

Abstract
DNAzymes are catalytically active DNA molecules, which have previously been described in solution. Here, we organize these molecules into a series of 2D arrays using a periodic arrangement of DNA structures based on the DNA double crossover (DX) motif. We demonstrate by means of atomic force microscopy that the DNAzymes are organized according to the design, and that they retain their activity when attached in linear strings within the context of the 2D array.
Keywords: DNAzymes, DNA self-assembly, 2D DNA arrays, Organizing Matter with DNA, Atomic Force Microscopy
DNA has been shown to be one of the most exciting molecules available for use in nanotechnology. The key attraction of this molecule is related to its capacity to contain and express information through Watson-Crick hydrogen bonding; the last 15 years have shown repeatedly that both its intramolecular structure and its intermolecular interactions can be programmed through these interactions.1 Conventional double helical DNA is not a valuable structure for molecular construction, because of the linear topology of its helix axis. Fortunately, it is straightforward to design sequences that lead to a variety of branched molecular motifs,2 for example, double crossover (DX),3 DX-Triangle,4 or 6-Helix Bundle5 structures. Such motifs can be tailed in sticky ends6 that facilitate their self-assembly into larger periodic7 two-dimensional arrays.
A separate thread in the development of DNA-based nanoscience has been the development of DNAzymes, starting in 1994.8 These catalytic DNA molecules, also called deoxyribozymes, have been shown to catalyze many of the same reactions as the better known ribozymes (e.g., ref. 9) including RNA/DNA cleavage8,10,11 and ligation.12,13 The endonuclease activity of DNAzymes is the function that has been explored the most. Similar to that of RNAzymes,9 the endonuclease activity makes DNAzymes promising anti-viral pharmaceutical agents against diseases such as AIDS and leukemia. For example, a version of the “10–23” DNAzyme14,15 has been targeted to destroy hepatitis B viral RNA.16 DNAzymes have also been used as highly sensitive and selective metal ion sensors.17 Divalent metal ions such as Mg2+, Mn2+, or Ca2+, are essential for the catalytic function of the majority of DNAzyme (or ribozyme) RNA endonucleases.18 RNA is often cleaved by attack of a metal-activated 2′ hydroxyl nucleophile on the siscile phosphate. Because DNA lacks a 2′ hydroxyl, it is much less susceptible to hydrolysis, and neither DNAzymes nor ribozymes have been shown to carry out efficient DNA cleavage in the presence of alkaline-earth-metal ions. Nevertheless, Carmi and Breaker have used in vitro selection to obtain Cu2+-dependent DNAzymes that can cleave DNA efficiently.19–21 The DNAzyme was minimized to a 46-mer, with conserved sequences defined. The metal-binding curve of this DNAzyme is Gaussian, and enzyme activity is maximized for a Cu2+ concentration of 30–60 μM.
Here, we seek to combine the structural and catalytic facets of DNA nanotechnology by integrating DNAzymes into 2D DNA arrays. To demonstrate the organization of a DNAzyme in a periodic array, it is convenient to use a molecule that is capable of self-cleavage in the presence of a cofactor. We use a modification of the Cu2+-dependent DNAzyme that undergoes auto-cleavage when Cu2+ is added to the solution. This cleavage event is readily visible in the atomic force microscope (AFM) so that both the presence of the DNAzyme and its cleavage in the context of an array are readily visible.
Figure 1 indicates the structures of both the reported19–21 46-nucleotide DNAzyme (a) and a bimolecular complex of 22-nucleotide and a 29-nucleotide strands (b) that self-cleave in the presence of Cu2+. The points of scission are indicated by arrows in both drawings. The portion of the molecule on the right can be regarded as a catalytic fragment and that on the left can be regarded as the substrate. We found that under our assay conditions, the cis-acting 46 nucleotide construct shown in Figure 1a had a relatively low extent of cleavage reaction (10–30%). We therefore sought to reengineer the ribozyme in order to achieve more complete cleavage. A number of constructs were tested in which the GAA loop was altered or removed, or in which the stem was lengthened. The construct with the greatest extent of cleavage (80% or greater) was the trans-acting variant shown in Figure 1b. Sample digestions are shown in the Supplementary Material. This deoxyribozyme was therefore chosen for incorporation into the 2D lattice.
Figure 1.
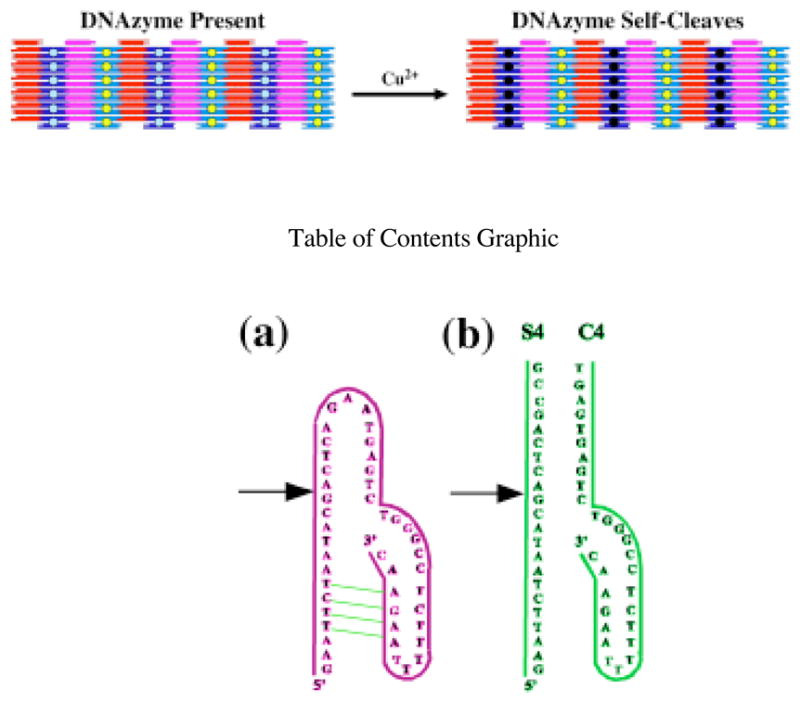
Schematic Drawings of Cu2+ Dependent DNAzymes. (a) This DNAzyme was the starting point for the studies reported here. The site of cleavage is indicated by the arrow. Watson-Crick complementarity is indicated by the green lines. A dodecamer sequence complementary to the B tile of the 2D array22 is attached to the 5′ end. In conditions where the 2D arrays are stable, cleavage was poor (<30%). A more successful DNAzyme (cleavage ~80%), shown in (b) was developed by trial and error.
We have previously described a system consisting of four different DX tiles that form a 2D lattice, and which is amenable to modification on its surface.22 We have shown that it is possible to add DNA hairpins to dodecamer sticky ends on one tile via hybridization, without ligating the sticky end, and to detect the presence of the sticky end by AFM. Hairpins that are inherently part of the array, or those that are added, appear as stripes in the image. In the same study, we demonstrated that it is possible to detect by AFM the removal of a hairpin by a restriction endonuclease, evidenced by the removal of the stripe. In this work, we have used the same approach, and have replicated the previous system to the greatest extent possible. A schematic of the 2D system is shown in Figure 2. We have added a 12-nucleotide sticky end to the 5′ end of the shorter molecule shown in Figure 1b, resulting in the addition of the DNAzyme to the lattice. The added DNAzyme is indicated schematically by the off-white filled circle on the B tile. We have modified the B tile from that used previously by removing a five-nucleotide double helix from the base of the sticky end; regardless of cleavage, the presence of the extra five-nucleotide double helix showed up as a striped feature in the AFM. In the previous work, we added material to sticky ends on both faces of the array, but here we have a fixed hairpin on the face of the tile opposite to the sticky end. The sequences of the various tiles are shown in the supplementary material.
Figure 2.
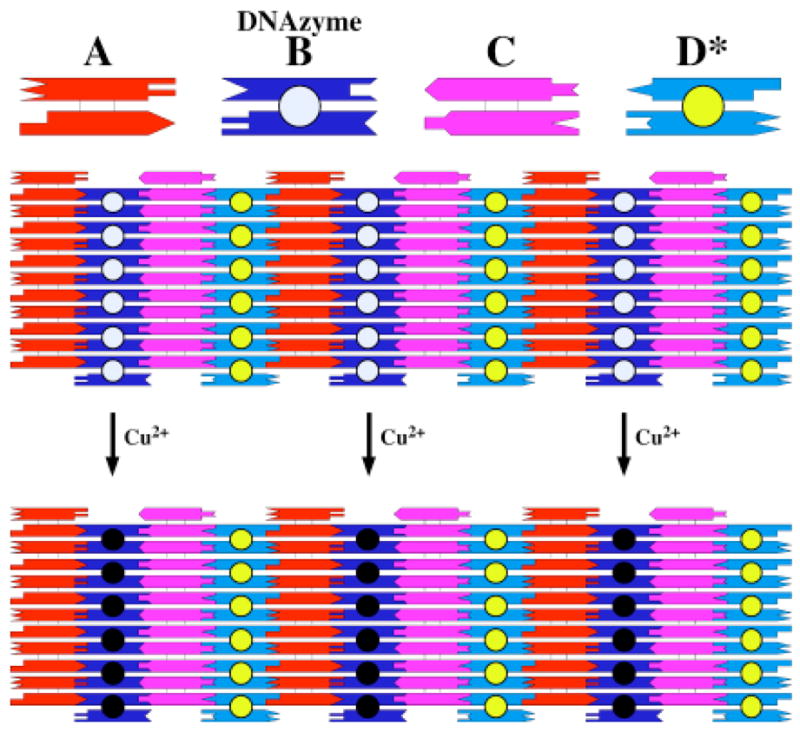
Schematic indicating the Nature of the Reaction. Four DX tiles are shown at the top, with their sticky ended complementarity represented geometrically. The B tile contains the intact Cu2+-dependent DNAzyme, which is represented by the off-white-filled circle. The D* tile contains a permanent hairpin represented by the yellow-filled circle. The four tiles have a horizontal dimension of about 16 nm each. The array below the four tiles shows that these tiles can fit together to form a 2D lattice, containing stripe-like features separated by 32 nm, owing to the protrusions on the B tile and on the D* tile. Treatment with Cu2+ leads to removal of the stripe-like feature that results from the presence of the intact DNAzyme. This is represented by the replacement o the off-white-filled circle with a black-filled circle. The spacing of the stripes is now about 64 nm.
As illustrated in Figure 2 the idea behind the experiment is that the addition of Cu2+ should result in the self-cleavage of the DNAzyme. This, in turn is intended to remove the stripe-like feature from the array, since the remaining interaction between the DNAzyme and the 5′ portion of the substrate is minimal and the former can therefore freely dissociate. In addition to the stripe that results from the presence of the DNAzyme, another stripe is present, owing to the presence of permanent hairpins on the D tiles. The length of each tile is 16 nm in the horizontal direction in Figure 2. Thus, in the presence of the DNAzyme, one expects an array with a repeating 32 nm striped feature, and after it has been removed, the length of the repeat should increase to 64 nm. Figure 3 illustrates that this occurs when the samples are treated with Cu2+. The stripes before the addition of Cu2+ are uniform, and their separation is about 31.5 nm. The image following treatment with Cu2+ is markedly different. Alternating rows are seen to be intense, and they are interspersed with faint rows. The separation of the intense rows from each other is about 61.5 nm, indicating that every other row has been removed by the autocatalytic cleavage of the DNAzyme.
Figure 3.

Atomic Force Micrographs of Two-Dimensional Arrays Containing the DNAzyme Before and After Treatment with Cu2+. The scale bars are 100 nm long. The image on the left has a stripe spacing of 31.5 nm, indicating the presence of the DNAzyme in every other row. The image on the right has a major stripe separated by about 61.5 nm. Close inspection of the image shows a smaller stripe halfway in between the major stripe, corresponding to nucleotides left after DNAzyme cleavage.
We have demonstrated that it is possible to organize DNAzymes in specific patterns using the self-assembly of DNA tiles that contain complementary sticky ends. Although a simple pattern has been used, more complex patterns are possible, and one can imagine using both algorithmic assembly23 and viral-based techniques24 to organize a variety of DNAzymes into patterns that are far more complex. In addition to organizing the DNAzyme into a specific pattern, we have demonstrated that it is functional when so arranged. Thus, one could take advantage of the diverse activities of different DNAzymes to create a variety of nanoscale patterns from a constant base, depending upon which ones were activated. Similarly, one could activate different DNAzymes in a programmed progression to create specific patterns over a particular time course, leading to a developmental series in nanotechnology, akin to the developmental progression seen in embryological development.
EXPERIMENTAL
DNA Cleavage Assays
DNA self-cleavage assays were conducted using trace amounts of radiolabeled precursor DNA (~100 pM) at 23 °C in buffer [50 mM HEPES (pH 7.0 at 23 °C), 0.5 M NaCl] containing 30–120 μM CuCl2, or in the presence of ascorbate. All solutions were prepared with deionized water without regard for dissolved oxygen content. Examinations of the DNA cleavage activity of unimolecular or bimolecular complexes were conducted under similar conditions using concentrations of substrate and catalyst DNA as indicated for each experiment. Either the substrate or the unimolecular component was 32P-labeled on the 5′ end for detection purposes. Cleavage products were separated by denaturing PAGE and imaged by Phosphorimager (Molecular Dynamics). Product yields were determined by quantitation of the corresponding precursor and product bands.
Formation of Hydrogen-Bonded Arrays
Complexes are formed by mixing a stoichiometric quantity of each strand, as estimated by OD260. Exact stoichiometry is determined, if necessary, by titrating pairs of strands designed to hydrogen bond together and visualizing them by non-denaturing gel electrophoresis; absence of monomer is taken to indicate the endpoint. All 21 strands are mixed in 50 mM HEPES (pH 7.0), 500 mM NaCl, 50 mM MgCl2, and 5 mM EDTA. The final concentration of DNA is 0.4 μM, and the final volume is 20 μL. The tube containing the DNA solution is put in about 2 L of boiled water and placed in a Styrofoam box for at least 40 h to facilitate hybridization.
Hydrogen Bonding of DNAzymes
Both DNAzymes, the unimolecular 46mer and bimolecular S4/C4contain a dodecamer sticky end complementary to the one on the B tile. The DNAzymes are added to a 20 μL aliquot of the annealed lattice. The final volume is 25 μL, the final DNA concentration of array components is 0.32 μM and the concentration of DNAzyme is 0.80 μM. The solution is incubated in about 2 L of water (37°C) and cooled slowly to room temperature. For samples that are to be visualized after DNAzyme activity, CuCl is added to the system to a final concentration of 60 μM and incubated overnight.
Supplementary Material
The sequences of the molecules used and sample digestions. This material is available free of charge via the Internet at http://pubs.acs.org/.
Acknowledgments
This research has been supported by grants GM-29554 from NIGMS, N00014-98-1-0093 from ONR, grants DMI-0210844, EIA-0086015, CCF-0432009, CCF-0523290 and CTS-0548774 from the NSF, 48681-EL from ARO and NTI-001 from Nanoscience Technologies, Inc to N.C.S., as well as grants to A.D.E. from the National Institutes of Health.
References
- 1.Seeman NC. Nature. 2003;421:427–431. doi: 10.1038/nature01406. [DOI] [PubMed] [Google Scholar]
- 2.Seeman NC. J Theor Biol. 1982;99:237–247. doi: 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- 3.Fu TJ, Seeman NC. Biochem. 1993;32:3211–3220. doi: 10.1021/bi00064a003. [DOI] [PubMed] [Google Scholar]
- 4.Ding B, Sha R, Seeman NC. J Am Chem Soc. 2004;126:10230–10231. doi: 10.1021/ja047486u. [DOI] [PubMed] [Google Scholar]
- 5.Mathieu F, Liao S, Mao C, Kopatsch J, Wang T, Seeman NC. NanoLett. 2005;5:661–665. doi: 10.1021/nl050084f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen SN, Chang ACY, Boyer HW, Helling RB. Proc Nat Acad Sci (USA) 1973;70:3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winfree E, Liu F, Wenzler LA, Seeman NC. Nature. 1998;394:539–544. doi: 10.1038/28998. [DOI] [PubMed] [Google Scholar]
- 8.Breaker RR, Joyce GF. Chem Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 9.Pyle AM, Chu VT, Jankowsky E, Boudvillain M. Methods Enzymol. 2000;317:140–146. doi: 10.1016/s0076-6879(00)17012-0. [DOI] [PubMed] [Google Scholar]
- 10.Breaker RR, Joyce GF. Chem Biol. 1995;2:655–660. doi: 10.1016/1074-5521(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 11.Li J, Zheng W, Kwon AH, Lu Y. Nucleic Acids Res. 2000;28:481–488. doi: 10.1093/nar/28.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuenoud B, Szostak JW. Nature. 1995;375:611–614. doi: 10.1038/375611a0. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Breaker RR. Methods. 2001;23:179–190. doi: 10.1006/meth.2000.1119. [DOI] [PubMed] [Google Scholar]
- 14.Santoro SW, Joyce GF. Proc Nat Acad Sci USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santoro SW, Joyce GF. Biochem. 1998;37:13330–13342. doi: 10.1021/bi9812221. [DOI] [PubMed] [Google Scholar]
- 16.Asahina Y, Ito Y, Wu CH, Wu GY. Hepatology. 1998;28:547–554. doi: 10.1002/hep.510280236. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Lu Y. J Am Chem Soc. 2000;122:10466–10467. [Google Scholar]
- 18.Pyle AM. Science. 1993;261:709–714. doi: 10.1126/science.7688142. [DOI] [PubMed] [Google Scholar]
- 19.Carmi N, Shultz LA, Breaker RR. Chem Biol. 1996;3:1039–1046. doi: 10.1016/s1074-5521(96)90170-2. [DOI] [PubMed] [Google Scholar]
- 20.Carmi N, Balkhi HR, Breaker RR. Proc Natl Acad Sci USA. 1998;95:2233–2237. doi: 10.1073/pnas.95.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carmi N, Breaker RR. Bioorg Med Chem. 2001;9:2589–2600. doi: 10.1016/s0968-0896(01)00035-9. [DOI] [PubMed] [Google Scholar]
- 22.Liu F, Sha R, Seeman NC. J Am Chem Soc. 1999;121:917–922. [Google Scholar]
- 23.Rothemund PWK, Papadakis N, Winfree E. PLOS Biology. 2004;2:2041–2053. doi: 10.1371/journal.pbio.0020424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rothemund PWK. Nature. 2006;440:297–302. doi: 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The sequences of the molecules used and sample digestions. This material is available free of charge via the Internet at http://pubs.acs.org/.