

Abstract
Expansion of the genetic alphabet has been a long time goal of chemical biology. A third DNA base pair that is stable and replicable would have a great number of practical applications and would also lay the foundation for a semi-synthetic organism. We have reported that DNA base pairs formed between deoxyribonucleotides with large aromatic, predominantly hydrophobic nucleobase analogs, such as propinyl isocarbostyril (dPICS), are stable and efficiently synthesized by DNA polymerases. However, once incorporated into the primer, these analogs inhibit continued primer elongation. More recently, we have found that DNA base pairs formed between nucleobase analogs that have minimal aromatic surface area in addition to little or no hydrogen-bonding potential, such as 3-fluoro benzene (d3FB), are synthesized and extended by DNA polymerases with greatly increased efficiency. Here we show that the rate of synthesis and extension of the self pair formed between two d3FB analogs is sufficient for in vitro DNA replication. To better understand the origins of efficient replication, we examined the structure of DNA duplexes containing either the d3FB or dPICS self pairs. We find that the large aromatic rings of dPICS pair in an intercalative manner within duplex DNA, while the d3FB nucleobases interact in an edge-on manner, much closer in structure to natural base pairs. We also synthesized duplexes containing the 5-methyl substituted derivatives of d3FB (d5Me3FB) paired opposite d3FB or the unsubstituted analog (dBEN). In all, the data suggest that structure, electrostatics and dynamics can all contribute to the extension of unnatural primer termini. The results also help explain the replication properties of many previously examined unnatural base pairs and should help design unnatural base pairs that are better replicated.
Introduction
Expansion of the genetic alphabet to include a third base pair would be a fundamental accomplishment that would not only have immediate utility for a number of applications, such as site-specific oligonucleotide labeling, but would also lay the foundation for an organism with an expanded genetic code. Efforts toward this goal were first reported by Benner and coworkers1, who designed nucleobase analogs to pair based on hydrogen-bonding (H-bonding) patterns that are complementary to each other, but not to any of the natural nucleobases. While these analogs have found practical applications and improvements continue to be reported, work from the Kool group has shown that H-bonds are not absolutely essential for polymerase-mediated base pair synthesis2–6. This work demonstrated that forces other than H-bonding could control polymerase-mediated base pair synthesis, and it has inspired a variety of novel nucleobase design strategies.
We,7–18 and others,19–30 have examined a large number of unnatural nucleotides bearing nucleobase analogs that pair based on packing and hydrophobic interactions rather than H-bonding. (While many of the analogs are not actually basic, we refer to them as nucleobases for simplicity). Packing and hydrophobic interactions have a well documented role in protein folding, structure, and stability and should be inherently orthogonal to the H-bonding forces that mediate pairing of the natural base pairs. Indeed, DNA containing simple hydrophobic nucleobase analogs, such as benzene rings,31 has been studied for years and more recent studies have shown that hydrophobic nucleobase analogs may be incorporated into duplex DNA without significant structural distortions.32,33 Our original efforts focused primarily on nucleobase analogs derived from relatively large bicyclic scaffolds with extended aromatic surface area. These analogs were designed to preserve duplex stability in the absence of interstrand H-bonding, by increasing intrastrand packing. We identified several heteropairs11–14,34 (i.e. formed by pairing two different analogs) and self pairs10–12,17 (i.e. formed by pairing two identical analogs) that are stable in duplex DNA and efficiently synthesized by polymerase-catalyzed insertion of the unnatural triphosphate opposite its cognate base in a DNA template. For example, the dPICS self pair (Figure 1) is stable in duplex DNA, and also synthesized (by insertion of the triphosphate opposite the analog in the template) by the exonuclease deficient Klenow fragment of E. coli DNA polymerase I (Kf) with reasonable efficiency and selectivity.10 These results demonstrated that packing and hydrophobicity are sufficient to mediate duplex stability and unnatural base pair synthesis.
Figure 1.

Unnatural nucleotides used in this study.
While many of the unnatural base pairs formed between the relatively large nucleobase analogs, such as dPICS, are stable and efficiently synthesized, continued primer extension is very inefficient in all cases examined, regardless of the DNA polymerase employed. Thus, the determinants of base pair stability and synthesis are different from those of extension. Rate limiting extension has also been observed during the replication of other unnatural base pairs. We have speculated that while large aromatic surface area may stabilize base pairing, as well as the transition state for unnatural nucleotide insertion, it may also result in a structure at the primer terminus that is poorly recognized by DNA polymerases. To probe the role of nucleobase aromatic surface area, we designed and evaluated nucleoside analogs bearing a wide variety of simple, derivatized phenyl rings.11,12,15–17 The shape, hydrophobicity, and electronic properties of the nucleobase analogs were systematically varied by derivatization with fluoro, bromo, cyano, and/or methyl substituents. Surprisingly, we found that large aromatic surface area is not required for stable and selective pairing within the duplex, nor for the relatively efficient synthesis of the unnatural base pairs. More importantly, while we observed that extension remained inefficient for the majority of the pairs, several were extended with a significantly higher efficiency. Most notably, d3FB (Figure 1) is incorporated opposite itself and then correctly extended by Kf with a rate that is within 100-fold of a natural base pair.17 This result is remarkable considering that the d3FB self pair lacks both H-bonds and extended aromatic surface area, which are thought to underlie the stability and replication of natural DNA.
In this work, we first demonstrate that the synthesis and extension efficiencies of d3FB are sufficient for Kf to synthesize long strands of DNA containing the self pair. Then, to better understand why the d3FB self pair is efficiently synthesized and extended, while similar pairs, or pairs formed between the larger nucleobase analogs are not, we examine the structure of DNA duplexes containing either the d3FB or dPICS self pair. In addition, the role of hydrophobic packing at the interface between the nucleobase analogs was further examined by comparing the replication and structure of the d3FB self pair with that of a heteropair formed between d3FB and its 5-methyl-3-fluoro derivative (d5Me3FB) or between d5Me3FB and a simple phenyl nucleotide (dBEN). The data suggest that structure, electrostatics, and dynamics each contribute to efficient unnatural base pair replication.
Results
Previously we demonstrated that the d3FB self pair is efficiently synthesized by Kf and then extended by the addition of a single dCTP opposite dG in the template.17 As discussed above, the efficient extension of the d3FB self pair, relative to others that have been examined, including the dPICS self pair, suggests that the d3FB self pair forms a more natural-like primer terminus. To determine whether the d3FB self pair is compatible with efficient full length synthesis, we examined the ability of Kf to synthesize a full length strand of DNA that contained d3FB, as well as an additional 10 nucleotides (Figure 2). d3FB has high selectivity against misincorporation of guanosine and cytosine, but only modest selectivity against adenine and thymine, thus the template contained only dG and dC nucleotides, and only the triphosphates dCTP, dGTP, and d3FBTP were added to the reaction mixture. Under these conditions, full length product is efficiently produced, demonstrating that the d3FB self pair does not interfere with the addition of downstream nucleotides into the growing primer strand. By comparison, attempts to replicate the dPICS self pair in the same sequence context resulted in complete termination of primer elongation after insertion of the unnatural triphosphate.
Figure 2.
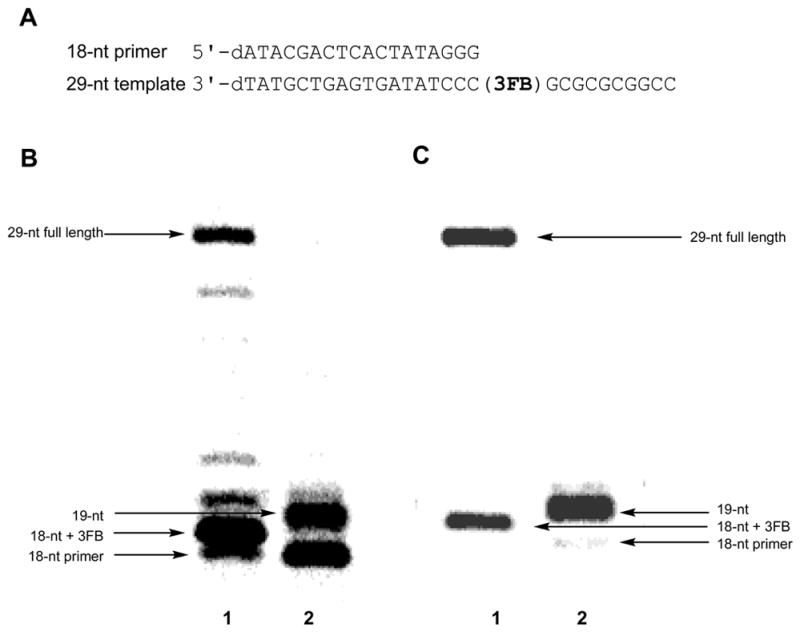
(A) Sequence of 18-nt primer and 29-nt template used for full length synthesis. (B) Full length synthesis by KF exo−; 40 nM primer-template, 1.23 nM enzyme, 50 mM Tris-HCl, pH 7.5, 1 mM DTT, 50 μg/mL acetylated BSA, 2 hours at 25 °C, 5 mM MgCl2, 1 mM MnCl2, and 50 μM of each dCTP and dGTP, with (lane 1) or without (lane 2) 200 μM d3FBTP. (C) Full length synthesis by KF exo+; 40 nM primer-template, 1.23 nM enzyme, 50 mM Tris-HCl, pH 7.5, 1 mM DTT, 50 μg/mL acetylated BSA, 2 hours at 25 °C, 10 mM MgCl2, 1 mM MnCl2, and 200 μM dCTP/dGTP, with (lane 1) or without (lane 2) 200 μM d3FBTP.
To identify possible determinants of efficient replication, and in particular, why the d3FB self pair is both efficiently synthesized and extended, while the dPICS self pair is efficiently synthesized but not extended, we examined the structure of duplex DNA containing these unnatural base pairs. The structure of DNA containing a dPICS self pair was determined in the duplex d(C1G2T3T4T5C6PICS7T8T9C10T11C12): d(G13A14G15A16A17PICS18G19A20A21A22C23G24) using NMR and restrained molecular modeling (Figure 3). Overall, the duplex assumes a canonical B-form structure and is not substantially distorted relative to natural DNA35 except at the site of the dPICS self pair. The dPICS propynyl groups are oriented into the major groove, while the carbonyl groups are positioned in the minor groove. This conformation, which is referred to as anti by analogy to natural nucleotides, likely minimizes repulsive electrostatic interactions between the nucleobase carbonyl oxygen and the oxygen and/or 5′ carbon of the ribose ring. The most notable feature of the structure is that the dPICS nucleotides interact through interstrand stacking: each nucleobase analog intercalates between the other and its flanking base pair, rather than pairing in an edge-on manner as observed with natural Watson-Crick base pairs. The positions of the bases were verified by NOE contacts between the edge of both dPICS nucleobases and the sugar of the opposite strand and demonstrate that only one of the two possibly intercalated structures is formed. The preference for the observed structure likely results from optimized packing interactions between the dPICS nucleobase and the flanking natural nucleobases. While we observed breaks in the normal NOE connectivities between the sugar and the flanking natural nucleobase at both dPICS18dG19 and dPICS7dT8, the imino proton resonances were observed for all Watson-Crick pairs, except the terminal dG:dC nucleotides, indicating that the hydrogen-bonds of the flanking DNA remain intact. The intrastrand phosphate distances are approximately 7.0 Å, which is typical of B-form DNA. The O4′-C1′-N1-C2 dihedral angles of the dPICS nucleotides are approximately −135°, also in the range for B-form DNA. The torsion angles about the exocyclic C4′–C5′ bonds, which position the 5′ phosphate group relative to the sugar and the nucleobase, are 60° to 70° for dPICS18 and the flanking natural nucleotides in the purine-rich strand. The same torsion angle is approximately 45° for dPICS7 in the pyrimidine-rich strand and approximately 70° for its flanking nucleotides, similar to that found with natural B-DNA. However the neighboring O5′-C5′ torsion angles for dPICS18 and dPICS7 are approximately 100° while normal B-DNA values are approximately 170°. These structural readjustments appear to help accommodate unnatural base pair intercalation. In the model, intrastrand packing appears to be better optimized within the purine-rich strand than the pyrimidine-rich strand, which is distorted by the buckling at the self pair.
Figure 3.

Structural characterization of the dPICS self pair in the duplex d(C1G2T3T4T5C6PICS7T8T9C10T11C12):d(G13A14G15A16A17PICS18G19A20A21A22C23G24). (A) Aromatic to H1′ region of a 200 ms NOESY spectrum. (B) Model of the duplex. The dPICS self pair is shown in the center of the duplex.
The structures of two different duplexes containing the d3FB self pair were determined, one using X-ray crystallography and the other using NMR spectroscopy (Figure 4). Figure 4A shows the central section of the DNA duplex d(C1G2BrC3G4A5A63FB7T8T9C10G11C12G13)2 containing a single d3FB self pair as determined by X-ray crystallography with a resolution of 2.8 Å (Table 1 and Supporting Information). Six copies of the duplex are present in the crystallographic asymmetric unit. Four copies are well ordered (chains A-H) and well defined by the electron density. The remaining two copies are less well ordered (chains I-L), and characterized by diffuse electron density; however the density was successfully fit using the bromine atoms in the Patterson maps (Supporting Information). Analysis of the four ordered duplexes using the 3DNA package36 revealed a right-handed B-form DNA conformation with a mean helix diameter of 19.9 Å, consistent with standard Watson-Crick base pairing (except at the 5′ and 3′ ends of the duplex, where the nucleobases intercalate into neighboring duplexes in the crystal to form two sets of semi-continuous helices). The root mean square deviation between the duplexes is 1.50 Å for backbone atoms and 0.84 Å for nucleobase atoms. The root mean square deviation between the average duplex and an ideal B-form duplex is 1.26 Å and 0.58 Å for sugar-phosphate backbone and nucleobase atoms, respectively. The d3FB nucleobases are oriented so that their fluorine atoms are positioned in the major groove of the duplex, separated by 9.8 Å (Figure 4B). At their closest approach, the nucleobase analogs are separated by an average carbon to carbon distance of 3.75 Å. This is slightly greater than the sum of the van der Waals radii (3.4 Å), which suggests that the nucleobase analogs are not optimally edge-to-edge packed. The unnatural base pairs adopt an average propeller twist of −12°, which is virtually identical to that of canonical B-form DNA. The mean distance between the d3FB nucleobase analog and the flanking natural nucleobases is 3.2 Å, which suggests that the analogs pack favorably with their flanking natural nucleobases. In fact, the only significant deviation from an ideal duplex geometry appears to be due to these stacking interactions, as the flanking natural nucleobases tilt in order to achieve optimal co-planarity with the unnatural nucleobases (Figure 4B).
Figure 4.

Structural characterization of the d3FB self pair. (A) X-ray structure of the DNA duplex d(C1G2C3BrG4A5A63FB7T8T9C10G11C12G13)2 at 2.8 Å resolution (PDB ID: 2PIS). The d3FB self pair is shown with flanking base pairs. Carbon atoms are shown in yellow, nitrogen in blue, oxygen in orange, phosphorous in magenta and fluorine in green. (B) 2Fo−Fc electron density map contoured at 1.2_ around the model of the d3FB self pair and its adjacent residues. The fluorine atoms are oriented into the major groove and are colored light cyan. Figure produced by Pymol (www.pymol.org). (C) NMR characterization of the DNA duplex d(C1G2C33FB4A5A6T7T83FB9G10C11G12)2. Shown is the aromatic to H2′/H2″ region of a 2D NOESY experiment with the NOE walk indicated by solid lines. Numbers indicate the aromatic to H2′ and H2″ intra-nucleotide peaks. Both the H2 and H6 protons of d3FB9 and of d3FB4 show cross peaks with the H2′ and H2″ protons of the preceding residue indicating rapid flipping of the d3FB rings. The four possible ring orientations of the d3FB used to model the NOE data are shown on the right. (D) 1H-19F NOESY spectrum of d(C1G2C33FB4A5A6T7T83FB9G10C11G12)2 acquired as described previously.46 The fluorine of d3FB9 resonates at −113.48 ppm while the chemical shift of the fluorine of d3FB4 is −113.64 ppm. 19F-1H NOE peaks are labeled according to Figure 4C. Experimental details and spectra are available in the Supporting Information.
Table 1.
Data collection, phasing and refinement statistics
| Remote | f″ | f′ | |
|---|---|---|---|
| Space Group | I4122 | ||
| Unit Cell Parameters (Å) | a=b=146.00, c=93.21 | ||
| Wavelength (Å) | 0.9050 | 0.92017 | 0.92030 |
| Resolution Range (Å) | 50.0-2.8 | 50.0-2.8 | 50.0-2.8 |
| Rsymm (in highest resolution shell) | 0.105(0.747) | 0.089 (0.59) | 0.099 (0.706) |
| No. Unique Refs (observed) | 12331 (186023) | 12382 (190046) | 12271 (92360) |
| Completeness (%) (Highest shell) | 97.7(87.5) | 97.8(88.5) | 97.1 (84.1) |
| Highest Resolution Shell (Å) | 2.9-2.8 | ||
| Mean I/σ(I) | 25.2 (2.9) | 19.6(1.5) | 12.0(3.1) |
| Phasing Statistics | |||
| No. of Br sites | 10 | ||
| Mean Figure of Merit | 0.42 (0.19) | ||
| Model and Refinement Statistics | |||
| No. of reflections (total) | 11082 | ||
| No of reflections (test) | 565 | ||
| Rcryst (Rfreea)b | 23.1,(30.8) | ||
| No. nucleic acid atoms | 2474 | ||
| No. hetero atoms | 678 | ||
| Stereochemical Parameters | |||
| rmsd bonds Å | 0.014 | ||
| rmsd angles ° | 1.742 | ||
| Average isotropic B-value (Å2) | 56.75 | ||
| ESU based on Rfree (Å)c | 0.483 | ||
Rcryst= Σ | Fo−Fc |/Σ | Fc |, where Fo and Fc are observed and calculated structure factors, respectively. Rfree was calculated from a test set (5%) omitted from the refinement.
Rfactor = Σ | Ii−<Ii>| |/Σ| Ii | where Ii is the scaled intensity of the ith measurement, and <Ii> is the mean intensity for that reflection.
Characterization of d(C1G2C33FB4A5A6T7T83FB9G10C11G12)2 by NMR spectroscopy and NOE restrained MD simulations (Supporting Information) also indicates a canonical B-form DNA duplex as demonstrated by characteristic NOE connectivities and intensities (Figure 4C). The base-sugar connectivities along each strand are not interrupted at the d3FB self pair (Figure 4C), unlike the connectivities observed for the dPICS self pair which show clear breaks between the dPICS sugars and their 3′ neighbor base protons. In addition, imino to imino and imino to adenine H2 NOE connectivities are observed throughout the DNA helix except at the terminal base pair (data not shown). The similarity of all proton chemical shifts to those reported for the fully natural DNA duplex (containing a dG4 and dC9, Table S1 and Figure S1)37 further demonstrates that both d3FB self pairs are accommodated within the double helix without substantial structural distortions.
Interestingly, the NMR data indicate that the d3FB self pair adopts multiple conformations that are related by simple ring flips about each C-glycosidic linkage (Figure 4C & D). This heterogeneity is demonstrated by NOE cross peaks from d3FB4 H2 and H6 to dC3 H2′/H2″, and from d3FB9 H2 and H6 to T8 H2′/H2″, respectively (Figure 4C), as well as by heteronuclear NOEs between the fluorine of d3FB4 and both dA5 H2 and dC3 H6, which are mutually exclusive for a single ring orientation (Figure 4D). An NOE between the d3FB9 fluorine and both dT8 CH3 and dT8 H6, as well as between the H4 and H5 protons of the d3FB4 further demonstrate that both nucleobase analogs undergo rapid ring flipping. Since single resonance lines are observed for the two fluorine atoms and for each aromatic proton, the rate of base flipping must be fast on the chemical shift time scale for these resonances, i.e. exchange lifetimes on the submillisecond time scale. This ring flipping model is also supported by NOE restrained MD simulations. When all 662 NOE restraints are applied, the d3FB self pair adopts conformations with large propeller twists causing distortions of the neighboring base pairs and high restraint violation energies (Table 2, Figures S2 and S3). Grouping NOEs into four separate sets, corresponding to each of the four possible self pair conformations eliminated these violations (Table 3 and S3) and distortions (Figure S4), suggesting that they arise from a superposition of signals corresponding to multiple conformations present in solution. The predicted energy of the conformation with both fluorine atoms oriented into the duplex, as well as the absence of an observable fluorine-fluorine NOE cross peak (Figure S5) suggest this conformation is not significantly populated. However, the NMR data suggests that all three other possible self pair conformations are populated (Figure 4C).
Table 2.
NOE-restrained MD calculations with d(C1G2C33FB4A5A6T7T83FB9G10C11G12)2 using all restraints. Identical restraints were applied to the four different families of d3FB conformers (Figure 4C).
| Self Pair Conformation a | # Structures | RMSD | RMSD from Mean Struct. | Average E- violation | Average E- AMBER | % of E- Amber due to E-violation | Total # of Restraints | # Restraints w/ave. viol. > 0.2 Å |
|---|---|---|---|---|---|---|---|---|
| F33 | 22 | 1.80 | 1.24 | 156.0 | −5184 | 3.0 | 662 | 26 |
| F35 | 17 | 2.06 | 1.41 | 197.9 | −5146 | 3.8 | 662 | 21 |
| F53 | 22 | 1.88 | 1.30 | 170.2 | −5160 | 3.3 | 662 | 21 |
| F55 | 21 | 1.93 | 1.34 | 183.3 | −5122 | 3.6 | 662 | 16 |
For a definition of d3FB self pair conformations, see Figure 4C.
Table 3.
NOE-restrained MD calculations with d(C1G2C33FB4A5A6T7T83FB9G10C11G12)2 after organizing the restraints into four sets consistent with the four different starting conformations (Figure 4C).
| Self Pair Conformationa | # Structures | RMSD | RMSD from Mean Struct. | Average E- violation | Average E- AMBER | % of E- Amber due to E-violation | Total # of Restraints | # Restraints w/ave. viol. > 0.2 Å |
|---|---|---|---|---|---|---|---|---|
| F33 | 18 | 1.97 | 1.35 | 19.87 | −5307 | 0.37 | 608 | 0 |
| F35 | 22 | 2.04 | 1.41 | 28.19 | −5261 | 0.54 | 610 | 0 |
| F53 | 20 | 1.93 | 1.33 | 21.79 | −5275 | 0.41 | 618 | 0 |
| F55 | 17 | 1.92 | 1.32 | 17.87 | −5242 | 0.34 | 616 | 0 |
For a definition of d3FB self pair conformations, see Figure 4C.
Spectral congestion precludes an accurate assessment of the population of the three self pair conformations, however, the d3FB self pair conformation, in which both fluorine atoms are positioned in the major groove, as seen in the X-ray structure, yields the lowest energy structure in the NOE restrained MD simulations (Table 3), suggesting that it may also be the most populated in solution. To approximate the actual populations of each self pair conformation we examined the NMR spectra of the d3FB in the d5Me3FB heteropair (see below). In contrast to the self pair, the signals of the d3FB:d5Me3FB heteropair are more resolved and the d3FB nucleotide appears to adopt the same conformations. The intensity of the intranucleotide aromatic to H2′/H2″ NOE peaks of the d3FB analog of the heteropair suggests that the fluorine atom is oriented into the major groove approximately 70% of the time, while the remainder of the time it is oriented into the interior of the duplex. If the same ratio is assumed for both nucleobases of the self pair, then one can conclude that the conformation with both fluorine atoms disposed in the major groove is populated approximately 40% of the time, while the two conformations where one fluorine atom is positioned in the major groove and the other within the duplex are each populated approximately 30% of the time (Figure 4C). These populations are consistent with the observed X-ray structure. In addition to rapid ring flipping, additional self pair motion is suggested by line broadening of intranucleotide NOE cross peaks of the flanking nucleobases dA5 and dG10, and dA5 imino resonance broadening which may result from enhanced solvent exchange, possibly due to increased solvent accessibility or base pair fluctuations.38 Such fluctuations may be coupled to the observed ring flipping of the d3FB analogs.
Taken together, the X-ray and NMR data indicate that the d3FB self pair does not perturb the structure of a B-form DNA duplex and that the nucleobase analogs are co-planar and well packed with their flanking natural nucleobases. However, while the d3FB self pair is significantly more stable than the mispairs of the natural bases,17 it is somewhat less stable than pairs formed between more highly substituted analogs, and the structures suggest that d3FB is not optimized for edge-on packing within the self pair. Less than optimal packing may underlie the self pair dynamics observed in the NMR experiments. To explore the effect of increased interbase packing, we synthesized and examined an analog with a methyl group at position 5 of the benzene ring, d5Me3FB (Figure 1). This base analog was incorporated into the duplex d(C1G2C35Me3FB4A5A6T7T83FB9G10C11G12)2, which pairs d5Me3FB opposite d3FB. NOE analysis unambiguously demonstrates that the d5Me3FB analog does not undergo base flipping and that its methyl group positioned into the duplex where it packs against the pairing d3FB (Figure S6). Consistent with a more optimally packed interface, the UV melting temperature of the duplex with the d3FB:d5Me3FB heteropair was ~2°C higher than that of the analogous duplex containing the d3FB self pair (Supporting Information). However, we cannot exclude the possibility that packing interactions between the methyl group and flanking nucleobases also contribute to the increased stability. While the d5Me3FB nucleobase is less dynamic, rotation of the d3FB nucleobase of the heteropair remains fast on the chemical shift timescale, and line broadening of the flanking nucleotides dA5 and dG10 is observed, as in the d3FB self pair. Similar observations were made when d5Me3FB was paired opposite dBEN (Figure 1) in the same sequence context (Figure S7). While d5Me3FB is locked into one orientation, the phenyl ring undergoes rapid flipping as indicated by the observation of only three aromatic resonances rather than the five expected for a static nucleobase analog in the magnetically non-symmetric environment of a DNA duplex. These data indicate that the addition of the methyl group restrains the modified nucleobase to a single, well defined conformation but that the pairing nucleobase remains dynamic. The data also confirm that sufficient space is available for at least a single fluorine within the interface between the nucleobase analogs and suggests that the positioning of the fluorine atoms of the d3FB self pair must result from other interactions, such as electrostatic or packing interactions with the flanking nucleobases.
To examine how a better packed nucleobase interface impacts DNA synthesis, we examined the ability of Kf to extend a primer terminating with d5Me3FB (paired with d3FB in the template) or terminating with d3FB (paired with d5Me3FB in the template). With d5Me3FB at the primer terminus, no extension product was observed (kcat/KM < 1 × 103 M−1min−1). In contrast, with d3FB at the primer terminus opposite d5Me3FB, the extension proceeded with a second order rate constant of 1.6 × 104 M−1min−1, which is only 30-fold reduced relative to the d3FB self pair. This data suggests that the most efficient extension results when d3FB at the primer terminus is free to adopt the conformation with its fluorine oriented into the duplex, as opposed to into the major groove.
Discussion
The expansion of the genetic alphabet requires an unnatural base pair that is efficiently replicated by DNA polymerases. The unnatural nucleoside triphosphate must be efficiently and selectively inserted opposite its partner in the template, and the resulting terminus must be efficiently extended by incorporation of the next correct nucleotide. Generally, we have found that hydrophobic packing interactions between large aromatic nucleobase analogs, such as the isocarbostyril group of dPICS, give rise to stable base pairs that are synthesized with reasonable efficiency, but not extended by further primer elongation. In contrast, the d3FB self pair is both synthesized and extended by Kf.
In order to understand the basis for the differences in behavior between the dPICS and d3FB self pairs, we characterized duplex DNA containing these unnatural base pairs. While both are accommodated within duplex DNA without significant loss of duplex stability, the large aromatic isocarbostyril rings of the dPICS pair in an intercalative, stacked manner. The intercalative mode of interaction within the dPICS self pair is consistent with its high stability as well as its efficient synthesis, as significant hydrophobic packing is expected to be manifest in the developing transition state for dPICSTP insertion. However, it is also likely that an intercalated structure at the primer terminus results in the mispositioning of the 3′OH in the enzyme active site, which may explain why the dPICS self pair is not efficiently extended. An intercalative mode of interaction has also been predicted by computational analysis of a related self pair39 and has been observed with other nucleotides that have large aromatic nucleobase analogs.40–43 Thus, stability, efficient synthesis, and poor extension are likely to be general features of unnatural base pairs formed between nucleotides with large aromatic nucleobase analogs, due to intercalative pairing.
In contrast to the dPICS self pair, the nucleobase analogs of the d3FB self pair are not sufficiently large to bridge the duplex and cannot intercalate. Instead, they interact in an edge-on manner similar to natural nucleobases. The d3FB self pair stability and synthesis are likely mediated by hydrophobic forces and may have contributions from intrastrand electrostatic interactions.
While a more natural terminal base pair structure is likely to be important for the extension of the d3FB self pair, other factors must also contribute, as a wide variety of similar self pairs are not efficiently extended. We have altered the number, position, and nature of the substituents of the phenyl ring scaffold and find that such modifications have a significant and generally detrimental impact on both synthesis and extension efficiencies. To date, the specific meta fluoro substitution pattern of d3FB is unique in its ability to facilitate replication. Similarly positioned methyl, bromo, and cyano substituents, or alternate fluoro substituent patterns result in self pairs with significantly reduced extension efficiencies.12,16,17 Moreover, we have shown here that while addition of a methyl group to the meta position of d3FB stabilizes pairing, it also prevents replication. Specifically, when the analog in the DNA template is modified with the methyl group it has only a small effect on the efficient extension, however, when the analog at the primer terminus is modified, extension is undetectable. Along with the NMR data, which suggests that the addition of the methyl group localizes the nucleobase to the conformation with the fluorine atom in the major groove, this data also suggests that efficient extension occurs only when the d3FB nucleotide at the primer terminus rotates to position its fluorine atom into the interbase interface. Thus, in addition to a natural-like primer terminus structure, base pair dynamics and dipole interactions appear to be critical for unnatural base pair extension.
Despite a complete absence of H-bonding and modest shape complementarity, the d3FB self pair is reasonably stable and replicable. This argues that precise shape complementarity and H-bonding are not unique in their ability to control the specific interbase interactions required for DNA stability and replication. The data suggest that replication may be optimized within base pairs with co-planar nucleobase analogs by careful manipulation of shape and electrostatic properties. The results also emphasize that overly stabilized unnatural base pairs might be localized to structures that are not well recognized at a primer terminus; in the absence of perfectly designed nucleobases flexibility may be advantageous for replication. These results, along with those of recent studies demonstrating that extension may also be facilitated by the addition of minor groove H-bond acceptors, help explain the replication properties of a large number of previously reported unnatural base pairs and should help design pairs that are more efficiently replicated.
Supplementary Material
Kinetic, thermodynamic, NMR, and X-ray crystallography methods and supporting figures and tables.
Acknowledgments
Funding was provided by the National Institutes of Health (GM60005 to F.E.R.).
References
- 1.Piccirilli JA, Krauch T, Moroney SE, Benner SA. Nature. 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 2.Morales JC, Kool ET. Nat Struct Biol. 1998;5:950–954. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]
- 3.Moran S, Ren RXF, Kool ET. Proc Natl Acad Sci USA. 1997;94:10506–10511. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moran S, Ren RXF, Rumney SI, Kool ET. J Am Chem Soc. 1997;119:2056–2057. doi: 10.1021/ja963718g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kool ET. Biopolymers. 1998;48:3–17. doi: 10.1002/(SICI)1097-0282(1998)48:1<3::AID-BIP2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 6.Kool ET. Curr Op Chem Biol. 2000;4:602–608. doi: 10.1016/s1367-5931(00)00141-1. [DOI] [PubMed] [Google Scholar]
- 7.Tae EL, Wu YQ, Xia G, Schultz PG, Romesberg FE. J Am Chem Soc. 2001;123:7439–7440. doi: 10.1021/ja010731e. [DOI] [PubMed] [Google Scholar]
- 8.Ogawa AK, Wu Y, McMinn DL, Liu J, Schultz PG, Romesberg FE. J Am Chem Soc. 2000;122:3274–3287. [Google Scholar]
- 9.Ogawa AK, Wu Y, Berger M, Schultz PG, Romesberg FE. J Am Chem Soc. 2000;122:8803–8804. [Google Scholar]
- 10.McMinn DL, Ogawa AK, Wu Y, Liu J, Schultz PG, Romesberg FE. J Am Chem Soc. 1999;121:11585–11586. [Google Scholar]
- 11.Matsuda S, Romesberg FE. J Am Chem Soc. 2004;126:14419–14427. doi: 10.1021/ja047291m. [DOI] [PubMed] [Google Scholar]
- 12.Matsuda S, Henry AA, Romesberg FE. J Am Chem Soc. 2006;128:6369–6375. doi: 10.1021/ja057575m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leconte AM, Matsuda S, Romesberg FE. J Am Chem Soc. 2006;128:6780–6781. doi: 10.1021/ja060853c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leconte AM, Matsuda S, Hwang GT, Romesberg FE. Angew Chem Int Ed. 2006;45:4326–4329. doi: 10.1002/anie.200601272. [DOI] [PubMed] [Google Scholar]
- 15.Kim Y, Leconte AM, Hari Y, Romesberg FE. Angew Chem Int Ed. 2006;45:7809–7812. doi: 10.1002/anie.200602579. [DOI] [PubMed] [Google Scholar]
- 16.Hwang GT, Romesberg FE. Nucleic Acids Res. 2006;34:2037–2045. doi: 10.1093/nar/gkl049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henry AA, Olsen AG, Matsuda S, Yu C, Geierstanger BH, Romesberg FE. J Am Chem Soc. 2004;126:6923–6931. doi: 10.1021/ja049961u. [DOI] [PubMed] [Google Scholar]
- 18.Henry AA, Romesberg FE. Curr Opin Chem Biol. 2003;7:727–733. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 19.Ishikawa M, Hirao I, Yokoyama S. Tetrahedron Lett. 2000;41:3931–3934. [Google Scholar]
- 20.Hirao I, Harada Y, Kimoto M, Mitsui T, Fujiwara T, Yokoyama S. J Am Chem Soc. 2004;126:13298–13305. doi: 10.1021/ja047201d. [DOI] [PubMed] [Google Scholar]
- 21.Mitsui T, Kimoto M, Harada Y, Yokoyama S, Hirao I. J Am Chem Soc. 2005;127:8652–8658. doi: 10.1021/ja0425280. [DOI] [PubMed] [Google Scholar]
- 22.Mitsui T, Kitamura A, Kimoto M, To T, Sato A, Hirao I, Yokoyama S. J Am Chem Soc. 2003;125:5298–5307. doi: 10.1021/ja028806h. [DOI] [PubMed] [Google Scholar]
- 23.Hirao I, Ohtsuki T, Fujiwara T, Mitsui T, Yokogawa T, Okuni T, Nakayama H, Takio K, Yabuki T, Kigawa T, Kodama K, Yokogawa T, Nishikawa K, Yokoyama S. Nat Methods. 2006;3:729–735. [Google Scholar]
- 24.Kincaid K, Beckman J, Zivkovic A, Halcomb RL, Engels JW, Kuchta RD. Nucleic Acids Res. 2005;33:2620–2628. doi: 10.1093/nar/gki563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiaramonte M, Moore CL, Kincaid K, Kuchta RD. Biochemistry. 2003;42:10472–10481. doi: 10.1021/bi034763l. [DOI] [PubMed] [Google Scholar]
- 26.Hirao I. Curr Opin Chem Biol. 2006;10:622–627. doi: 10.1016/j.cbpa.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 27.Matray TJ, Kool ET. Nature. 1999;399:704–708. doi: 10.1038/21453. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Lee I, Berdis AJ. Biochemistry. 2005;44:13101–13110. doi: 10.1021/bi050585f. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Lee I, Zhou X, Berdis AJ. J Am Chem Soc. 2006;128:143–149. doi: 10.1021/ja0546830. [DOI] [PubMed] [Google Scholar]
- 30.Morales JC, Kool ET. J Am Chem Soc. 1999;121:2323–2324. doi: 10.1021/ja983502+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Millican TA, Mock GA, Chauncey MA, Patel TP, Eaton MAW, Gunning J, Cutbush SD, Neidle S, Mann J. Nucleic Acids Res. 1984;12:7435–7453. doi: 10.1093/nar/12.19.7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guckian KM, Krugh TR, Kool ET. J Am Chem Soc. 2000;122:6841–6847. doi: 10.1021/ja994164v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smirnov S, Matray TJ, Kool ET, de los Santos C. Nucleic Acids Res. 2002;30:5561–5569. doi: 10.1093/nar/gkf688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu YQ, Ogawa AK, Berger M, McMinn DL, Schultz PG, Romesberg FE. J Am Chem Soc. 2000;122:7621–7632. [Google Scholar]
- 35.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 36.Lu XJ, Olson WK. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hare DR, Wemmer DE, Chou SH, Drobny G, Reid BR. J Mol Biol. 1983;171:319–336. doi: 10.1016/0022-2836(83)90096-7. [DOI] [PubMed] [Google Scholar]
- 38.Guckian KM, Krugh TR, Kool ET. Nat Struct Biol. 1998;5:954–959. doi: 10.1038/2930. [DOI] [PubMed] [Google Scholar]
- 39.Reha D, Hocek M, Hobza P. Chem Eur J. 2006;12:3587–3595. doi: 10.1002/chem.200501126. [DOI] [PubMed] [Google Scholar]
- 40.Zahn A, Brotschi C, Leumann CJ. Chem Eur J. 2005;11:2125–2129. doi: 10.1002/chem.200401128. [DOI] [PubMed] [Google Scholar]
- 41.Brotschi C, Mathis G, Leumann CJ. Chem Eur J. 2005;11:1911–1923. doi: 10.1002/chem.200400858. [DOI] [PubMed] [Google Scholar]
- 42.Brotschi C, Haberli A, Leumann CJ. Angew Chem Int Ed. 2001;40:3012–3014. doi: 10.1002/1521-3773(20010817)40:16<3012::AID-ANIE3012>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 43.Brotschi C, Leumann CJ. Angew Chem Int Ed. 2003;42:1655–1658. doi: 10.1002/anie.200250516. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Methods, Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Tickle IJ, Laskowski RA, Moss DS. Acta Cryst. 1998;D54:243–252. doi: 10.1107/s090744499701041x. [DOI] [PubMed] [Google Scholar]
- 46.Scott LG, Geierstanger BH, Williamson JR, Hennig M. J Am Chem Soc. 2004;126:11776–11777. doi: 10.1021/ja047556x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Kinetic, thermodynamic, NMR, and X-ray crystallography methods and supporting figures and tables.