bands 2 and 3 in the IPNS spectrum correspond well with the weak d→d ligand field (LF) transitions (Bands 1 and 2) in the Fe-EDTA-NO spectrum. Band 3 in the Fe-EDTA-NO spectrum is very weak and is located in an energy region that is overlapped in the Fe-IPNS-ACV-NO spectrum by the presence of Band 5. Bands 1 and 5 have no counterpart in the Fe-EDTA-NO spectrum and are therefore new features induced by the binding of the ACV thiolate.
From the ACV thiolate interaction with FeIII in the Fe-IPNS-ACV-NO complex one would expect two additional transitions: 1) a S π to Fe dx2−y2 CT transition and 2) a S pseudo-σ to Fe dz2 CT transition, with the S π to Fe d CT transition significantly lower in energy. While the new Bands 1 and 5 potentially match the number of anticipated for thiolate CT, the rules governing MCD intensity can be used to rigorously assign the new transitions. The MCD spectrum of Fe-IPNS-ACV-NO possesses a unique feature in that the transition at 19,800 cm−1 has negative intensity, creating a pattern of alternating positive and negative intensity referred to as a Pseudo-A Term, which has certain electronic structure requirements in order to occur in the MCD spectrum.42 First, the complex must have two spin-orbit coupled (SOC) excited states with perpendicular transition dipole moments. These two charge transfer excited states must only differ by one electron on a single center, so they must share either a common donor or acceptor orbital. Finally, the orbitals which differ by one electron must rotate into each other by spin-orbit coupling Li in a direction i perpendicular to the two transition moments.
In the Fe-IPNS-ACV-NO complex, the ACV thiolate S is bound cis to the NO, so any thiolate S → Fe d CT and NO → Fe d CT transitions will have perpendicular transition dipole moments. Using the Kohn-Sham ground state orbitals from the DFT calculations (Figure 6), the electron configurations of the S pseudo-σ → Fe d and S π → Fe d CT excited states can be compared to those of the NO π* → Fe d CT excited states to look for transitions with the same acceptor orbital. (The donor orbitals are different.) The acceptor orbital for the S pseudo-σ transition is the Fe dz2 orbital; however, no NO π* → Fe d CT transitions excite an electron into that orbital. Therefore no SO mechanism exists to couple the S pseudo-σ → Fe dz2 transition with the NO π* →Fe d CT transitions. The other possible assignment of Band 5 is the S π → Fe dx2−y2 CT transition. As the two excited states associated with the S π → Fe dx2−y2 CT transition and the NO π* (ip) → Fe dx2−y2 CT transitions both excite an electron into the Fe dx2−y2 orbital, these excited states differ by one electron in donor orbitals, potentially creating a Pseudo-A term.
Strong covalent interactions mix dx2−y2 and dxz character into the S π and NO π* (ip) donor orbitals, respectively, which allows the Fe-based SO operator to couple these two CT excited states. Because the LMCT transitions are polarized along the molecular axes of the complex, the x and y axes were rotated by 45° to be colinear with the Fe-ligand bonds (x along the Fe-S bond and y along the Fe-NHis bond). Using these newly defined axes, the Fe components of the donor orbitals are transformed from dx2−y2 to dx′y′ and from dxz to dx′z′ + dy′z′.
In order to create a pseudo A-term, the two orthogonal LMCT transitions must SOC in the third orthogonal direction. The S π → Fe dx2−y2 CT transition occurs in the x′ direction and the NO π* (ip) → Fe dx2−y2 CT transition occurs in the z′ direction. Because Ly′ rotates dx′y′ into dy′z′, the S π → Fe d CT transition and the NO π* (ip) → Fe d CT transition will couple to produce a pseudo A-Term feature in the Fe-IPNS-ACV-NO MCD spectrum. From this MCD intensity analysis, the negative band at 19,800cm−1 is assigned as the S π → Fe dx2−y2 CT transition.
B. ΔSCF Calculations
In order to confirm the assignment of Band 5 as the S π → Fe dx2−y2 CT transition and to identify the source of the low energy Band 1 introduced by binding the ACV thiolate ligand, SCF calculations were performed for the NO π* → Fe d and S → Fe d CT transitions (Figure 7). Using the Kohn-Sham ground state generated from the optimized Fe-IPNS-ACV-NO complex, the energies of 5 CT transitions were calculated: NOπ*(in-plane)→FeIIIdx2−y2, NO π*(in-plane)→FeIIIdxz, NO π*(out-of-plane)→FeIIIdyz, Sπ→FeIIIdx2−y2 and S pseudo-σ →FeIIIdxy and the calculated energies and their correlation to Fe-[Me3TACN]-(N3)2-NO are given in Table 2.
Figure 7.
ΔSCF charge transfer transition for the Fe-IPNS-ACV-NO complex calculated using ADF/BP86/TZV with Zeff = 25.6.
Table 2.
Calculated ΔSCF/ADF and Experimental Energies of CT transitions in {FeNO}7 complexes
| Transition | Fe-[Me3TACN]-NO Experiment | Fe-[Me3TACN]-NO SCF/ADF | IPNS-ACV-NO Experiment | IPNS-ACV-NO SCF/ADF |
|---|---|---|---|---|
| NO π*(ip) → dx2−y2 | 17, 400 cm−1 | 15,975 cm−1 | 13, 100 cm−1 | 13, 280 cm−1 |
| NO π*(ip) → dxz | 19, 650 cm−1 | 19, 565 cm−1 | 18, 500 cm−1 | 17, 420 cm−1 |
| NO π*(op) → dyz | 22, 580 cm−1 | 19, 950 cm−1 | 22, 100 cm−1 | 21, 320 cm−1 |
| S π → dx2−y2 | N/A | N/A | 19, 800 cm−1 | 22, 980 cm−1 |
| S ps.σ → dz2 | N/A | N/A | -- | 30,600 cm−1 |
The energy of Sπ→FeIIIdx2−y2 CT transition is calculated to be 22, 980 cm−1, in reasonable agreement with the experimental energy of Band 5 in the MCD spectrum, further confirming the MCD Pseudo A-Term assignment of this band. The S pseudo-σ→FeIIIdxy CT transition is calculated to be substantially higher in energy, as would be expected from the one electron orbital diagram, and this can be excluded as an assignment of either Band 1 or Band 5. It is interesting to note that from the Kohn-Sham ground state diagram the Sπ→FeIIIdx2−y2 CT transition involves promotion from the β HOMO to LUMO and would be expected to be lower in energy than the NO π*→FeIIId CT transitions. However, Sπ→FeIIIdx2−y2 CT transition is calculated to be higher in energy than the NO π*(ip)→FeIIIdxz transition and similar in energy to NO π*(op)→FeIIIdyz transition as observed experimentally. This reordering of transition energies from the ground state prediction results from the differences in orbital covalency of the donor/acceptor pairs for the Sπ→FeIIIdx2−y2 and the NO π*→FeIIId CT transitions. The Sπ→FeIIIdx2−y2 transition involves excitation of an electron from a donor largely centered on the S ligand to an acceptor largely centered on Fe, which increases the electron repulsion in the excited state. The donor and acceptor orbitals for the NO π*→FeIIId CT transitions are much more covalent, so the electron repulsion in their ground and excited states is similar. As a result of this difference in electron repulsion, the Sπ→FeIIIdx2−y2 CT transition is shifted higher in energy than would be expected from the energy splitting in the one electron Kohn-Sham ground state diagram.
From Table 2, the energy of the NOπ*(in-plane)→FeIIIdx2−y2 is calculated to be 13,280cm−1, in good agreement with the energy of Band 1 in the MCD spectrum of Fe-IPNS-ACV-NO. The calculated energy for the NOπ*(in-plane)→FeIIIdx2−y2 is substantially lower than both the experimental and calculated energy for the same transition in Fe-[Me3TACN]-(N3)2-NO. From the VTVH MCD data, Band 1 is half z-polarized, which would correspond with a NOπ*→FeIIId CT transition, but the x,y-polarization which is also present would correspond to S character mixed into the transition. Examination of the donor and acceptor orbitals for the NOπ*(in-plane)→FeIIIdx2−y2 transition shows significant S character in both the NOπ*(ip) and FeIIIdx2−y2 orbitals (10.5% and 9.5% S, respectively, Figure 5, Contours α151 and β147). This S character will mix x,y-polarized intensity into the NOπ*(ip)→FeIIIdx2−y2 CT transition, and lead to the increased intensity in this band relative to the very weak NOπ*(ip)→FeIIIdx2−y2 CT band in the Fe-[Me3TACN]-(N3)2-NO spectra. Configuration interaction of the S pseudo-σ MO with the NOπ*(ip) MO raises the energy of the donor orbital, lowering the transition in energy.
The two remaining NOπ*(ip)→FeIIIdxz and NO π*(op)→FeIIIdyz transitions were calculated at similar energy to the experimental and calculated values of the same transitions in Fe-[Me3TACN]-(N3)2-NO model complex. It is interesting to note that while the NOπ*(ip)→FeIIIdx2−y2 and NOπ*(ip)→FeIIIdxz transitions share the same donor orbital, only the NOπ*(ip)→FeIIIdx2−y2 transition is shifted to significantly lower energy. The FeIIIdxz acceptor orbital has more antibonding ligand character than the FeIIIdx2−y2 orbital, shifting it to higher energy. Thus while configuration interaction shifts the donor orbital up in energy, the acceptor orbital for the NOπ*(in-plane)→FeIIIdxz transition is also shifted up in energy and the transition energy is not greatly changed relative to the model complex.
3.4. {FeNO}7→{FeO2}8
A. Geometry Optimization of Fe-IPNS-ACV-O2
In order to explore the reaction coordinate of Fe-IPNS-ACV with O2, the NO moiety of the Fe-IPNS-ACV-NO complex was replaced with O2 in the crystal structure geometry and the complex was optimized. As O2 could theoretically bind to Fe-IPNS-ACV to give complexes of S=1, 2 or 3, the complex was optimized in each of the three spin states with the S=2 complex lower in energy than the S=1 and S=3 complexes by 2.2 and 3.7kcal/mol, respectively. Relevant geometric parameters for the optimized S=2 Fe-IPNS-ACV-O2 complex and the optimized Fe-[Me3TACN]-(N3)2-O2 complex28 are given in Table 3. Comparison of the Fe-O and O-O bond distances and the Fe-O-O bond angle in the two complexes reveal similar Fe-O2 geometries (Table 3). The Fe-S bond distance is relatively unchanged by the replacement of NO with O2 (2.33 versus 2.35Å).
Table 3.
Geometric parameters for {FeO2}8 unit of Fe-IPNS-ACV-O2 and related model complex
| Geometric Parameter | Fe-IPNS-ACV-O2 Optimized Complex | Fe-[Me3TACN]-O2-(N3)2 Optimized Complex |
|---|---|---|
| Fe-O | 1.87 Å | 1.88 Å |
| O-O | 1.31 Å | 1.29 Å |
| Fe-S | 2.33 Å | N/A |
| Fe-O-O | 134° | 130° |
B. Electronic Structure of Fe-IPNS-ACV-O2
Previous calculations on the Fe-[Me3TACN]-(N3)2-O2 complex describe an electronic structure of a high-spin FeIII antiferromagnetically paired to O2− superoxide to give a S=2 complex.28 O2 has one more electron than NO, so when it bonds to FeII this extra electron fills one of the unoccupied π* orbitals relative to the {FeNO}7. The ground state wave function of the FeIII-superoxide complex is characterized by five unoccupied β Fe d MOs and one unoccupied α O2 π* MO. The MOs and the energy level diagram for the optimized Fe-IPNS-ACV-O2 complex are given in Figures 8 and 9, respectively. As found in the Fe-[Me3TACN]-(N3)2-O2 complex, the two lowest unoccupied β MOs of Fe-IPNS-ACV-O2 have mostly Fe d character, with some contribution from the O2 π* and ACV thiolate orbitals. In the α manifold, the lowest unoccupied MO contains mostly O2 2π* in-plane (ip) character with additional small contributions from the Fe d orbitals and the ACV thiolate. In the optimized Fe-IPNS-ACV-O2 complex the spin densities on the Fe and O2 are 3.79 and −0.22, respectively, which is consistent with the values calculated for the Fe-[Me3TACN]-(N3)2-O2 model complex.
Figure 8.
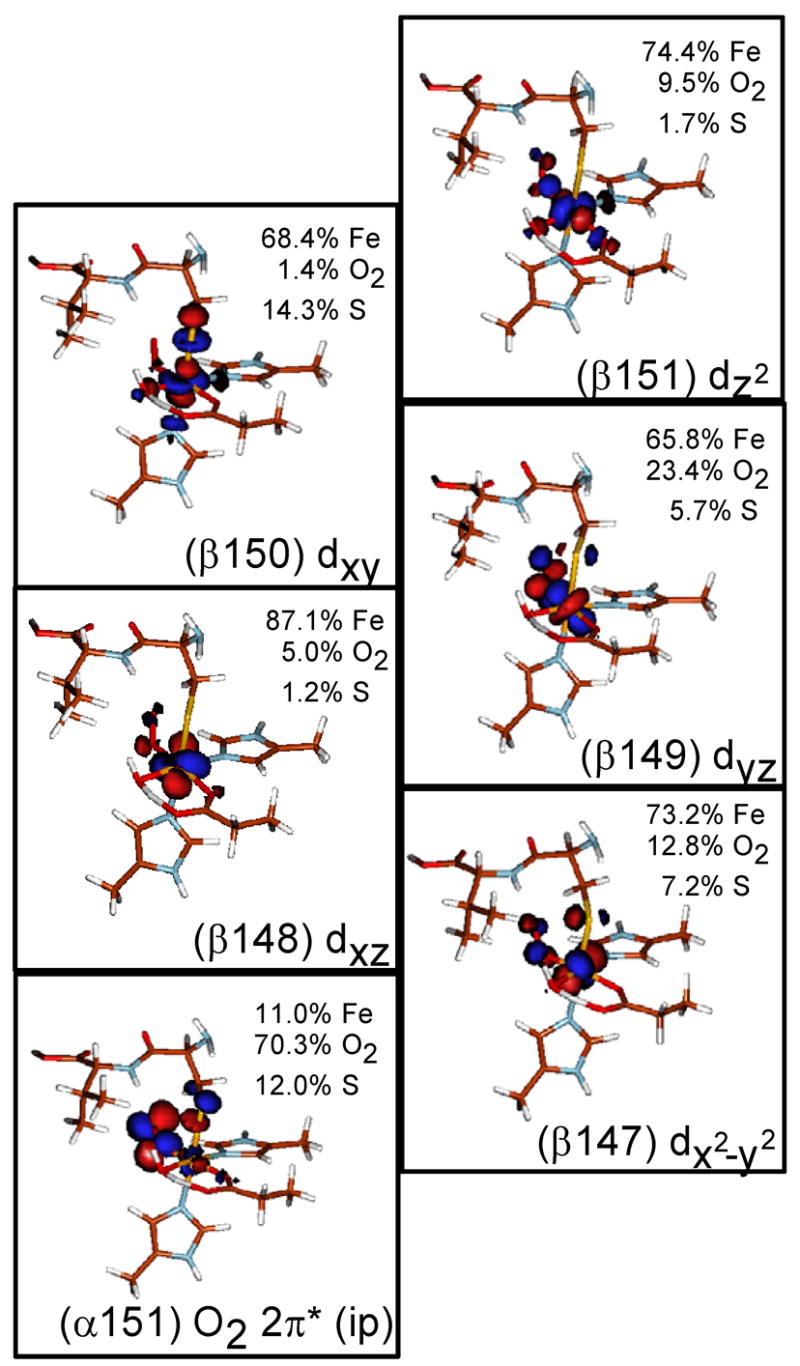
Contours of the lowest unoccupied α and β molecular orbitals from the ground state wavefunction of Fe-IPNS-ACV-O2
Figure 9.

Molecular orbital diagram of Fe-IPNS-ACV-O2. The 5 lowest unoccupied β orbitals are comprised of mostly Fe d character, while the lowest unoccupied α orbital is comprised of mostly O2 π* character, consistent with an electronic structure description of a high-spin FeIII antiferromagnetically coupled to O2.−.
However, the electronic structure of the Fe-IPNS-ACV-O2 complex differs from that of the model complex in one important aspect. In the Fe-[Me3TACN]-(N3)2-O2 complex, the lowest unoccupied α MO is the O2 π* (op) orbital, while in the IPNS complex, the lowest unoccupied α MO is the O2 π* (ip) orbital (Figure 8, Contour α151). From the electronic structure of the Fe-IPNS-ACV-NO complex, there is a configuration interaction between the thiolate S pseudo-σ and the NOπ* (ip) orbital, which raises the π* (ip) orbital in energy. When an extra electron is added in going from {FeNO}7 to {FeO2}8 complex, this electron occupies the lower energy O2 π* (op) orbital. The occupation of the O2 π* (op) has direct mechanistic implications that are examined in the Discussion.
C. Energetics of NO/O2 Binding to Fe-IPNS-ACV: Comparison to Non-Thiolate Liganded Non-Heme Fe
While the electronic structure of the Fe-IPNS-ACV-O2 is best described as a high-spin FeIII-superoxide complex, the energetics of the formation of this complex must be explored to better understand the enzyme reaction mechanism. Previous studies of the energetics of NO and O2 binding to the Fe-[Me3TACN]-(N3)2-O2 determined that the binding of NO to non-heme FeII is highly exergonic (~−20kcal/mol) compared to O2 binding, which is thermoneutral in the model complex.28 In order to determine the source of the different binding energies of NO/O2, the reaction was separated into the transfer of an electron from FeII to NO/O2 to give FeIII and NO−/O2.− at infinite distance and the subsequent formation of the FeIII-NO−/O2− bond. The transfer of the electron to NO/O2 is highly endergonic, with the reduction of O2 approximately 5kcal/mol more favorable than that of NO. However the FeIII-NO− bond formed is much stronger than the FeIII-O2.− bond, providing the driving force for the reaction. In the mononuclear non-heme Fe enzyme DHBD, the binding of O2 is calculated to be even less favorable than in the model complex, with a highly endergonic energy of formation for the FeIII-O2.− complex (ΔG ≥ +20kcal/mol).45 Thus non-heme FeII complexes are reasonably stable in O2 in the absence of a redox active cofactor or substrate to supply additional electrons for the favorable two electron reduction of O2.
In order to estimate the influence of the ACV thiolate coordination on the binding of NO and O2 to Fe-IPNS-ACV, calculations were performed comparing the NO/O2 binding energy of Fe-IPNS-ACV to a non-thiolate liganded mononuclear non-heme Fe site. The crystal structure of the resting form of Phenylalanine hydroxylase (PAH)46 was used as a starting structure for FeII coordinated by the facial triad of two histidines and one monodentate aspartate, with two waters filling the remaining coordination positions in the five-coordinate (5C) resting complex. The 5C FeII structures of IPNS-ACV and PAH were optimized using UBP86+10%HF/6–311G*/6–31G*, and subsequently an electron was removed and the 5C FeIII complexes were optimized. Finally, the PAH-NO and PAH-O2 complexes were optimized for comparison with the Fe-IPNS-ACV-NO and O2 complexes. In order to properly account for the diffuse nature of the S atom of the ACV thiolate and to avoid any basis set superposition error that may be introduced in calculating the formation energy of a bimolecular reaction, the energy of each species was calculated using the 6–311+G(2d,p) basis set with the PCM solvent model (ε = 4.0). The energy profiles for NO/O2 binding to Fe-IPNS-ACV and Fe-PAH are given in Figure 10.
Figure 10.

Energy profiles of NO/O2 binding to Fe-IPNS-ACV and Fe-PAH.
As with the model complex and previous DHDB enzyme calculations,28,45 the binding of NO to Fe-PAH is exergonic (ΔG = −5.7kcal/mol) while the binding of O2 to Fe-PAH is highly endergonic (ΔG = 18.9kcal/mol). In the Fe-IPNS-ACV complex, the binding of NO is stabilized relative to Fe-PAH-NO and is much more exergonic (ΔG = −22.2kcal/mol) Importantly, the binding of O2 to Fe-IPNS-ACV has also been stabilized relative to Fe-PAH-O2, to the extent that the formation of Fe-IPNS-ACV-O2 is exergonic (ΔG = −7.0kcal/mol). Examination of the energy profile shows that the FeIII complex is stabilized by 49.1kcal/mol in Fe-IPNS-ACV relative to Fe-PAH, which corresponds to a 2.1eV stabilization in the oxidation of FeII to FeIII in the presence of the ACV thiolate ligand. The Fe-S bond length decreases from 2.40 to 2.24Å upon oxidation of the FeII to FeIII, indicative of greater thiolate donation to the Fe upon oxidation. Also, the charge on the Fe in the ferric IPNS-ACV complex is 1.45, compared with 1.67 in the ferric PAH complex, again showing donation of negative charge from the ACV thiolate to stabilize the positive charge on the FeIII and render the formation of the IPNS-ACV FeIII-superoxide complex energetically favorable.
4. Discussion
We have used a combination of EPR, absorbance, CD, MCD, and VTVH-MCD spectroscopies in conjunction with DFT calculations to study the {FeNO}7 complex of Isopenicillin N-synthase bound with its substrate ACV. Comparison of the MCD spectrum of Fe-IPNS-ACV-NO to that of previously studied {FeNO}7 model complexes reveals two new spectroscopic features associated with the binding of the ACV thiolate to the {FeNO}7 unit of Fe-IPNS-ACV-NO at approximately 13,100 and 19,800cm−1, assigned as the NO π*(ip) → Fe dx2−y2 and S π→ Fe dx2−y2 CT transitions, respectively. The calculated ground state wavefunction of the Fe-IPNS-ACV-NO complex and VTVH-MCD data collected on the 13,100 cm−1 band reveal a configuration interaction between the S pseudo-σ and NO π*(ip) orbitals, which shifts the NO π*(ip) → Fe dx2−y2 transition to lower energy and increases its intensity relative to the {FeNO}7 model complexes. When we replace NO with O2 in the Fe-IPNS-ACV complex, the optimized Fe-IPNS-ACV-O2 complex is best described as high-spin FeIII antiferromagnetically coupled to superoxide, as is calculated for {FeO2}8 model complexes. However, unlike the model complexes, the configuration interaction between the S pseudo-σ and O2 π*(ip) orbitals observed both computationally and spectroscopically in the Fe-IPNS-ACV-NO complex lifts the degeneracy of the π* orbitals in the Fe-IPNS-ACV-O2 complex. In going from {FeNO}7 to {FeO2}8, the extra electron occupies the lower energy O2 π*(op) orbital in the Fe-IPNS-ACV-O2 complex, leaving the O2 π*(ip) unoccupied and available as a low lying Frontier Molecular Orbital (FMO).
This FMO has direct mechanistic implications in that the first proposed step in the reaction with substrate is the abstraction of the methylene H-atom from the cysteine β carbon17; therefore an unoccupied FMO with the proper orientation and character on the distal O must be available to overlap with the methylene H and accept an electron. The unoccupied O2 π* (ip) orbital in the Fe-IPNS-ACV-O2 complex is oriented such that there is σ overlap with respect to the methylene H with high distal O character (Figure 11). The VTVH MCD data on Fe-IPNS-ACV-NO give direct experimental evidence for the mixing of S character into the NOπ*(ip)→FeIIIdx2−y2 CT transition. In the O2 complex this configuration interaction of the S pseudo-σ and the O2 π* (ip) orbital insures this is the unoccupied FMO with proper orientation for H-atom abstraction.
Figure 11.
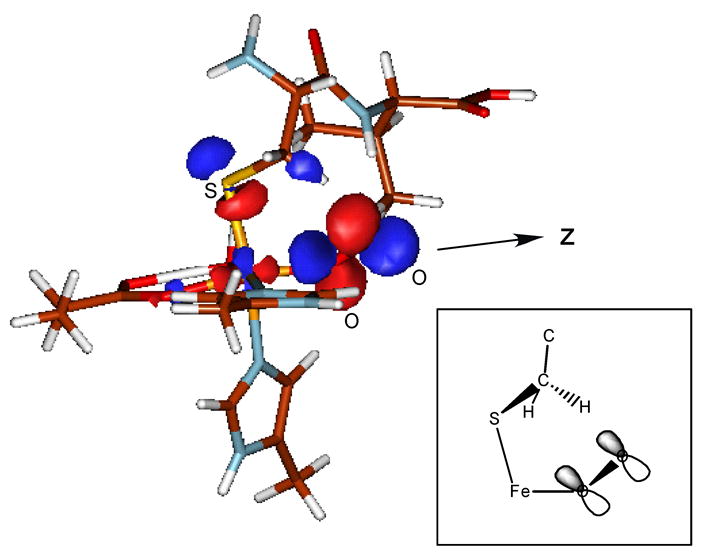
Fe-IPNS-ACV-O2 FMO with proper orientation for H-atom abstraction of cysteine β-methylene H from ACV.
In order to determine how the binding of the ACV thiolate affects the formation energies of the Fe-IPNS-ACV-NO and –O2 complexes, we have calculated the energetics of NO/O2 binding to the IPNS-ACV complex and to a non-thiolate liganded non-heme Fe complex modeled on Phenylalanine hydroxylase.46 In comparison to the PAH model, the ACV thiolate stabilizes NO binding to Fe-IPNS-ACV ΔG = −22.2kcal/mol) and, importantly, stabilizes the energy of O2 binding to Fe-IPNS-ACV to the extent that the formation of Fe-IPNS-ACV-O2 becomes exergonic (ΔG = −7.0kcal/mol). By separating the oxidation of FeII and reduction of NO/O2 from the subsequent FeIII-NO−/O2− bond formation (Figure 10), we have determined that the charge donation from the ACV thiolate stabilizes the oxidation of FeII to FeIII, allowing the IPNS-ACV complex to form a now energetically favorable FeIII-superoxide complex. Through calculations, we have demonstrated the FeIII-IPNS-ACV-superoxide complex is energetically accessible and therefore a reasonable description of the IPNS mechanism is that it proceeds through H atom abstraction by an FeIII-superoxide species.
The thiolate stabilization of the FeIII-IPNS-ACV-superoxide complex has major mechanistic implications for enzyme reactions. Many mononuclear non-heme Fe enzymes bind O2 and activate it for reaction with an organic substrate; however, most of these enzymes overcome the unfavorable one electron reduction of O2 to superoxide by obtaining additional electron(s) from a redox active substrate or co-factor.3 The additional electron is generally gained by O2 binding in a bridging fashion to both the Fe center and the redox active cofactor or cosubstrate, as is proposed to occur in the αKG-dependent enzymes, pterin-dependent enzymes and diol dioxygenases. Oxygen insertion occurs in the dioxygenases through this bridged binding of O2 reduced to the peroxide level to both the Fe and either the substrate, as in the extradiol catechol dioxygenases, or the cosubstrate, as in the αKG- and pterin-dependent enzymes (Figure 12). With the stabilization gained by the charge donation of the ACV thiolate, IPNS can form an energetically favorable FeIII-superoxide complex without having to bind O2 in a bridging manner to gain the extra electron for peroxide formation. From our spectroscopic and DFT studies, this Fe only centered O2 reactivity is directly controlled by the binding of the ACV thiolate to the Fe-IPNS-ACV complex. The binding of the ACV thiolate lowers the potential and drives the reaction at the Fe center, allowing for oxidase reactivity in IPNS without the oxygen atom insertion into the cosubstrate.
Figure 12.

Comparison of plausible intermediates for mononuclear non-heme Fe oxygenases (αKG-dependent dioxygenases, shown top) and oxidases (IPNS, shown bottom).
The formation of the IPNS-ACV FeIII-superoxide complex is both energetically favorable and presents the correct FMO for H-atom abstraction from the Cysteine methylene C for the first step of the IPNS reaction with ACV. While one of the proposed IPNS-ACV reaction mechanisms17 calls for H-atom abstraction by an FeO2 complex, this study presents the first experimental/computational evidence for the formation of an FeIII-superoxide complex in IPNS. We are currently using this Fe-IPNS-ACV-O2 model to further explore the reaction coordinate of IPNS and determine the influence of the ACV thiolate binding on later steps in the reaction mechanism.
Supplementary Material
Cartesian coordinates of the geometry optimized model of the Fe-IPNS-ACV-NO active site complex with Gaussian BP86+10% Hartree-Fock (Table S1); Relevant structural parameters for the optimized Fe-IPNS-ACV-NO complex (Table S2); Cartesian coordinates of the geometry optimized model of the Fe-IPNS-ACV-O2 active site complex with Gaussian BP86+10% Hartree-Fock (Table S3); Relevant structural parameters for the optimized Fe-IPNS-ACV-O2 complex (Table S4); Cartesian coordinates of the geometry optimized models of the Fe-IPNS-ACV, Fe-PAH, Fe-PAH-NO and Fe-PAH-O2 active sites complexes used to calculated NO/O2 binding (Tables S5-S8); Complete Ref. 31. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by NIH Grant GM40392 (E.I.S.) and NIH Grant GM24689 (J. D. L.). We thank Michael Mbughuni for assistance in preparation of IPNS.
References
- 1.Feig AL, Lippard SJ. Chem Rev. 1994;94:759–805. [Google Scholar]
- 2.Que L, Ho RYN. Chem Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 3.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 4.Hegg EL, Que L., Jr Eur J Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin JE, Adlington RM, Moroney SE, Field LD, Ting HH. J Chem Soc, Chem Commun. 1984:984–986. [Google Scholar]
- 6.Baldwin JE, Abraham E. Nat Prod Rep. 1988;5:129–145. doi: 10.1039/np9880500129. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin JE, Bradley M. Chem Rev. 1990;90:1079–1088. [Google Scholar]
- 8.Rocklin AM, Tierney DL, Kofman V, Brunhuber NMW, Hoffman BM, Christoffersen RE, Reich NO, Lipscomb JD, Que L., Jr Proc Natl Acad Sci USA. 1999;96:7905–7909. doi: 10.1073/pnas.96.14.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J, Rocklin AM, Lipscomb JD, Que LJr, Solomon EI. J Am Chem Soc. 2002;124:4602–4609. doi: 10.1021/ja017250f. [DOI] [PubMed] [Google Scholar]
- 10.Rocklin AM, Kato K, Liu H-w, Que L, Lipscomb JD. J Biol Inorg Chem. 2004;9:171–182. doi: 10.1007/s00775-003-0510-3. [DOI] [PubMed] [Google Scholar]
- 11.Chen V, Orville A, Harpel M, Frolik C, Surerus K, Munck E, Lipscomb J. J Biol Chem. 1989;264:21677–21681. [PubMed] [Google Scholar]
- 12.Orville AM, Chen VJ, Kriauciunas A, Harpel MR, Fox BG, Munck E, Lipscomb JD. Biochemistry. 1992;31:4602–4612. doi: 10.1021/bi00134a010. [DOI] [PubMed] [Google Scholar]
- 13.Randall CR, Zang Y, True AE, Que L, Jr, Charnock JM, Garner CD, Fujishima Y, Schofield CJ, Baldwin JE. Biochemistry. 1993;32:6664–6673. doi: 10.1021/bi00077a020. [DOI] [PubMed] [Google Scholar]
- 14.Scott RA, Wang S, Eidsness MK, Kriauciunas A, Frolik CA, Chen VJ. Biochemistry. 1992;31:4596–4601. doi: 10.1021/bi00134a009. [DOI] [PubMed] [Google Scholar]
- 15.Roach PL, Clifton IJ, Fulop V, Harlos K, Barton GJ, Hajdu J, Andersson I, Schofield CJ, Baldwin JE. Nature (London) 1995;375:700–704. doi: 10.1038/375700a0. [DOI] [PubMed] [Google Scholar]
- 16.Roach PL, Clifton IJ, Hensgens CMH, Shibata N, Long AJ, Strange RW, Hasnain SS, Schofield CJ, Baldwin JE, Hajdu J. Eur J Biochem. 1996;242:736–740. doi: 10.1111/j.1432-1033.1996.0736r.x. [DOI] [PubMed] [Google Scholar]
- 17.Roach PL, Clifton IJ, Hensgens CMH, Shibta N, Schofield CJ, Hajdu J, Baldwin JE. Nature (London) 1997;387:827–830. doi: 10.1038/42990. [DOI] [PubMed] [Google Scholar]
- 18.Chen P, Solomon EI. J Am Chem Soc. 2004;126:4991–5000. doi: 10.1021/ja031564g. [DOI] [PubMed] [Google Scholar]
- 19.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 20.Francisco WA, Blackburn NJ, Klinman JP. Biochemistry. 2003;42:1813–1819. doi: 10.1021/bi020592t. [DOI] [PubMed] [Google Scholar]
- 21.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM., Jr Proc Natl Acad Sci USA. 2006;103:6130–6135. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arciero D, Lipscomb J, Huynh B, Kent T, Munck E. J Biol Chem. 1983;258:14981–14991. [PubMed] [Google Scholar]
- 23.Westcott BL, Enemark JH. Transition Metal Nitrosyls. In: Solomon EI, Lever ABP, editors. Inorganic Electronic Structure and Spectroscopy. Applications and Case Studies. II. John Wiley and Sons; New York: 1999. pp. 403–450. [Google Scholar]
- 24.Han AY, Lee AQ, Abu-Omar MM. Inorg Chem. 2006;45:4277–4283. doi: 10.1021/ic060478p. [DOI] [PubMed] [Google Scholar]
- 25.Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. J Am Chem Soc. 2003;125:7056–7066. doi: 10.1021/ja0214126. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Oldfield E. J Am Chem Soc. 2004;126:9494–9495. doi: 10.1021/ja0401242. [DOI] [PubMed] [Google Scholar]
- 27.Brown CA, Pavlosky MA, Westre TE, Zhang Y, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1995;117:715–732. [Google Scholar]
- 28.Schenk G, Pau MYM, Solomon EI. J Am Chem Soc. 2004;126:505–515. doi: 10.1021/ja036715u. [DOI] [PubMed] [Google Scholar]
- 29.Samson SM, Belagaje R, Blankenship DT, Chapman JL, Perry D, Skatrud PL, VanFrank RM, Abraham EP, Baldwin JE, Queener SW, Ingolia TD. Nature (London) 1985;318:191–194. doi: 10.1038/318191a0. [DOI] [PubMed] [Google Scholar]
- 30.Kriauciunas A, Frolik CA, Hassell TC, Skatrud PL, Johnson MG, Holbrook NL, Chen VJ. J Biol Chem. 1991;266:11779–11788. [PubMed] [Google Scholar]
- 31.Frisch MJ, et al. 2004 Gaussian03, Revision C. 03, http://www.gaussian.com, Wallinford, CT.
- 32.Becke AD. Phys Rev A: Gen Phys. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 33.Perdew JP. Phys Rev B: Cond Mater. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 34.Gorelsky SI. AOMix program, rev. 6.04. Available from: < http://www.sg-chem.net/>.
- 35.Gorelsky SI, Lever ABP. J Organomet Chem. 2001;635:187–196. [Google Scholar]
- 36.Schaftenaar G, Noordik JH. J Comp-Aided Mol Des. 2000;14:123–134. doi: 10.1023/a:1008193805436. [DOI] [PubMed] [Google Scholar]
- 37.Cramer CJ, Truhlar DG. Chem Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m. [DOI] [PubMed] [Google Scholar]
- 38.Gte Velde FMB, Baerends EJ, Fonseca Guerra C, Avan Gisbergen SJ, Snijders JG, Ziegler T. J Comput Chem. 2001;22:931–967. [Google Scholar]
- 39.Guerra CF, Snijders JG, Velde Gt, Baerends EJ. Theor Chim Acta. 1998;V99:391–403. [Google Scholar]
- 40.ADF2004.01, SCM. Theoretical Chemistry. Vrije Universiteit; Amsterdam, The Netherlands: http://www.scm.com. [Google Scholar]
- 41.Due to the temperature instability of the Fe-IPNS-ACV-NO glass, VTVH data points were obtained from full MCD spectra collected at different temperatures and fields. This resulted in fewer data points and larger errors than normal for the VTVH data due to a temperature-dependent CD background.
- 42.Neese F, Solomon EI. Inorg Chem. 1999;38:1847–1865. doi: 10.1021/ic981264d. [DOI] [PubMed] [Google Scholar]
- 43.Hauser C, Glaser T, Bill E, Weyhermuller T, Wieghardt K. J Am Chem Soc. 2000;122:4352–4365. [Google Scholar]
- 44.The π interaction results from overlap of the 3p orbital perpendicular to the Fe-S-C plane. The pseudo-σ interaction results from overlap in the plane, which is tilted off the Fe-S axes because the Fe-S-C is greater than 90°.
- 45.Davis MI, Wasinger EC, Decker A, Pau MYM, Vaillancourt FH, Bolin JT, Eltis LD, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2003;125:11214–11227. doi: 10.1021/ja029746i. [DOI] [PubMed] [Google Scholar]
- 46.Andersen OA, Flatmark T, Hough E. J Mol Biol. 2001;314:279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cartesian coordinates of the geometry optimized model of the Fe-IPNS-ACV-NO active site complex with Gaussian BP86+10% Hartree-Fock (Table S1); Relevant structural parameters for the optimized Fe-IPNS-ACV-NO complex (Table S2); Cartesian coordinates of the geometry optimized model of the Fe-IPNS-ACV-O2 active site complex with Gaussian BP86+10% Hartree-Fock (Table S3); Relevant structural parameters for the optimized Fe-IPNS-ACV-O2 complex (Table S4); Cartesian coordinates of the geometry optimized models of the Fe-IPNS-ACV, Fe-PAH, Fe-PAH-NO and Fe-PAH-O2 active sites complexes used to calculated NO/O2 binding (Tables S5-S8); Complete Ref. 31. This material is available free of charge via the Internet at http://pubs.acs.org.