

Abstract
The enormous scope of natural human genetic variation is now becoming defined. To accurately annotate these variants, and to identify those with clinical importance, is often difficult to assess through functional assays. We explored systematic annotation by using homologous recombination to modify a native gene in hemizygous (wt/Δexon) human cancer cells, generating a novel syngeneic variance library (SyVaL). We created a SyVaL of BRCA2 variants: nondeleterious, proposed deleterious, deleterious, and of uncertain significance. We found that the null states BRCA2Δex11/Δex11 and BRCA2Δex11/Δ3308X were deleterious as assessed by a loss of RAD51 focus formation on genotoxic damage and by acquisition of toxic hypersensitivity to mitomycin C and etoposide, whereas BRCA2Δex11/Y3308Y, BRCA2Δex11/P3292L, and BRCA2Δex11/P3280H had wild-type function. A proposed phosphorylation site at codon 3291 affecting function was confirmed by substitution of an acidic residue (glutamate, BRCA2Δex11/S3291E) for the native serine, but in contrast to a prior report, phosphorylation was dispensable (alanine, BRCA2Δex11/S3291A) for BRCA2-governed cellular phenotypes. These results show that SyVaLs offer a means to comprehensively annotate gene function, facilitating numerical and unambiguous readouts. SyVaLs may be especially useful for genes in which functional assays using exogenous expression are toxic or otherwise unreliable. They also offer a stable, distributable cellular resource for further research.
Introduction
The scope of natural human genetic variation is now becoming evident but is enormous (1, 2). To accurately annotate these variants and to identify which of them have clinical importance through functional assays may be difficult for several reasons. Natural cell lines expressing the variants homozygously are often unavailable and do not allow isogenic experimental controls. Artificially introduced null states can be inviable. For some genes, such as the clinically important BRCA2 gene, even the exogenous expression of wild-type (wt) gene fragments can cause interference with endogenous gene function (squelching) unless assays are performed with extreme technical care and multiple controls (3–6).
The BRCA2 gene exhibits allelic differences in its sequence among members of the human population. Some of the variants are deleterious, causing inherited susceptibility to breast, ovarian, and other cancer types (7, 8). Unfortunately, in many patients who are tested and have a BRCA2 variant identified, the alteration is subtle, changing a single amino acid (9). For many of these variants, it is not possible to use epidemiologic data or laboratory studies of protein function to determine unambiguously nor efficiently whether one should classify the variant as either deleterious or benign.
The BRCA2 gene on chromosome 13q12-13 encodes a 3,418–amino acid protein. Its sequence reveals only a few clues to its cellular function and thus variation in the sequence is not readily interpreted. The role of the protein in DNA repair specifically involves homologous recombination, mediated by its interaction with RAD51 (10–12). BRCA2 interacts with RAD51 through highly conserved BRC repeats in exon 11 and an interaction domain mapped to exon 27 (12, 13). The interaction of RAD51 with exon 27 has been proposed to be down-regulated by cyclin-dependent kinase–dependent phosphorylation of S3291, which is reduced during S phase and after DNA damage (4). However, most functional studies of BRCA2 have been hampered by the lack of well-controlled human cancer cellular models (14). Such absence was in the past overcome by using methods of considerable technical difficulty (5).
To accurately annotate sequence variations of the BRCA2 gene in an unambiguous and technically facile model, we performed targeted disruption of BRCA2 exon 11 by homologous recombination, yielding the first available syngeneic human cancer BRCA2 knockout cell line. Using hemizygous BRCA2 cells, we constructed a library of syngeneic exon 27 genetic variant lines (a SyVaL). By functional evaluation of multiple clones harboring individual mutations, we were able to classify sequence variants as deleterious, hypomorphic, or neutral and to create a pseudophosphorylated BRCA2 variant replacing the normal gene. Our data support the importance of exon 27 in recombination-mediated DNA repair and the role of S3291 in phosphorylation-regulated BRCA2-RAD51 interaction.
Materials and Methods
Cell lines and cell culture
DLD1 cells were obtained from the American Type Culture Collection and cultured in conventional medium supplemented with 10% FCS, l-glutamine, and penicillin/streptomycin.
Targeted disruption by homologous recombination
We first disrupted exon 11 of BRCA2 by homologous recombination using the technique of Kohli and colleagues (15). The targeting construct was designed to excise a part of exon 11, containing a known mutation and including BRC repeat 6, thus causing a premature stop codon after BRC repeat 5. The construct, which consisted of a selection cassette flanked on both sides by loxP sites and ∼ 1 kb of genomic sequences adjoining the targeted part of exon 11, was ligated into pAAV (Stratagene). It was then contransfected with pRC and pHelper into HEK 293 cells using Lipofectamine (Invitrogen). Two days later, virus was harvested form HEK 293 cells and used for gene targeting of DLD1 cells. Infected cells were split into 96-well plates, and 3 wk later, hygromycin-resistant clones having homologous integration of the targeting construct were identified by PCR using a primer inside the resistance gene and a primer outside of the homology arms (primer pairs SF/SR and SF2/SR2; Fig. 1). The selection cassette was removed by transiently expressing exogenous Cre recombinase. A recombinant hemizygous clone, identified by PCR using primers flanking the deletion (primer pair OF/OR; Fig. 1), was used for second allele targeting. Reusing the same exon 11 targeting construct, clones acquiring biallelic disruption of BRCA2 were identified by PCR screening using a combination of primers described above.
Figure 1.
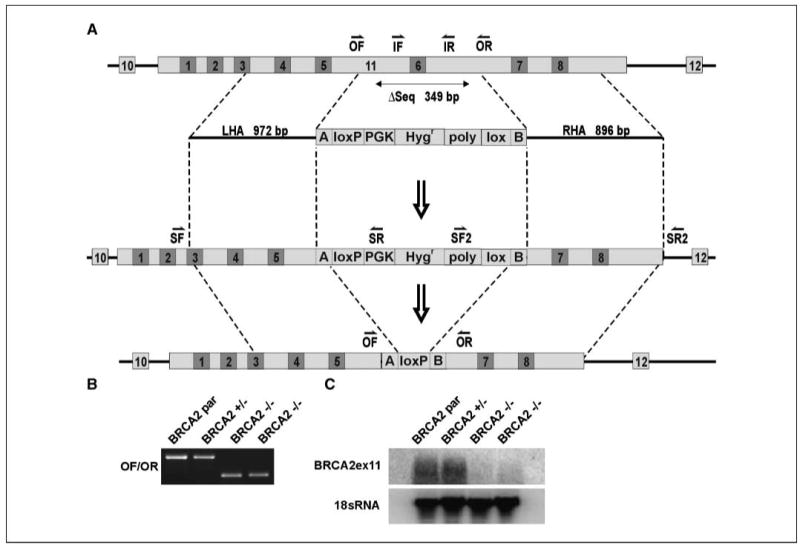
Structural and functional demonstration of BRCA2 gene disruption. A, targeting scheme. OF and OR, primers outside the deleted sequence; IF and IR, primers inside the deleted sequence; SF, SR, SF2, and SR2, screening primer pairs; LHA and RHA, left and right homology arms. Numbers on light background indicate exons, and numbers on dark background indicate BRC repeats. B, PCR detecting the wt and targeted alleles using genomic DNA as template. C, Northern blot detecting the targeted exon 11 mRNA transcript.
Alternately, exon 27 was targeted in the hemizygous cells using a construct whose right homology arm included intact exon 27 and individual sequence variants (Fig. 4). Homologous integration of the construct, the presence of desired sequence variants, and allelic phase of the construct (see below) were determined by allele-specific PCR and sequencing. An analogous vector design was used by Hurley and colleagues (16) to introduce an ATR mutation into alleles of a human cell and by Konishi and colleagues (17) to convert a wt KRAS gene to a mutant allele in a nontumorigenic human cell line.
Figure 4.
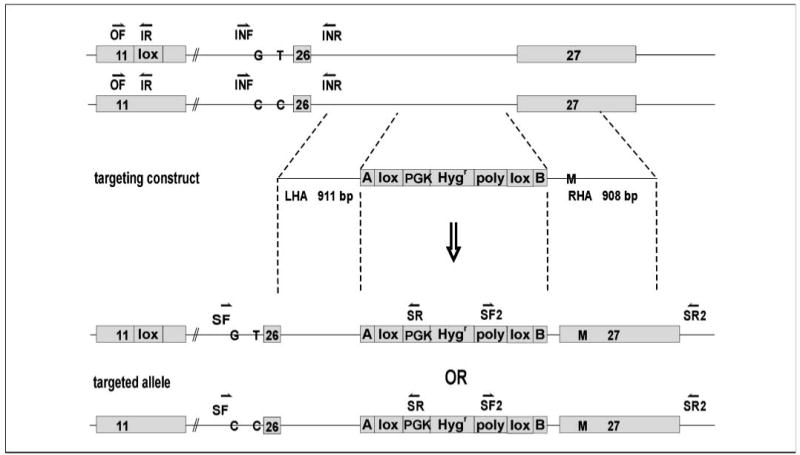
Structural demonstration of BRCA2 exon 27 targeting. Targeting scheme. OF, primer outside the deleted sequence; IR, primer inside the deleted sequence; SF, SR, SF2, and SR2, screening primer pairs; INF and INR, intron 25 spanning primers; LHA and RHA, left and right homology arms.
Distinguishing homologous recombination events affecting the active or inactive allele (“phase” determination)
The hemizygous BRCA2wt/Δex11 cells, having one active and one inactive allele, were used for subsequent experiments (Fig. 4). In preparation to target exon 27, residing at a large genomic distance from exon 11, we devised a means to readily distinguish long-distance cis relations of each allele. We first performed automated sequencing of the heterozygous cell line to identify natural, preexisting heterozygous nucleotide polymorphisms within intron 25. We next distinguished whether the intronic polymorphisms were on the active or inactive allele (termed the phase) by determining whether they were in cis or in trans to the deleted exon using limiting dilution of the template DNA and digital duplex PCR (18). Primers were specific for the exon 11 deletion sequence (primer pair OF/IR; Fig. 4) or flanked the identified polymorphisms (primer pair INF/INR; Fig. 4). Sequencing was performed to identify the allele amplified in each well.
Subsequently, after exon 27 targeting and clone isolation, the presence and the phase of the introduced exon 27 variant sequence were in turn assessed in relation to the known phase of the natural intron 25 polymorphic site by sequencing a PCR product incorporating both sites (primer pair SF, upstream of the intron 25 polymorphism, and SR, specific for the targeting construct; Fig. 4).
DNA and RNA isolation, PCR, and sequencing
Genomic DNA was isolated from cells using Lyse-N-Go PCR Reagent (Pierce Biotechnology). RNA was isolated using RNeasy Mini kit (Qiagen). Automated sequencing was performed by a core facility. PCR conditions and primer sequences are available on request.
Allele-specific PCR
The presence of the targeting construct and its subsequent Cre-mediated removal enabled selective amplification from one of two alleles using primers specific for either the construct or the deleted sequence.
Digital PCR
Digital PCR was used to readily distinguish the active and inactive alleles at a distance from exon 11 (in intron 25) in the hemizygous BRCA2wt/Δex11 cells. It was performed essentially as described previously (18). In a duplex reaction, primers specific for deletion-specific sequences at exon 11 and spanning intron 25 were combined (Fig. 4).
Site-directed mutagenesis
Mutated targeting constructs were generated using the QuikChange Site-Directed Mutagenesis kit (Stratagene) using the manufacturer's protocol.
Northern blotting
To confirm absence of exon 11 transcript, Northern blotting was performed. Random primer-labeled cDNA was used to detect BRCA2 exon 11 mRNA, and end-labeled oligomers were used to detect 18S rRNA (loading control; ref. 19).
Cell cycle analysis
Cells were treated with mitomycin C (MMC) for 48 h, processed as described previously (20), and analyzed by flow cytometry.
Chromosome aberration assay
Cells were treated with equitoxic doses of MMC (80 nmol/L for BRCA2wt/Δex11 and 6 nmol/L for BRCA2Δex11/Δex11) for 24 h. For each sample, 50 metaphases were analyzed by the cytogenetics core facility at Johns Hopkins according to standard protocols.
Cell proliferation assay
In each well of 96-well plates, 1,000 cells were plated, allowed to adhere, and treated with various drugs. The drugs were obtained from Sigma except for SJG-136 (NSC 694501), which was generously provided by Ipsen, Inc. and the National Cancer Institute, NIH. The cells were washed after 6 d and lysed in 100 μL of water. Fluorescence was measured after addition of 0.5% PicoGreen (Molecular Probes) using a fluorometer (Fusion, Perkin-Elmer). Three independent experiments were performed per drug.
Colony formation assay
Cells were plated in six-well plates, allowed to adhere, and exposed to 137Cs γ-rays. After 14 d, cells were washed and stained with crystal violet and colonies were counted. For each dose, cells were plated at three different concentrations in duplicate. Three independent experiments were performed.
Immunofluorescence
Cells were treated with MMC (2.4 μg/mL) or γ-irradiation and fixed in PBS/2% paraformaldehyde after 24 h of incubation. Following permeabilization in PBS/0.5% Triton X-100 and blocking for 30 min in PBS/1% bovine serum albumin/0.15% glycine, the cells were incubated with an anti-RAD51 antibody (1:200; Calbiochem) for 2 h. Cells were then washed and incubated with a rabbit IgG secondary antibody (1:200, Alexa Fluor 488; Molecular Probes) for 1 h. After washing, slides were mounted and analyzed by a fluorescent microscope (Zeiss Axiovert 135) and MetaMorph 4.6 software (Universal Imaging). Pictures were acquired, keeping exposure time and software settings constant for all samples.
Baseline or induced RAD51 focus formation was considered “negative” when less than 5% or 20%, respectively, of cells had more than five nuclear foci and “positive” when more than 5% or 30%, respectively, of cells had more than five nuclear foci. Intermediate phenotypes were not observed.
Results
Targeted disruption of BRCA2
We first attempted to disrupt the remaining wt allele in a BRCA2 heterozygous, p53 wt cell line RKO (BRCA2wt/5355delA), but we could not obtain viable BRCA2-null clones (21). We then tried the diploid p53-deficient cancer cell line, DLD1. DLD1 originally harbored two nonsense mutations of BRCA2 (22). Both mutations are present on one allele (data not shown). In the first round of BRCA2 targeting, we deleted exon 11 of this allele, ensuring a permanent null state without a possibility of the reversion of the mutations (23). The other allele in the hemizygous clone remained wt. In the second round of BRCA2 targeting, 81 viable clones having homologous integration of the targeting construct were obtained in several independent targeting attempts, of which only one clone had integrated the construct in the wt allele, producing a homozygous BRCA2Δex11/Δex11 knockout. The other 80 clones had reintegrated the construct into the already-knocked-out allele.
PCR and Northern blot analyses confirmed the heterozygous and homozygous gene disruption (Fig. 1). Northern blot revealed the expression of exon 11 transcript in the parental and heterozygous populations. There was no detectable transcript in the homozygous cells.
Functional analysis of BRCA2Δex11/Δex11 cells
Both engineered and natural cells harboring truncated BRCA2 exhibit a proliferative impediment that may worsen with successive passages (24, 25). As expected, we also confirmed the proliferation rate in our BRCA2 Δex11/Δex11 cells usually to be slower than syngeneic controls using daily direct cell counting and confirmation by assaying DNA content of the culture (data not shown). For example, in three replicate cultures of each cell type (parental, heterozygote, and biallelic knockout) counted on each day of day 0 to day 5, the following calculated doubling times for each 5-day period were obtained: for parental BRCA2 wt/mut × 2, 0.64, 1.06, and 1.07 days; for heterozygous knockout BRCA2 wt/Δex11, 0.59, 0.83, and 1.6 days; and for biallelic knockout BRCA2 Δex11/Δex11, 1.61, 1.22, and 1.58 days. This modest proliferation defect seemed stable with successive passage in culture.
RAD51 focus formation
To investigate whether the BRCA2Δex11/Δex11 cells were defective in homologous recombination, we examined their ability to form RAD51 foci on DNA damage induction. Treatment with MMC induced RAD51 foci (more than five foci) in about 60% to 80% of BRCA2-proficient cells and, with irradiation, in about 50% to 60% of cells. BRCA2Δex11/Δex11 cells had reduced levels of foci in untreated cells, and no induction of foci was observed on DNA damage (Fig. 2A).
Figure 2.
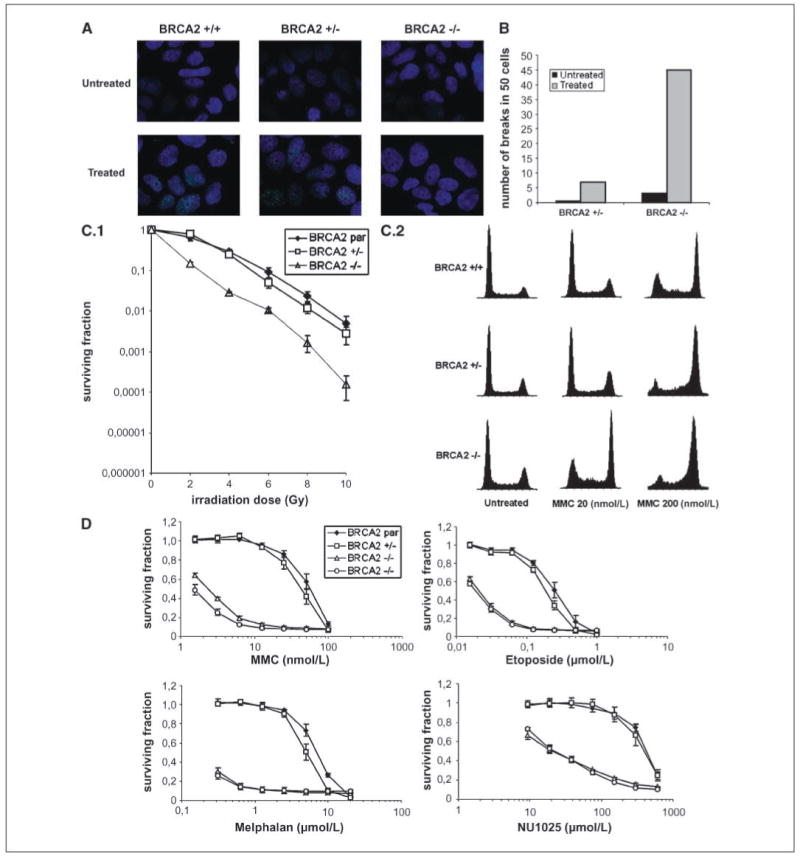
Characterization of BRCA2-null cells. A, immunofluorescence of RAD51 nuclear foci in untreated and treated cells (2.4 μg/mL MMC). BRCA2 +/−, BRCA2wt/Δex11; BRCA2−/−, BRCA2Δex11/Δex11. B, analysis of 50 metaphases for chromosomal aberrations in untreated and treated (equitoxic doses of MMC) heterozygous and knockout clones. C.1, colony formation assays on irradiation. BRCA2 par, parental BRCA2wt/mut × 2. Points, mean of three independent experiments; bars, SE. C.2, cell cycle analysis 48 h after MMC treatment. Representative cell cycle profiles obtained after treatment with indicated concentrations of MMC. D, cell proliferation following treatment with selected drugs. Two BRCA2 −/− (BRCA2Δex11/Δex11) subclones were analyzed. Points, mean of three independent experiments; bars, SE.
Chromosomal instability
BRCA2-deficient cells display increased chromosomal instability, which is further enhanced after treatment with MMC (26). Treatment-induced breakage serves as a diagnostic test for the Fanconi anemia (FA) syndrome, including the FA-D1/BRCA2 group (27). BRCA2Δex11/Δex11 displayed much higher rates of chromosomal aberrations than BRCA2wt/Δex11 cells, including breaks and radials, on MMC treatment at equitoxic doses (Fig. 2B).
Cell survival after irradiation
There has been conflicting data about the sensitivity of BRCA2-deficient cells to irradiation (24, 26). Therefore, we assessed the effect of BRCA2 disruption on survival after treatment with X-rays using colony formation assays. We observed a 10-fold decrease in relative survival of BRCA2Δex11/Δex11 cells compared with their controls (Fig. 2C.1).
Cell proliferation on drug treatment
The role of BRCA2 in DNA repair and replication fork maintenance is indicated by the hypersensitivity of BRCA2-defective cells to DNA-damaging agents. We assayed survival following exposure to several clinically relevant drugs in parental cells and heterozygous and homozygously deleted cells. BRCA2-deficient cells exhibited greatly increased sensitivity to various interstrand cross-linking (ICL) agents, including MMC (20-fold), melphalan (25-fold), cisplatin (16-fold), carboplatin (18-fold), oxaliplatin (10-fold), and SJG-136 (NSC 694501; 22-fold; Figs. 2D and 3; data not shown).
Figure 3.
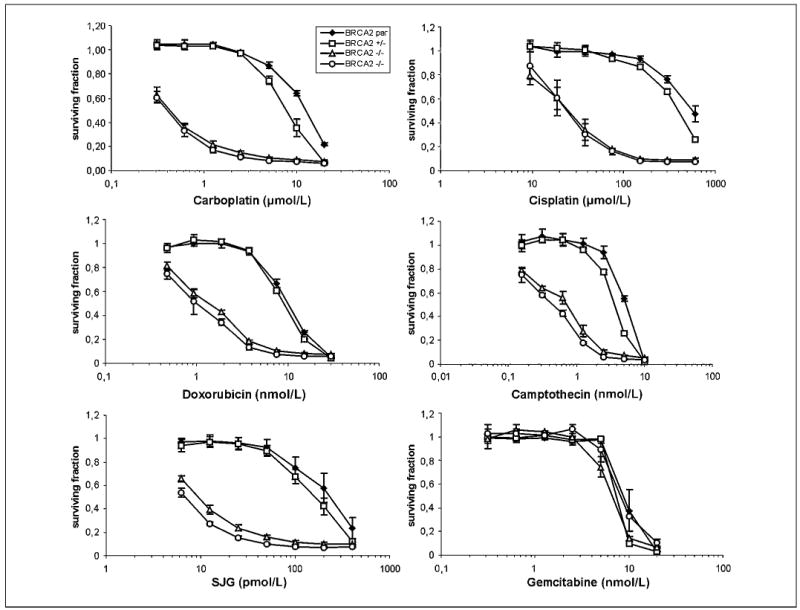
Drug sensitivity studies. Cell proliferation on treatment with various drugs at the indicated concentrations compared with untreated cells. Points, mean of three independent experiments; bars, SE.
Topoisomerase inhibitors affect enzymes incising either one (topoisomerase I inhibitors) or two (topoisomerase II inhibitors) strands of DNA in the process of DNA replication. Our BRCA2-deficient cells displayed increased sensitivity to topoisomerase II inhibitors etoposide (10-fold) and doxorubicin (10-fold) and to a smaller extent to topoisomerase I inhibitor camptothecin (6-fold; Figs. 2D and 3). Etoposide hypersensitivity was additive to, but not synergistic with, other anticancer agents (data not shown; ref. 28).
Hypersensitivity of BRCA2-deficient cells to poly(ADP-ribose) polymerase (PARP) inhibitors has been reported (29, 30). Our cells having an acute disruption of BRCA2 were more sensitive (20-fold) to NU1025 than control cells (Fig. 2D).
We observed no differences in sensitivities to gemcitabine and vinblastine as well as to agents that have various DNA-damaging potential, such as norethindrone (31), apigenin, curcumin, and hesperidin (data not shown).
The increased genotype-dependent toxicity to MMC was confirmed by flow cytometry, indicating a profound G2-M arrest of the cell cycle (Fig. 2C.2).
Introducing sequence variants into exon 27
The heterozygous clone used for exon 27 targeting was verified to have intact BRCA2 function by showing an absence of MMC hypersensitivity and the formation of RAD51 foci on DNA damage (Fig. 2A and D). A series of mutations was introduced by site mutagenesis into a targeting construct whose original right homology arm included wt exon 27 (Table 1; Fig. 4). As a positive control, a known deleterious nonsense mutation (10152C→G, Y3308X) was used. As a negative control, a synonymous mutation was introduced in the same codon (10152C→T, Y3308Y). Two naturally occurring missense human sequence variants (10067C→A, P3280H; 10103C→T, P3292L) reported from several cancer families and listed in the BRCA2 mutation database4 as variants of uncertain significance were examined (Table 1).
Table 1.
Characterization and proposed classification of BRCA2 SyVaL clones
| Clone | RAD51 | MMC | Etoposide | Chromosomal instability | Classification |
|---|---|---|---|---|---|
| BRCA2wt/Δex11 | Pos (16.7 ± 1.5/59.0 ± 0.7) | No Δ | No Δ | Neg | Neutral |
| BRCA2Δex11/Δex11 | Neg (2.5 ± 0.8/3.3 ± 2.8) | 20-fold Δ | 10-fold | Pos | Deleterious* |
| BRCA2Δex11/Y3308X | Neg (1.8 ± 0.5/1.6 ± 0.3) | 20-fold Δ | 10-fold | – | Deleterious |
| BRCA2Δex11/Y3308Y | Pos (12.4 ± 0.6/34.1 ± 1.1) | No Δ | No Δ | – | Neutral |
| BRCA2Δex11/P3292L | Pos (13.6 ± 2.4/41.6 ± 4.7) | No Δ | No Δ | – | Neutral |
| BRCA2Δex11/P3280H | Pos (12.2 ± 1.3/41.7 ± 4.5) | No Δ | No Δ | – | Neutral |
| BRCA2Δex11/S3291E | Neg (3.6 ± 0.6/12.4 ± 0.9) | 3-fold Δ | 3-fold Δ | – | Hypomorphic |
| BRCA2Δex11/S3291A | Pos (23.8 ± 4.1/47.6 ± 9.7) | No Δ | No Δ | – | Neutral |
NOTE: Numbers in parentheses represent mean percentage of untreated/treated cells with more than five RAD51 foci ± SE. Fold Δ refers to the ratio of the IC50 of cells having the hemizygous wt allele to the IC50 of cells having the mutated allele.
Abbreviations: Pos, positive; Neg, negative; —, not done; Δ, change.
The extreme rarity of knockout clones may indicate an especially severe deleterious effect (21).
The significance of the recently reported phosphorylation site in exon 27, S3291, was investigated by introducing a glutamate (mimicking constitutive phosphorylation of the protein) and alanine (to prevent phosphorylation) at codon 3291.
On average for each mutation, 20 clones having homologously integrated the targeting construct were identified by PCR; between 30% and 80% of these clones harbored the introduced mutation. The phase of the construct was subsequently determined in these clones by PCR and sequencing (data not shown). Clones having integrated the mutated construct into the inactive allele, as well as clones having integrated the targeting construct at an unrelated genomic site, were used as controls in the functional assays.
Functional evaluation of SyVaL clones
The robust and reproducible differences between wt BRCA2wt/mut × 2 and BRCA2Δex11/Δex11 cells in drug treatment assays and in RAD51 focus formation assays (Table 1; Fig. 2) suggested that these tests were well suited to evaluate the effect of introduced mutations on BRCA2 function.
MMC and etoposide sensitivity in the SyVaL
BRCA2Δex11/Y3308X had profound sensitivity to MMC and etoposide (20- and 10-fold, respectively), whereas BRCA2Δex11/Y3308Y did not differ from control cells. BRCA2Δex11/P3280H and BRCA2Δex11/P3292L were not significantly more sensitive than the corresponding controls. BRCA2Δex11/S3291E, but not BRCA2Δex11/S3291A, cells were 3-fold more sensitive than controls (Table 1).
RAD51 focus formation in the SyVaL
RAD51 focus formation was drastically diminished in untreated and treated BRCA2Δex11/Y3308X (a negative assay result) but robust in BRCA2Δex11/Y3308Y cells (a positive, normal result). No significant reduction in focus formation was observed in BRCA2Δex11/P3280H and BRCA2Δex11/P3292L (each, positive). A significant decrease in focus formation on MMC treatment was seen in BRCA2Δex11/S3291E (negative) but not BRCA2Δex11/S3291A cells (positive; Table 1).
Discussion
We sought to develop a model in which human gene function could be evaluated based on comparative assessments of the functional effect of individual in situ sequence alterations of one allele. We believe that this has not been previously done. We constructed a syngeneic stable library of sequence variants of BRCA2 exon 27. This enabled us to functionally classify BRCA2 genomic sequence variants of previously unknown significance in a robust manner that may help to predict the cancer risk associated with presence of the same mutations in heterozygous patients. Furthermore, we interrogated the protein based on functional evaluation of subtle protein residue substitutions.
To show the utility of SyVaLs, we chose BRCA2 as our initial target gene. First, as a positive control for functional evaluation of our SyVaL clones, we generated BRCA2-null cells. To date, no BRCA2 engineered human gene knockout cells have been reported. BRCA2 knockout models exist in mouse, rat, hamster, and chicken cells. The pancreatic cancer cell line CAPAN1, the only BRCA2-deficient human cancer cell line (7), is cumbersome for its low transfectability, poor clonogenicity, and lack of isogenic controls (14). Our BRCA2Δex11/Δex11 cells now provide a stable human cancer syngeneic BRCA2 knockout model.
Our knockout cells had features of BRCA2 deficiency, being defective in homologous recombination as assessed by absence of induction of RAD51 foci on DNA damage, having chromosomal instability after treatment with MMC, and being modestly more sensitive to irradiation.
To further confirm the BRCA2 phenotype and explore potential treatment options for patients having BRCA2-deficient tumors, we tested an informative panel of drugs in our cells. We confirmed increased sensitivity to various ICL agents and the PARP inhibitor NU1025, producing pharmacogenetic windows between 10 and 25 (28).
In this well-controlled human cancer model, we confirmed the etoposide hypersensitivity of BRCA2-deficient cells, first reported by Wiegant and colleagues (23) and Abbott and colleagues (32) in CAPAN1 cells and syngeneic hamster cells, and showed that the difference in sensitivity may be even greater (10-fold) than recently reported by Treszezamsky (2.75-fold; ref. 33) in fibroblasts from a Fanconi D1 patient. In yeast, the requirement of homologous recombination for reducing topoisomerase-associated toxicity was shown by Flores and colleagues (34). Not all models have been in agreement. For example, no increased etoposide sensitivity was observed in BRCA2-deficient mouse thymocytes (35) or small intestine (36). The findings thus distinguish cells having defects of the distal (BRCA2, etoposide hypersensitive) and proximal (no etoposide hypersensitivity) FA pathway (37). We extended our experiments to additional topoisomerase inhibitors. Given the rather low side effect profile of some of these drugs and our measured pharmacogenetic window of up to 10 (28), we concur that they represent candidate agents for rational treatment of patients with BRCA2-deficient tumors.
The BRCA2Δex11/Δex11 cells together with various SyVaL clones thus also represent an optimal model suitable for high-throughput drug screening of compound libraries to identify novel agents having selective toxicity in cells containing pathogenic mutations. We have not seen any intermediate phenotype of our heterozygous clones, suggesting that use of these drugs may be permissible in cancer patients who are carriers of heterozygous germ-line BRCA2 mutations.
The lack of understanding of the functional importance and associated cancer risk of sequence variants in the BRCA2 gene represents a serious clinical problem. The strongest interpretive evidence tends to come from segregation analyses and observed co-occurrence with known pathogenic mutations, both of which require information that is often not available (9). Functional studies of human BRCA2 have proven very difficult. Prior cell-based in vitro assays depended on expression of either partial or complete wt or mutant proteins. Testing of partial gene and protein sequences may not fully reveal the influence of the variant on the activity of the intact protein, and findings obtained by their use were not always confirmed using independent types of assay (38). The use of full-length wt or mutant constructs has been mostly limited to transient expression because it has proven difficult and at times impossible to generate stably expressing lines (4). Expression of exogenous BRCA2, including the BRC repeats, may result in BRCA2 deficiency, perhaps due to squelching (titrating out) of free RAD51 (3, 4, 6). Studies using cells having endogenous wt BRCA2 alleles depend on the presumption that the codominant or dominant-negative effects of exogenous mutated constructs will govern the assay results. Thus, evaluating BRCA2 function using exogenous expression constructs is of extreme technical difficulty.
We present here a new approach to evaluate genomic variants by applying a simple and fast homologous recombination replacement technique to produce syngeneic clones. This syngeneic replacement strategy was approximately as rapid as the stable transfection of routine expression plasmids, a technique with which it shares many invariant steps. Such approach allows an unambiguous readout by generating multiple clones of stable cell lines having the endogenous gene altered.
We applied a panel of functional assays to our syngeneic knockout cell pair and selected two robust assays that in combination provided a clear and unambiguous readout of BRCA2 function that we applied to our library of variants: RAD51 focus formation and MMC sensitivity. By applying these two assays, we observed our positive control to have a detrimental phenotype, our negative control to have a neutral phenotype (same as proficient cells), and the two variants of uncertain significance to have a neutral phenotype.
The high ratio, relating the number of clones having integrated a construct into the already targeted allele and the clones having integrated a construct in the wt allele, observed for the BRCA2Δex11/Δex11 cells (80:1) was not observed for any of the exon 27 variants. We believe that the ratio is influenced by selection against emergence of clones with a detrimental phenotype (21). In addition, the ratio may be potentially influenced by differences in the targeting construct as well as sequence variation or regional differences of chromatin at the targeted locus. This interesting aspect of the technique deserves further investigation.
SyVaLs can also be used to test the function of individual in situ residues of a protein or regulatory code, unrestricted by the limited number of known natural variants. For such a purpose, we selected the 3291 serine residue recently evaluated for its phosphorylation. Cyclin-dependent phosphorylation of this site was shown to inhibit both the interaction of BRCA2 with RAD51 and with recombination repair. This site was discovered and initially evaluated using expression of partial protein sequences (4). By mimicking constitutive phosphorylation by substituting serine with glutamate, we observed a hypomorphic phenotype characterized by a decrease in induced RAD51 focus formation and a slight increase in sensitivity to MMC. When we replaced serine with alanine to prevent phosphorylation, neither a decrease in RAD51 focus formation nor increased sensitivity to MMC was observed.
Our findings of inhibition of RAD51 focus formation by the glutamate, and no effect of the alanine, do support the hypothesis of phosphorylation-dependent inhibition of RAD51 interaction. Yet, the use of variant endogenous BRCA2 genes provided a nuanced relation that was not previously determinable. We found that this inhibition is partial, not complete. In addition, where the prior authors saw a surprising inhibitory effect on RAD51 binding also of alanine 3291 when partial peptides were tested, our studies suggest differently. We find that phosphorylation of S3291 is dispensable for BRCA2-governed cellular phenotype as judged by the intact function of the alanine 3291 variant, as assayed by a set of robust tests used here. Our syngeneic lines thus serve as a starting point in addressing the teleologic purpose, if any, of cell cycle regulation of the RAD51-BRCA2 interaction.
In conclusion, we propose the creation of SyVaLs as a technically facile, ethically acceptable, means to compare the effects of subtly altering human genomes. It could be applied to any other clinically relevant genes, thus permitting high-throughput annotation of natural human genetic variation. SyVaLs also enable the interrogation of individual codons and regulatory codes in human chromosomes while avoiding many common artifacts of introducing exogenous genes and gene fragments.
Acknowledgments
Grant support: National Cancer Institute grant CA62924, NIH Specialized Program of Research Excellence in Breast Cancer grant CA88843, and Mary Stewart Trust.
Footnotes
Disclosure of Potential Conflicts of Interest: The Johns Hopkins University School of Medicine has property rights in the materials (cell lines) reported in this article. T. Hucl and S.E. Kern are entitled as inventors to a share of the royalty received by the University for licensed uses of the materials by commercial entities. The terms of licensing agreement(s) are being managed by the Johns Hopkins University School of Medicine Committee on Conflict of Interest in accordance with its conflict of interest policy. (Licensing agreements covering the materials in this article total and inventors' interest of less than $10,000 in total.) The other authors disclosed no potential conflicts of interest.
References
- 1.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 2.Greenman C, Wooster R, Futreal PA, Stratton MR, Easton DF. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics. 2006;173:2187–98. doi: 10.1534/genetics.105.044677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies AA, Masson JY, McIlwraith MJ, et al. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7:273–82. doi: 10.1016/s1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- 4.Esashi F, Christ N, Gannon J, et al. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- 5.Wu K, Hinson SR, Ohashi A, et al. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005;65:417–26. [PubMed] [Google Scholar]
- 6.Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59:3547–51. [PubMed] [Google Scholar]
- 7.Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–4. [PubMed] [Google Scholar]
- 8.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–6. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 9.Frank TS, Deffenbaugh AM, Reid JE, et al. Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol. 2002;20:1480–90. doi: 10.1200/JCO.2002.20.6.1480. [DOI] [PubMed] [Google Scholar]
- 10.Connor F, Bertwistle D, Mee PJ, et al. Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat Genet. 1997;17:423–30. doi: 10.1038/ng1297-423. [DOI] [PubMed] [Google Scholar]
- 11.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 12.Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997;272:31941–4. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- 13.Sharan SK, Morimatsu M, Albrecht U, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–10. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- 14.Gallmeier E, Kern SE. Targeting Fanconi anemia/BRCA2 pathway defects in cancer: the significance of preclinical pharmacogenomic models. Clin Cancer Res. 2007;13:4–10. doi: 10.1158/1078-0432.CCR-06-1637. [DOI] [PubMed] [Google Scholar]
- 15.Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hurley PJ, Wilsker D, Bunz F. Human cancer cells require ATR for cell cycle progression following exposure to ionizing radiation. Oncogene. 2007;26:2535–42. doi: 10.1038/sj.onc.1210049. [DOI] [PubMed] [Google Scholar]
- 17.Konishi H, Karakas B, Abukhdeir AM, et al. Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras mediated transformation. Cancer Res. 2007;67:8460–7. doi: 10.1158/0008-5472.CAN-07-0108. [DOI] [PubMed] [Google Scholar]
- 18.Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci U S A. 1999;96:9236–41. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23:3092–102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vindelov LL, Christensen IJ, Nissen NI. A detergent-trypsin method for the preparation of nuclei for flow cytometric DNA analysis. Cytometry. 1983;3:323–7. doi: 10.1002/cyto.990030503. [DOI] [PubMed] [Google Scholar]
- 21.Gallmeier E, Hucl T, Calhoun ES, et al. Gene-specific selection against experimental Fanconi anemia gene inactivation in human cancer. Cancer Biol Ther. 2007;6:654–60. doi: 10.4161/cbt.6.5.3978. [DOI] [PubMed] [Google Scholar]
- 22.Ikediobi ON, Davies H, Bignell G, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–12. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiegant WW, Overmeer RM, Godthelp BC, van Buul PP, Zdzienicka MZ. Chinese hamster cell mutant, V-C8, a model for analysis of Brca2 function. Mutat Res. 2006;600:79–88. doi: 10.1016/j.mrfmmm.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Godthelp BC, van Buul PP, Jaspers NG, et al. Cellular characterization of cells from the Fanconi anemia complementation group, FA-D1/BRCA2. Mutat Res. 2006;601:191–201. doi: 10.1016/j.mrfmmm.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Patel KJ, Yu VP, Lee H, et al. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1:347–57. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- 26.Kraakman-van der Zwet M, Overkamp WJ, van Lange RE, et al. Brca2 (XRCC11) deficiency results in radio-resistant DNA synthesis and a higher frequency of spontaneous deletions. Mol Cell Biol. 2002;22:669–79. doi: 10.1128/MCB.22.2.669-679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auerbach AD. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp Hematol. 1993;21:731–3. [PubMed] [Google Scholar]
- 28.Hucl T, Gallmeier E, Kern SE. Distinguishing rational from irrational applications of pharmacogenetic synergies from the bench to clinical trials. Cell Cycle. 2007;6:1336–41. doi: 10.4161/cc.6.11.4359. [DOI] [PubMed] [Google Scholar]
- 29.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 30.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 31.Gallmeier E, Winter JM, Cunningham SC, Kahn SR, Kern SE. Novel genotoxicity assays identify norethindrone to activate p53 and phosphorylate H2AX. Carcinogenesis. 2005;26:1811–20. doi: 10.1093/carcin/bgi132. [DOI] [PubMed] [Google Scholar]
- 32.Abbott DW, Freeman ML, Holt JT. Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst. 1998;90:978–85. doi: 10.1093/jnci/90.13.978. [DOI] [PubMed] [Google Scholar]
- 33.Treszezamsky AD, Kachnic LA, Feng Z, Zhang J, Tokadjian C, Powell SN. BRCA1- and BRCA2-deficient cells are sensitive to etoposide-induced DNA double-strand breaks via topoisomerase II. Cancer Res. 2007;67:7078–81. doi: 10.1158/0008-5472.CAN-07-0601. [DOI] [PubMed] [Google Scholar]
- 34.Flores KG, McAllister KA, Greer PK, Wiseman RW, Hale LP. Thymic model for examining BRCA2 expression and function. Mol Carcinog. 2002;35:103–9. doi: 10.1002/mc.10081. [DOI] [PubMed] [Google Scholar]
- 35.Hay T, Patrick T, Winton D, Sansom OJ, Clarke AR. Brca2 deficiency in the murine small intestine sensitizes to p53-dependent apoptosis and leads to the spontaneous deletion of stem cells. Oncogene. 2005;24:3842–6. doi: 10.1038/sj.onc.1208533. [DOI] [PubMed] [Google Scholar]
- 36.Walker JV, Nitiss KC, Jensen LH, et al. A mutation in human topoisomerase IIα whose expression is lethal in DNA repair-deficient yeast cells. J Biol Chem. 2004;279:25947–54. doi: 10.1074/jbc.M312314200. [DOI] [PubMed] [Google Scholar]
- 37.Gallmeier E, Calhoun ES, Rago C, et al. Targeted disruption of FANCC and FANCG in human cancer provides a preclinical model for specific therapeutic options. Gastroenterology. 2006;130:2145–54. doi: 10.1053/j.gastro.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–44. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]