

Abstract
Glucagon counterregulation (GCR) is a key protection against hypoglycemia that is compromised in diabetes via an unknown mechanism. To test the hypothesis that α-cell-inhibiting signals that are switched off during hypoglycemia amplify GCR, we studied streptozotocin (STZ)-treated male Wistar rats and estimated the effect on GCR of intrapancreatic infusion and termination during hypoglycemia of saline, insulin, and somatostatin. Times 10 min before and 45 min after the switch-off were analyzed. Insulin and somatostatin, but not saline, switch-off significantly increased the glucagon levels (P = 0.03), and the fold increases relative to baseline were significantly higher (P < 0.05) in the insulin and somatostatin groups vs. the saline group. The peak concentrations were also higher in the insulin (368 pg/ml) and somatostatin (228 pg/ml) groups vs. the saline (114 pg/ml) group (P < 0.05). GCR was pulsatile in most animals, indicating a feedback regulation. After the switch-off, the number of secretory events and the total pulsatile production were lower in the saline group vs. the insulin and somatostatin groups (P < 0.05), indicating enhancement of glucagon pulsatile activity by insulin and somatostatin compared with saline. Network modeling analysis demonstrates that reciprocal interactions between α- and δ-cells can explain the amplification by interpreting the GCR as a rebound response to the switch-off. The model justifies experimental designs to further study the intrapancreatic network in relation to the switch-off phenomenon. The results of this proof-of-concept interdisciplinary study support the hypothesis that GCR develops as a rebound pulsatile response of the intrapancreatic endocrine feedback network to switch-off of α-cell-inhibiting islet signals.
Keywords: pancreatic endocrine network, feedback control, insulin, somatostatin
pancreatic glucagon secretion is a key protection against hypoglycemia that restricts glucose nadirs and stimulates quick recovery from rapid transient drops in glucose concentration (22, 40). Consequently, the reduction (or complete absence) of glucagon response to hypoglycemia associated with insulin-dependent diabetes is a major limiting factor to achieving optimal glycemic control through intensive insulin therapy (19, 20), especially if combined with an acquired loss of epinephrine counterregulation, which are part of a syndrome referred to as hypoglycemia-associated autonomic failure (HAAF). Therefore, understanding the mechanisms that mediate glucagon counterregulation (GCR) secretion and its compromise is a major challenge in the struggle for a better treatment of type 1 diabetes. However, the mechanisms that mediate the GCR and its compromise are not well understood.
Numerous pieces of evidence suggest that an intrapancreatic network of interactions relating different cell types and hormones regulates the pancreatic output and, in particular, directs the glucagon response to different stimuli, including hypoglycemia. For example, experiments involving pancreas perfusion with antibodies have suggested that the blood within the islets of Langerhans flows from β- to α- to δ-cells in dogs, rats, and humans and vascularly borne insulin inhibits glucagon, which in turn regulates the release of somatostatin (77, 78, 83, 84). Other data suggest the possibility of paracrine intraislet control exerted by somatostatin, since δ-cells are in close proximity to both β- and α-cells in rat and human islets and δ-cell processes were observed to extend into α-cell clusters in rat islets (11, 63). However, the high complexity of intraislet interactions obscures the analysis and understanding of system functionality. The recently formulated “switch-off” hypothesis envisions that α-cell activation during hypoglycemia requires both the availability and rapid decline of intraislet insulin (3) and attributes the defect in glucagon response to hypoglycemia in insulin-deficient diabetes to loss of an (insulin) “switch-off” inhibitory signal from the β-cells. Recent in vivo experiments support this hypothesis by demonstrating that in streptozotocin (STZ)-treated rats, GCR is impaired but can be restored if their deficient intraislet insulin is locally reestablished and switched off simultaneously with the decline of the blood glucose (BG) concentration (98). Additional in vitro and in vivo evidence to support the switch-off hypothesis has been reported (51, 99). However, the mechanisms behind the observed effect are unclear, and whether insulin is the switch-off trigger of GCR has recently been challenged by new results showing that zinc ions, not the insulin, provide the switch-off signal to trigger glucagon release during hypoglycemia (100).
In this work we provide experimental evidence and theoretical modeling results to support the concept that GCR develops, at least partially, as a rebound response to disinhibition of the α-cells, which are under the control of at least one β-cell-independent feedback mechanism. In particular, we show that the glucagon response to hypoglycemia in β-cell-deficient STZ-treated rats can be enhanced by local infusion into the pancreas and switch-off (at hypoglycemia) of somatostatin, thereby supporting the notion that GCR is a general disinhibition phenomenon and can be enhanced by switch-off of α-cell-suppressing intrapancreatic signals other than insulin (or zinc). We show that in the face of substantial reduction in β-cell mass the GCR is pulsatile, suggesting that at least one intrapancreatic feedback mechanism not mediated by β-cells that regulates the glucagon response to hypoglycemia. Finally, we use a mathematical model to establish the consistency of the hypothesis that a minimal pancreatic network exists based on α-cell-δ-cell reciprocal interactions and this network contributes to the local control of GCR.
METHODS
Outline of the Methodology
The approach in this work is a “hybrid” between experimental and model-based studies. Although not presented chronologically, it starts with a model-based analysis of the enhancement of GCR by switch-off of insulin (or zinc) reported elsewhere (98–100). This analysis proposes a control network to explain the switch-off phenomenon by interpreting GCR as a rebound and predicts that if this hypothesis is correct 1) in STZ-treated rats multiple α-cell-suppressing signals should enhance GCR if they are terminated during hypoglycemia and 2) the switch-off-triggered GCR must be pulsatile. These model-based results motivate the design of in vivo experiments to confirm the predictions. Finally, additional simulations are performed to facilitate the design and interpretation of future experiments to dissect the network connectivity controlling the GCR.
Animals
All animal experiments were approved by the University of Virginia Institutional Animal Care and Use Committee. Groups of 200-g male Wistar rats were housed with free access to food and water. Diabetes was induced by an intraperitoneal injection of 80 mg/kg STZ. Animals were treated with insulin pellets (LinShin) inserted under the skin (1–2 pellets/rat; release rate ∼2 U·24 h−1·implant−1). Only STZ-treated rats with documented diabetes of at least 2-wk duration were used in the study. The animals weighed ∼250–350 g at the time of the experiment.
Surgery
We followed the surgery procedure described in Ref. 98. On the experimental day the animals were anesthetized (ketamine-xylazine) and the right jugular vein was cannulated for intravenous insulin infusion and blood sampling. The upper abdomen was opened, and the superior pancreaticoduodenal artery was cannulated via the hepatic artery for infusion of switch-off signals directly into the pancreas.
Infusion Protocols
The infusion protocol is similar to the design proposed in Ref. 98. 1) After surgery, BG concentrations were monitored every 10 min and reduced by several small intravenous insulin injections (0.5 U/kg each) to ∼100–110 mg/dl. 2) When euglycemia was achieved, a constant-rate infusion of a switch-off signal [saline (50 μl/min), insulin (regular insulin, 0.5 U/ml, 50 μl/min), or somatostatin (5 μg/ml, 50 μl/min; Sigma)] into the superior pancreaticoduodenal artery was initiated; blood samples (100 μl) were collected every 5 min until the end of the experiment. 3) Ten minutes after the start of the infusion of the switch-off signal, a large insulin bolus (12 U/kg) was administered in the jugular catheter to induce hypoglycemia. 4) The intrapancreatic infusion was switched off at hypoglycemia, when plasma glucose fell below 60 mg/dl (switch-off point). 5) Blood collection continued for the next 45 min.
Assays
Glucagon concentrations were measured with a radioimmunoassay (RIA) kit [Linco Research, St. Louis, MO; 20 pg/ml sensitivity (100-μl sample size), within-/between-assay coefficients of variation: 6.8/13.5, 4.0/12.7, 4.6/13.4, and 4.0/7.3 at concentrations of 60, 65, 90, and 220 pg/ml, respectively]. Glucose concentrations were measured at the time of sample collection with a glucose meter (OneTouch Ultra, LifeScan).
Data Analysis
Glucagon pulsatility.
Measurements of glucagon concentrations reflect a combination of pulsatile secretory events and endogenous metabolic clearance. Multiparameter deconvolution analysis is a computer-based mathematical technique that analyzes the profile of changes over time in observed hormone levels to characterize the properties of the underlying secretory and clearance events. The secretory event variables are duration, rate (amplitude), and temporal position of the bursts. A secretory event is approximated algebraically by a Gaussian distribution of instantaneous molecular secretory rates centered on a particular point in time and dispersed with a finite standard deviation (for more details, see Refs. 16 and 57–59). We used deconvolution to determine whether the switch-off signal-triggered GCR is pulsatile and to compare the characteristics of the pulses between the different animal groups. In the analysis we utilized the new automated deconvolution algorithm AutoDecon (57).
Statistical analysis.
Group comparisons were performed with nonparametric tests (Mann-Whitney or Wilcoxon signed ranks test). Before the switch-off, glucagon concentrations were estimated by averaging the concentrations at t = 0 (time of switch-off), t = −5 min, and t = −10 min. The GCR response was estimated by averaging the glucagon concentrations at t = 25, 30, 35, 40, and 45 min. A P level of 0.05 was considered significant. Results are reported as means ± SE.
Dynamic Network Modeling of GCR
Minimal control network of GCR.
Figure 1 represents a postulated minimal network of glucagon secretion control (minimal control network, MCN) via selected interactions between plasma glucose and the β-, α-, and δ-cells.
Fig. 1.

Minimal control network assumed to provide intrapancreatic regulation of glucagon counterregulation (GCR) in normal physiology.
The postulated connectivity is based on the following.
α-CELL INHIBITION OF β-CELLS.
Early experiments involving anterograde and retrograde pancreas perfusion with antibodies to insulin, somatostatin, and glucagon suggested that the blood within the islets of Langerhans flows from β- to α- to δ-cells in dogs, rats, and humans (77, 78, 83, 84). It was then proposed that within the islet vascularly borne insulin regulates the release of glucagon. Various signals released from the β-cells provide rapid and potent inhibitory stimuli to the α-cells and suppress glucagon secretion. For example, signals originating from upstream β-cells include the cosecreted insulin, zinc, GABA, and amylin, all of which have been implicated to suppress glucagon release (38, 54, 55, 74, 75, 77, 93, 96, 97). Insulin in particular can directly suppress the release of glucagon by binding to receptors for insulin-like growth factor that have been located on α-cells (93) and/or indirectly potentiate the inhibitory effects of GABA by inducing activation of GABAA receptors as shown in Ref. 97. It should be noted that the inhibitory effect of β-cell zinc on glucagon release (54) and its possible role in GCR control suggested in Ref. 100 is unclear in view of one study reporting that zinc ions do not suppress glucagon secretion in the mouse (73). In particular, where insulin is largely lacking, substances not ordinarily important could become regulators of α-cell secretion.
δ-CELL INHIBITION OF α-CELLS.
Exogenous somatostatin is a potent inhibitor of both insulin and glucagon secretion in vivo and in vitro (12, 13, 15, 62–64, 67, 72, 79, 85, 90). However, the involvement of the endogenous hormone in the regulation of pancreatic endocrine outflow remains controversial. The concept that δ-cells are downstream to both α- and δ-cells favors the perception that, in vivo, intraislet somatostatin cannot directly suppress α- or β-cell function through the islet microcirculation (77, 78, 83, 84). Accordingly, somatostatin was assumed to be neutral in regard to intraislet cell signaling because of its place in the sequence of cellular perfusion. On the other hand, the majority of pancreatic cells express at least one of the five known somatostatin receptors (SSTR1–5) (67, 72), and recent studies involving somatostatin immunoneutralization in the perfused human pancreas (13) or application of highly selective antagonists to different somatostatin receptors suggest that α-cell somatostatin inhibits the release of glucagon (and insulin) (15). In addition, δ-cells are shown to be in close proximity to α-cells in rat and human islets, and δ-cell processes were observed to extend into α-cell clusters in rat islets (11, 63). Therefore, in view of the topographical proximity of δ-cells to α-cells, somatostatin may exert its action via common gap junctions or by diffusion through the islet interstitium.
α-CELL STIMULATION OF δ-CELLS.
The ability of locally released glucagon to stimulate somatostatin secretion is supported by one immunoneutralization study in which administration of glucagon antibodies in the perfused human pancreas during both low- and high-glucose conditions resulted in inhibition of somatostatin release that approached significance (13). Earlier immunoneutralization experiments involving perfusion of the rat or dog pancreas also supported the notion that glucagon directly stimulates somatostatin secretion from islet δ-cells (83, 84). In another study, the glucagon receptor was colocalized with 11% of immunoreactive somatostatin cells (25), suggesting that some of the α-cells may regulate subpopulations of δ-cells. Large doses of glucagon have been shown to stimulate the release of somatostatin (13, 14, 28, 92). Another α-cell signal that may stimulate the δ-cells is glutamate, which is cosecreted with glucagon under low-glucose conditions (47) and has been shown to stimulate somatostatin release from diencephalic neurons in primary culture (89).
GLUCOSE STIMULATION OF β- AND δ-CELLS.
It is well established that glucose controls the endocrine pancreas by direct stimulation of the β-cells, which react almost instantaneously to BG changes (2, 4, 26, 80). Additionally, it has been proposed that δ-cells have a glucose-sensing mechanism similar to those in β-cells (37, 43), and consequently somatostatin release is increased in response to glucose stimulation (27, 49), possibly via a Ca2+-dependent mechanism (49).
Model Formulation
Using our established dynamic technique (29–33), we formalized the network shown in Fig. 1 with a set of coupled nonlinear differential equations that describe the evolution of the system assuming significant β-cell deficiency (as in STZ-treated rats). The model equations are:
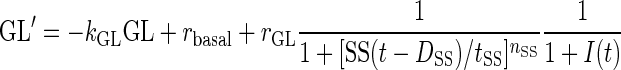 |
(1) |
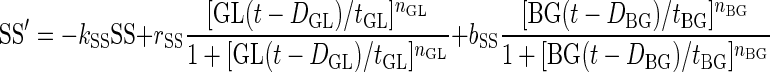 |
(2) |
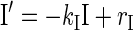 |
(3) |
Here, GL(t), SS(t), BG(t), and I(t) denote time-dependent concentrations of glucagon, somatostatin, blood glucose, and exogenous switch-off signal, respectively; the derivative is the rate of change with respect to the time t.
Model Parameters
The half-life of glucagon was assumed to be ∼2 min to match the results of the pulsatility analysis. Therefore, we fixed the parameter kGL = 20 h−1. There is no information on the half-life of somatostatin in the pancreas and of the functional half-life of the switch-off signal. Therefore these parameters were functionally determined, and we chose the half-life of somatostatin to be similar to that of glucagon (kSS = 20 h−1). The functional half-life of the infused switch-off signal was assumed to be fourfold longer then the half-life of the endogenously released hormones, thereby accounting for possible slower delivery of the “functional” signal. Accordingly, we posed kI = 2.5 h−1. The delays in the system were functionally determined (together with the potencies and sensitivities, below) to guarantee that glucagon pulses occur at intervals of 15–20 min to correspond to the number of pulses after the switch-off point detected in the pulsatility analysis: DBG = 1/20 h, DSS = 1/20 h, DGL = 1/40 h. The remaining parameters used in the simulations were determined functionally, and the concentrations presented below are in arbitrary units. These units, however, can be easily rescaled to match real concentrations. In particular, we used release rates rSS = 110, bSS = 40, rGL = 80, and rbasal = 0.2 for the basal secretion of glucagon (concentration/hour); potencies tBG = 80, tSS = 1.3, and tGL = 0.07 (concentration); and sensitivities (Hill coefficients) nBG = 3, nSS = 3 , nGL = 3. The infusion rate for the switch-off signal rI (concentration/hour) was assumed ≠ 0 (rI = 35) only from 94:15 to 95:00. To integrate the equations we used a Runge-Kutta 4 algorithm and performed numerical simulations to test the system response to external switch-off signals that suppress and release α-cell activity under different conditions.
Simulation of Combined Infusion Experiments
Infusion experiments were simulated as follows: 1) glucose infusion: increase of the function of BG(t) by a constant parameter; 2) glucagon infusion: addition of a new constant term to Eq. 1 (emulating basal glucagon secretion); 3) somatostatin infusion: addition of a new constant term to Eq. 2; 4) infusion of somatostatin antibody: increase of the clearance constant of somatostatin by a constant factor; 5) BG levels were superimposed to mimic a decline into hypoglycemia followed by a recovery.
RESULTS
Experimental Observations
GCR response to switch-off signals.
To estimate the GCR we compared the pre-switch-off glucagon concentration levels [Pre = average glucagon concentrations at t = 0 (time of switch-off), t = −5 min, and t = −10 min] to the post-switch-off glucagon levels and to the average response from t = 25 min to t = 45 min (Post = average concentrations at t = 25, 30, 35, 40, and 45 min).
Figure 2 illustrates the mean fold increase at each time point (t = −10, … , 45 min) relative to the Pre levels. The values were computed for each animal individually and then averaged across all animals in the corresponding group.
Fig. 2.

Fold increase at each time point (t = −10, … , 45 min) relative to the pre-switch-off levels in the Saline (n = 7), Somatostatin (n = 6), and Insulin (n = 6) groups. Data are means ± SE averaged across the 3 groups.
In the three groups, the fold increases in Post relative to Pre were 1.16 ± 0.17-, 2.85 ± 0.24-, and 3.05 ± 0.65-fold in the Saline (n = 7), Somatostatin (n = 6), and Insulin (n = 6) groups, respectively, and the increases in the Insulin and Somatostatin groups were significantly higher vs. the control group (P = 0.032 and P = 0.003, respectively; Mann-Whitney nonparametric test). The Insulin and Somatostatin groups were not different in this metric (P = 0.87).
In absolute terms, in the control group there was no significant difference between the Pre and Post glucagon levels (Wilcoxon signed ranks test: P = 0.24, n = 7). In the Insulin and Somatostatin groups the Pre and Post levels were significantly different at P = 0.03 (n = 6 in both groups). In all three groups the Pre levels were not different (97.5 ± 20.4, 83.3 ± 17.1, and 127.9 ± 17.2 pg/ml in Saline, Somatostatin, and Insulin groups, respectively). The Post glucagon concentrations in the Somatostatin and Insulin groups (227.9 ± 42.9 and 368.3 ± 86.2 pg/ml, respectively) were significantly higher (P < 0.05) compared with the control group (113.9 ± 29.3 pg/ml), and there was no significant difference between them, even though the response to insulin switch-off was ∼30% higher (Fig. 3).
Fig. 3.

Glucagon concentration from t = −10 to t = 0 min before the switch-off point (Pre) and from t = 25 to t = 45 min after the switch-off (Post) in the Saline (n = 7), Somatostatin (n = 6), and Insulin (n = 6) groups. Data are means ± SE. *Significantly different, P < 0.05.
Pulsatility analysis of GCR in response to a switch-off signal.
The 5-min profiles of glucagon were mathematically modeled by deconvolution analysis (59) to examine the underlying patterns of glucagon secretory activity in the 45-min post-switch-off interval after the switch-off point. The analysis was performed with the new automated deconvolution algorithm AutoDecon (57) without restrictions on the parameters, which were allowed to vary and were fitted for each animal individually.
In 17 of 19 animals the deconvolution analysis converged and demonstrated that in these rats GCR is pulsatile. In the remaining two animals (1 in the Insulin group and 1 in the Somatostatin group) the analysis did not converge and the post-switch-off increase in glucagon was not demonstrated to be pulsatile.
In the post-switch-off interval, both the number of secretory events and the total pulsatile production were significantly lower in the Saline group compared with either the Insulin or the Somatostatin group. In particular, we detected that the average number of secretory events was 0.71 ± 0.29 in the Saline group, 2.40 ± 0.51 in the Insulin group (different from Saline, P = 0.02), and 2.60 ± 0.40 in the Somatostatin group (different from Saline, P = 0.01). The average total pulsatile production (area under the curve) in the 45-min interval was 387 ± 161 pg·ml−1·45 min−1 in the Saline group, 7,792 ± 2,877 pg·ml−1·45 min−1 in the Insulin group (different from Saline, P = 0.003), and 2,878 ± 1,282 pg·ml−1·45 min−1 in the Somatostatin group (different from Saline, P = 0.01).
The basal secretion was significantly different only between the Saline and Insulin groups (28 ± 12 vs. 103 ± 34 pg·ml−1·min−1; P = 0.03). This parameter in the Somatostatin group was 43 ± 12 pg·ml−1·min−1. The reconstructed apparent average half-life was 1.9 ± 0.4 min (all animals), which agrees with published data (39, 56, 71).
Model-Assisted Analysis
Mechanism of the glucagon response to a switch-off signal in the face of low-glucose conditions.
We tested the model response to a brief intrapancreatic infusion of α-cell-suppressing signal switched off during BG decline. The infusion was initiated at time 94:15 (after the start of the simulations) and switched-off at 95:00. Simulated gradual BG decline started at 94:30, and BG was below 6 concentration units (or 60 mg/dl after rescaling) at the switch-off point. The model response is depicted in Fig. 4, bottom, and illustrates a rebound glucagon secretion after the switch-off point and gradual recovery toward the normal state of the system. For comparison, Fig. 4, top, depicts the GCR response in a representative animal from the Somatostatin switch-off group. From a mechanistic perspective, the model explains the observed effects as follows. During the basal state, the reciprocal interaction between glucagon and somatostatin yields pulsatile behavior (with spikes recurring every 0.25 h), as explained elsewhere (29). After the system is perturbed the diminished glucagon output stimulates less somatostatin secretion. Therefore, both glucagon and somatostatin remain low during the enforced suppression of α-cell activity. Thus the first postsuppression glucagon pulse evolves during lower than typical somatostatin nadir, but still in the presence of a decaying exogenous suppressor, which explains why the second postsuppression glucagon spike is higher than the first spike. The model prediction agrees with our experimental results, with those reported in Refs. 98 and 99 showing a restoration of GCR by an insulin/zinc switch-off signal, and with other results showing glucagon pulsatility during hypoglycemia (39, 56).
Fig. 4.

Bottom: model-predicted glucagon rebound response to a switch-off signal (94:30–95:00) in the face of low-glucose conditions in streptozotocin (STZ)-treated rats. Top: GCR response in a representative animal from the Somatostatin switch-off group aligned in a way that the times of the switch-off in the 2 plots coincide (t = 95:00). Vertical dotted lines mark the time period studied in the experiment. BG, blood glucose.
It is important to note that even though the reciprocal interactions between glucagon and somatostatin (theoretically) can drive pulsatile secretion during the basal state, here based on the simulations we predict only that oscillations arising during counterregulation are due to glucagon-somatostatin feedback interactions and cannot exclude the possibility that other mechanisms also contribute to the pulsatility of islet hormones (6, 7, 9).
Restriction of glucagon response to a switch-off signal by high-glucose conditions in STZ-treated rats.
A key point in the reconstruction of the experiments in Refs. 98 and 99 is to predict the lack of glucagon response during high-glucose conditions. Accordingly, we tested the model response to a switch-off signal (same as in Fig. 4) in the face of euglycemia. The model output shown in Fig. 5 agrees with the results reported in Ref. 98 and predicts that high BG blocks the glucagon response to a switch-off signal by preventing the low nadirs in somatostatin release that otherwise would permit high-amplitude glucagon spikes during the rebound.
Fig. 5.

Model-predicted restriction of the rebound response to a switch-off signal by high-glucose conditions in STZ-treated rats.
Lack of glucagon response to glucose decline in the face of absent switch-off signal in STZ-treated rats.
The graphs in Fig. 6 show the predicted lack of glucagon response to BG decline if the switch-off signal is missing—another key observation reported above in the experimental results and in Refs. 98 and 99. The system responds with ∼15% increase of glucagon pulse amplitude and slight increase in glucagon nadirs. The results agree well with our experimental observations: see Figs. 2 and 3 and Fig. 6, top, which shows the GCR response to a saline switch-off signal in a representative animal from the Saline group.
Fig. 6.

Bottom: model-predicted lack of glucagon response to glucose decline alone in STZ-treated rats. Top: GCR response in a representative animal from the Saline switch-off group aligned in a way that the times of the switch-off in the 2 plots coincide (t = 95:00). Vertical dotted lines mark the time period studied in the experiment.
The model explains the absence of significant increase in glucagon pulse amplitude by the inability of glucose decline alone to cause a significant perturbation in the system and cause a rebound.
Model-based predictions.
To design experiments that probe whether the glucagon response to α-cell-inhibiting switch-off signal in STZ-treated rats is mediated and amplified by a glucagon-somatostatin intraislet feedback loop, we simulated the system response to a switch-off signal in the face of altered glucagon-somatostatin relationships.
First, we tested the effect of a constant glucagon infusion administered simultaneously with the switch-off signal into the pancreas. To this end, we increased the basal glucagon secretion rate (rbasal) threefold in Eq. 1. Model-generated glucagon and somatostatin profiles are shown in Fig. 7, top (the glucagon profile is the sum of endogenous and exogenous glucagon).
Fig. 7.

Model-predicted attenuation of the switch-off effect due to disruption of glucagon-somatostatin interactions via constant infusion of glucagon (top), somatostatin (middle), and somatostatin antibody (bottom).
From a mechanistic perspective, the glucagon rebound response is suppressed since, as postulated in the MCN, exogenous glucagon stimulates somatostatin outflow and prevents low-somatostatin nadirs, which otherwise would permit the appearance of large glucagon spikes. Analogous reasoning explains the observed attenuation of the rebound glucagon response to a switch-off signal caused by constant small infusion of somatostatin (Fig. 7, middle). These simulations were performed by adding a constant (20 pg·ml−1·h−1) to the right-hand side of Eq. 1. Finally, we simulated partial intrapancreatic somatostatin neutralization by increasing the rate of elimination kSS twofold. The model response to a switch-off signal (94:30–95:00) is summarized in Fig. 7, bottom. Somatostatin removal prevents the feedback interactions and suppresses the hormone oscillations and rebound response, regardless of the fact that the overall glucagon concentration is elevated.
DISCUSSION
The findings of this proof-of-concept interdisciplinary study strongly support the hypothesis that α-cell-inhibiting intrapancreatic signals that are terminated during hypoglycemia amplify GCR via an intrapancreatic feedback mechanism. Our approach is a “hybrid” between experimental and modeling studies. Chronologically, it starts with a model-based analysis of the enhancement of GCR by switch-off of insulin (98–100) to show that α-cell-δ-cell reciprocal interactions within a putative MCN (Fig. 1) can explain the switch-off effect by interpreting GCR as a rebound. It predicts that the postulated MCN is consistent with the following: 1) in β-cell deficiency, multiple α-cell-suppressing signals should enhance GCR if they are terminated during hypoglycemia, and 2) the switch-off-triggered GCR must be pulsatile. These analytical outcomes motivate the design of our in vivo experiments, which confirm the model predictions by showing that in STZ-treated male Wistar rats intrapancreatic infusion of insulin and somatostatin followed by their switch-off during hypoglycemia enhances the pulsatile GCR. Finally, additional simulations predict the outcome of future experiments designed to confirm that the postulated MCN unifies the dominant controls of GCR.
At least three hypotheses have been proposed to explain the glucagon defense against hypoglycemia (88): 1) glucose decline directly stimulates the α-cells; 2) the response is mediated by removal of an inhibitory insulin β-cell signal (“switch-off” hypothesis); 3) central and/or local autonomic inputs direct α-cell activation. Decline of GCR is associated with progression of type 1 (36, 42, 50) and (possibly) type 2 (81) diabetes, but the underlying mechanism is not known. The “switch-off” hypothesis (3) attributes the defect in GCR in insulin-deficient diabetes to loss of an insulin switch-off signal from the β-cells. Major motivation for this work comes from in vivo experiments supporting the “switch-off” hypothesis by showing that in STZ-treated rats GCR is impaired but can be restored if their deficient intraislet insulin is locally reestablished and switched off at hypoglycemia (98). Additional in vitro and in vivo evidence was reported in Refs. 51 and 99. However, whether insulin is the switch-off trigger of GCR in these studies has been recently challenged by results by the same group, in which zinc ions, and not the insulin molecule itself, were shown to provide the switch-off signal to initiate GCR (100).
In this study we accept the general concept of the “switch-off” hypothesis that GCR develops as a disinhibition. However, we challenge the premise that the specific trigger is necessarily β-cell insulin and consider the GCR defects as a failure of a putative dynamic MCN to respond adequately to a hypoglycemic stimulus. The concept of network control of GCR is supported by studies that document the pulsatility of pancreatic hormones (39, 44, 56, 76), which suggest existence of a feedback control loop (29), and by results showing that insulin and somatostatin pulses are in phase (56, 68), pulses of insulin and glucagon recur with a phase shift (44), and pulses of somatostatin and glucagon appear in antisynchronous fashion (76). The results in Ref. 76 (see also Ref. 70) also suggest that insulin pulses are independent of the α- and δ-cells but might entrain their oscillations. These findings are consistent with the postulated MCN and suggest intrapancreatic ensemble control, a concept as yet almost completely unexplored in relation to GCR.
In this study, the switch-off effects were evaluated by comparing group differences between the pre- and post-switch-off glucagon levels (Pre and Post, respectively). We studied the immediate glucagon response in the 45-min interval after hypoglycemia since glucagon is responsible for the early defenses against hypoglycemia in concert with epinephrine and in conjunction with the cessation of insulin secretion (in less insulin-deficient models) (21, 22). In addition, the need for a frequent sampling paradigm renders it impossible to sample a longer time period. In the Saline group, hypoglycemia had no significant effect on the glucagon levels and there was no difference between Pre and Post. When the hypoglycemic stimulus was combined with the effect of a switch-off signal (insulin or somatostatin) Pre and Post became significantly different (P = 0.03). In addition, in all three groups the Pre levels were not different (Fig. 3), but the fold increases in Post relative to Pre in the Insulin and Somatostatin switch-off groups were significantly higher compared with the increase in the control group (Fig. 2). Finally, the absolute increase in glucagon after the switch-off was higher in the Insulin and Somatostatin groups compared with the control group (Fig. 3). It may be expected that both the relative increase and absolute level of glucagon secretion in response to a switch-off could be important. The absolute levels will matter if there is a threshold for glucagon required to exert a biological change. If such a threshold does not exist within the range of endogenous glucagon concentrations, the amplitude of relative change would be critical, especially given the fact that other factors act in concert with glucagon to maintain glucose homeostasis.
Our experimental results show that, as predicted by the model, the α-cell-suppressing signals somatostatin and insulin enhance the GCR if they are infused locally and switched off during hypoglycemia. They confirm the GCR-stimulating effect of insulin (or zinc) demonstrated in Ref. 98, since we used similar surgery technique and infusion paradigms. However, we utilized a more frequent sampling over a shorter time period to obtain a reliable estimate of the pre- and post-switch-off glucagon levels and apply deconvolution to analyze the GCR pulsatility. To our knowledge, the ability of somatostatin to enhance the GCR has not been tested directly before and is controversial in view of studies in normal men in which reboundlike insulin, but not glucagon, release after short-term somatostatin administration has been reported (66). Based on the putative MCN, the lack of glucagon rebound in normal physiology might be explained with β-cell activation by the switch-off of somatostatin, which could in turn suppress the α-cells.
A seemingly unexpected result is the lack of difference in the pre-switch-off levels between the control and switch-off groups. However, what matters for a rebound response is the individual decrease in glucagon in response to the infusion of the switch-off signal, and we were not able to estimate this effect. Moreover, it is possible that the exogenous switch-off signal exerts its suppressive effect only on a subpopulation of the α-cells, thereby masking the decrease in secretion in these cells. Future studies including frequent portal blood sampling before the start of the infusion of the switch-off signal should be able to confirm that GCR develops as a rebound.
In a different approach to evaluating the experimental results, the 5-min profiles of glucagon were mathematically analyzed by deconvolution to examine the glucagon secretory activity in the 45-min interval after the switch-off point. The analysis was performed without any restrictions on the parameters, which were fit simultaneously for each animal individually. In 17 of 19 animals the GCR was pulsatile, supporting the concept that in these STZ-treated rats the secretion of glucagon is controlled by at least one β-cell-independent feedback control loop. After the switch-off, the number of secretory events were significantly lower in the Saline group vs. the Insulin and Somatostatin groups (P = 0.02 and 0.01, respectively). In addition, the average total pulsatile production in the 45-min interval was also lower in the control group (P = 0.003 and 0.01, respectively). These results indicate enhancement of glucagon pulsatility by insulin/somatostatin switch-off compared with saline switch-off.
Even though the deconvolution was successful and the reconstructed apparent half-life (1.9 ± 0.4 min) agrees with published data (39, 56, 71), in two animals (1 in the Insulin group and 1 in the Somatostatin group) the post-switch-off increase in glucagon was not demonstrated to be pulsatile. This outcome reflects a limitation of the present study related to the lack of more frequently sampled data. If such data were available (e.g., 1-min samples to account for pulsatility specifics related to the short half-life of glucagon), the analysis undoubtedly would be more robust. Such data can be obtained in future experiments by collecting the samples from the portal circulation instead of the venous catheter.
The theoretical considerations utilize dynamic modeling, which has never been applied to the pancreatic endocrine system either alone or in combination with experimental work, even though the methods are well accepted and groundbreaking in other aspects of endocrine physiological investigation (5, 17, 29–33, 65). The goal of the analysis is to show that the MCN is consistent and can explain the in vivo glucagon response to switch-off signals in STZ-treated rats observed in this study and elsewhere (98, 99). The GCR is viewed as a rebound of a feedback structure incorporated into the model via perturbed oscillatory interaction between α-cell glucagon and δ-cell somatostatin. In biological terms, the perturbing mechanism is the switch-off signal (insulin/zinc or somatostatin), and the frequency of the putative glucagon-somatostatin oscillator depends on the aggregate time latency in the system conduits. Time delays within the pancreas are not known currently. However, the mean interburst interval would reflect the sum of the feedforward time delay for glucagon to promote sufficient somatostatin outflow and the feedback time lag for somatostatin to block α-cell glucagon. Frequent portal venous sampling of somatostatin and glucagon in response to a switch-off signal is not available to delineate the precise dynamics of glucagon and somatostatin release. In the absence of such data, the parameters of the interactions between somatostatin and glucagon are not definitive, albeit empirically suited to sustain an oscillating system that would rebound in response to a switch-off. They were functionally determined to generate pulses recurring every 15–20 min.
The interpretation of the GCR as a rebound permits the explanation of the system response to a switch-off signal observed here and in Refs. 98 and 99. From a mechanistic perspective, the model predicts that after the system is perturbed by the α-cell suppressing signal the reduced glucagon output stimulates less somatostatin secretion. Therefore, both glucagon and somatostatin remain low during the administration of the switch-off signal. When the suppression is removed, high glucagon pulses evolve because of lower than typical somatostatin nadirs. The dynamics of the response depend on the rate of removal of the suppression, and the model predicts that slow functional decay of the switch-off signal is necessary to explain why the second postsuppression glucagon spike is higher than the first spike (Fig. 4). These predictions agree with results showing GCR pulsatility (39, 56).
An important consequence of linking the switch-off effect to α-cell-δ-cell reciprocal interaction is the proposed explication of two key observations (98, 99). For example, the reduced glucagon response to a switch-off signal by high-glucose conditions in STZ-treated rats is attributed to elevated BG-driven somatostatin nadirs that prevent high-amplitude glucagon oscillations. In addition, the observed (here and in Refs. 98 and 99) lack of GCR in the face of absent switch-off signal in STZ-treated rats is attributed to the inability of glucose decline alone to cause a rapid removal of a strong suppression of the feedback system to cause a rebound.
The approximation of the MCN allows for several testable model-based predictions to probe whether the glucagon response to a switch-off signal is mediated and amplified by the putative α-cell-δ-cell loop. Accordingly, we simulated the system response to a switch-off in the face of altered α-cell-δ-cell relationships. The latter is achieved via infusion of glucagon, somatostatin, or somatostatin neutralization. In all three cases we predict attenuation of the response due to change in the properties of the feedback (Fig. 7). Thereby, the analysis provides justification for selected experiments designed to test the amplifying effect of the MCN interactions on the GCR.
To determine the model parameters, we have assumed that the α-cell-δ-cell interactions drive pulses recurring every 15–20 min as detected by deconvolution (2.4–2.6 pulses per 45 min). Even though these interactions can cause pulsatility in the basal state, it is possible that they generate oscillations only during the rebound state (29). Therefore, based on the simulations we predict only that oscillations during counterregulation are due to glucagon-somatostatin feedback interactions. We cannot exclude the possibility that other central or local mechanisms also contribute to the islet hormone pulsatility (1, 6, 7, 9, 39). Also, it is possible that the secretory events observed here are composite volleys consisting of two (or more) spikes, as suggested by in vitro studies detecting glucagon spikes recurring every 3–4 min (44, 76).
The postulated MCN is consistent with the concept that reduction in the switch-off signal would impair the GCR. Additional simulations (results not shown) demonstrated that gradual reduction of the strength of the α-cell suppression by the switch-off signal leads to dose-dependent decrease in GCR. The reduction begins from a full response (Fig. 4) and gradually declines to almost complete lack of a response (Fig. 6). These results suggest that decline of the strength of the suppression of the α-cells caused by reduction of β-cell mass would account for decrease in the amplitude of the rebound and loss of immediate glucagon response to hypoglycemia.
From a modeling perspective, the unification of the experimental findings with the concept that β-cell failure is the cause of loss of glucagon response to hypoglycemia poses two requirements. First, a β-cell-independent feedback loop involving the α-cells is necessary to explain the rebound after a switch-off and loss of GCR response to hypoglycemia in β-cell deficiency. Second, this feedback must be under BG regulation to account for the reduced response to a switch-off by high glucose in STZ-treated rats (98). The postulated α-cell-δ-cell loop is supported by most of the publications (methods), but other possibilities exist, like for example the α-cell autofeedback control suggested in Ref. 60. In the present model, BG affects the feedback via the δ-cells, but it is possible for BG to control the α-cells directly (41), which together with the putative α-cell autofeedback is an alternative δ-cell-independent construct that might explain the system behavior. The simulated experiments (Model-based predictions) are designed to disclose whether the δ-cells are part of the GCR control mechanism.
Other pathways may be also involved in GCR control. For example, a β-cell-α-cell loop is possible in view of results suggesting that 1) the β-cells express glucagon receptors (52, 61), 2) glucagon stimulates insulin (13, 52), and 3) α-cell-released glutamate stimulates insulin and GABA (8, 53, 91). However, it is unlikely that this loop is the mechanism behind the rebound since its failure in STZ-treated rats will result in lack of response to a switch-off. This was not observed here and in Ref. 98, where the GCR was amplified without restoration of the putative β-cell-α-cell loop.
To delineate the similarities and differences between the experimental results and the model predictions, we note that the simulations explain certain general features of the in vivo system rather than fitting the data. Therefore, the simulated profiles do not match exactly the individual hormone dynamics. For example, the experimental and simulated glucagon profiles in Figs. 4 and 6 match in the tendency to oscillate and rebound (or not) after a switch-off, but there is little point-to-point correspondence between the two. A logical next step would be to use the model to fit experimental data to reconstruct the MCN dose-response interactions. This requires in vivo experiments involving frequent portal sampling for glucagon and somatostatin and in vitro perfusion studies.
In conclusion, the experimental results in this proof-of-concept study support the hypothesis that α-cell-inhibiting intrapancreatic signals switched off during hypoglycemia enhance the pulsatile GCR via a local feedback mechanism. The proposed mathematical construct offers a new objective analytical platform to study the GCR regulation complementary to the experimental approach. The model-based analysis predicts that the mechanism behind the switch-off involves a rebound glucagon release triggered by the disinhibition of a putative α-cell-δ cell feedback network. Thereby, defects in GCR related to β-cell deficiency can be attributed to failure of the pancreatic network to respond adequately to a hypoglycemic stimulus due to decrease in the strength of the switch-off signal. Future in vivo and/or in vitro perfusion studies involving simultaneous frequent portal blood sampling for islet signals in STZ-treated and normal rats teamed with model-based analysis of the outcomes will be required to further explore the dynamics of the intrapancreatic network relationships and their impact on GCR control.
GRANTS
The study was supported in part by National Institutes of Health Grants R21-DK-072095 (to L. S. Farhy, Z. Du, Q. Zeng, M. L. Johnson, and A. L. McCall), R01-RR-019991 (to L. S. Farhy, M. L. Johnson, and P. P. Veldhuis), R01-DK-076037 (to L. S. Farhy and M. L. Johnson), P30-DK-063609 (to L. S. Farhy, Z. Du, Q. Zeng, and A. L. McCall), R01-DK-51562 (to L. S. Farhy), and U54-HD-28934, R01-RR-00847, and R25-DK-064122 (to M. L. Johnson).
Acknowledgments
The authors are thankful to Dr. Paul Robertson for critical advice on the experimental design of the study.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Ahren B, Taborsky GJ Jr, Havel PJ. Differential impairment of glucagon responses to hypoglycemia, neuroglycopenia, arginine, and carbachol in alloxan-diabetic mice. Metabolism 51: 12–19, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft FM, Proks P, Smith PA, Ammala C, Bokvist K, Rorsman P. Stimulus-secretion coupling in pancreatic beta cells. J Cell Biochem 55, Suppl: 54–65, 1994. [DOI] [PubMed] [Google Scholar]
- 3.Banarer S, McGregor VP, Cryer PE. Intraislet hyperinsulinemia prevents the glucagon response to hypoglycemia despite an intact autonomic response. Diabetes 51: 958–965, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Bell GI, Pilkis SJ, Weber IT, Polonsky KS. Glucokinase mutations, insulin secretion, and diabetes mellitus. Annu Rev Physiol 58: 171–186, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol Endocrinol Metab Gastrointest Physiol 236: E667–E677, 1979. [DOI] [PubMed] [Google Scholar]
- 6.Bergsten P Pathophysiology of impaired pulsatile insulin release. Diabetes Metab Res Rev 16: 179–191, 2000. [DOI] [PubMed] [Google Scholar]
- 7.Bergsten P Role of oscillations in membrane potential, cytoplasmic Ca2+, and metabolism for plasma insulin oscillations. Diabetes 51, Suppl 1: S171–S176, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Bertrand G, Gross R, Puech R, Loubatieres-Mariani MM, Bockaert J. Evidence for a glutamate receptor of the AMPA subtype which mediates insulin release from rat perfused pancreas. Br J Pharmacol 106: 354–359, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berts A, Liu YJ, Gylfe E, Hellman B. Oscillatory Ca2+ signaling in somatostatin-producing cells from the human pancreas. Metabolism 46: 366–369, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Biggers DW, Myers SR, Neal D, Stinson R, Cooper NB, Jaspan JB, Williams PE, Cherrington AD, Frizzell RT. Role of brain in counterregulation of insulin-induced hypoglycemia in dogs. Diabetes 38: 7–16, 1989. [DOI] [PubMed] [Google Scholar]
- 11.Brelje TC, Scharp DW, Sorenson RL. Three-dimensional imaging of intact isolated islets of Langerhans with confocal microscopy. Diabetes 38: 808–814, 1989. [DOI] [PubMed] [Google Scholar]
- 12.Brunicardi FC, Atiya A, Moldovan S, Lee TC, Fagan SP, Kleinman RM, Adrian TE, Coy DH, Walsh JH, Fisher WE. Activation of somatostatin receptor subtype 2 inhibits insulin secretion in the isolated perfused human pancreas. Pancreas 27: e84–e89, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Brunicardi FC, Kleinman R, Moldovan S, Nguyen TH, Watt PC, Walsh J, Gingerich R. Immunoneutralization of somatostatin, insulin, and glucagon causes alterations in islet cell secretion in the isolated perfused human pancreas. Pancreas 23: 302–308, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Brunicardi FC, Elahi D, Andersen DK. Splanchnic neural regulation of somatostatin secretion in the isolated perfused human pancreas. Ann Surg 219: 258–266, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cejvan K, Coy DH, Efendic S. Intra-islet somatostatin regulates glucagon release via type 2 somatostatin receptors in rats. Diabetes 52: 1176–1181, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Chapman IM, Hartman ML, Pezzoli SS, Thorner MO. Enhancement of pulsatile growth hormone secretion by continuous infusion of a growth hormone-releasing peptide mimetic, L-692,429, in older adults—a clinical research center study. J Clin Endocrinol Metab 81: 2874–2880, 1996. [DOI] [PubMed] [Google Scholar]
- 17.Cobelli C, Brier DM, Ferrannini E. Modeling glucose metabolism in man: theory and practice. Horm Metab Res Suppl 24: 1–10, 1990. [PubMed] [Google Scholar]
- 18.Coiro V, Passeri M, Volpi R, Rossi G, Camellini L, Davoli D, Marchesi M, Muzzetto P, Minelli R, Bianconi L. Effect of muscarinic and nicotinic-cholinergic blockade on the glucagon response to insulin-induced hypoglycemia in normal men. Horm Metab Res 21: 102–103, 1989. [DOI] [PubMed] [Google Scholar]
- 19.Cryer PE Hypoglycemia is the limiting factor in the management of diabetes. Diabetes Metab Res Rev 15: 42–46, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Cryer PE Hypoglycemia the limiting factor in the glycaemic management of type I and type II diabetes. Diabetologia 45: 937–948, 2002. [DOI] [PubMed] [Google Scholar]
- 21.Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care 26: 1902–1912, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Cryer PE, Gerich JE. Relevance of glucose counterregulatory systems to patients with diabetes: critical roles of glucagon and epinephrine. Diabetes Care 6: 95–99, 1983. [DOI] [PubMed] [Google Scholar]
- 23.Davis SN, Colburn C, Dobbins R, Nadeau S, Neal D, Williams P, Cherrington AD. Evidence that the brain of the conscious dog is insulin sensitive. J Clin Invest 95: 593–602, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis SN, Dunham B, Walmsley K, Shavers C, Neal D, Williams P, Cherrington AD. Brain of the conscious dog is sensitive to physiological changes in circulating insulin. Am J Physiol Endocrinol Metab 272: E567–E575, 1997. [DOI] [PubMed] [Google Scholar]
- 25.Dumonteil E, Magnan C, Ritz-Laser B, Ktorza A, Meda P, Philippe J. Glucose regulates proinsulin and prosomatostatin but not proglucagon messenger ribonucleic acid levels in rat pancreatic islets. Endocrinology 141: 174–180, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Dunne MJ, Harding EA, Jaggar JH, Squires PE. Ion channels and the molecular control of insulin secretion. Biochem Soc Trans 22: 6–12, 1994. [DOI] [PubMed] [Google Scholar]
- 27.Efendic S, Nylen A, Roovete A, Uvnas-Wallenstein K. Effects of glucose and arginine on the release of immunoreactive somatostatin from the isolated perfused rat pancreas. FEBS Lett 92: 33–35, 1978. [DOI] [PubMed] [Google Scholar]
- 28.Epstein S, Berelowitz M, Bell NH. Pentagastrin and glucagon stimulate serum somatostatin-like immunoreactivity in man. J Clin Endocrinol Metab 51: 1227–1231, 1980. [DOI] [PubMed] [Google Scholar]
- 29.Farhy LS Modeling of oscillations in endocrine networks with feedback. Methods Enzymol 384: 54–81, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Farhy LS, Straume M, Johnson ML, Kovatchev B, Veldhuis JD. A construct of interactive feedback control of the GH axis in the male. Am J Physiol Regul Integr Comp Physiol 281: R38–R51, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Farhy LS, Straume M, Johnson ML, Kovatchev B, Veldhuis JD. Unequal autonegative feedback by GH models the sexual dimorphism in GH secretory dynamics. Am J Physiol Regul Integr Comp Physiol 282: R753–R764, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Farhy LS, Veldhuis JD. Putative GH pulse renewal: periventricular somatostatinergic control of an arcuate-nuclear somatostatin and GH-releasing hormone oscillator. Am J Physiol Regul Integr Comp Physiol 286: R1030–R1042, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Farhy LS, Veldhuis JD. Joint pituitary-hypothalamic and intrahypothalamic autofeedback construct of pulsatile growth hormone secretion. Am J Physiol Regul Integr Comp Physiol 285: R1240–R1249, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Fox CR, Farhy LS, Evans WS, Johnson ML. Measuring the coupling of hormone concentration time series using polynomial transfer functions. Methods Enzymol 384: 82–94, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Frizzell RT, Jones EM, Davis SN, Biggers DW, Myers SR, Connolly CC, Neal DW, Jaspan JB, Cherrington AD. Counterregulation during hypoglycemia is directed by widespread brain regions. Diabetes 42: 1253–1261, 1993. [DOI] [PubMed] [Google Scholar]
- 36.Fukuda M, Tanaka A, Tahara Y, Ikegami H, Yamamoto Y, Kumahara Y, Shima K. Correlation between minimal secretory capacity of pancreatic beta-cells and stability of diabetic control. Diabetes 37: 81–88, 1988. [DOI] [PubMed] [Google Scholar]
- 37.Fujitani S, Ikenoue T, Akiyoshi M, Maki T, Yada T. Somatostatin and insulin secretion due to common mechanisms by a new hypoglycemic agent, A-4166, in perfused rat pancreas. Metabolism 45: 184–189, 1996. [DOI] [PubMed] [Google Scholar]
- 38.Gedulin BR, Rink TJ, Young AA. Dose-response for glucagonostatic effect of amylin in rats. Metabolism 46: 67–70, 1997. [DOI] [PubMed] [Google Scholar]
- 39.Genter P, Berman N, Jacob M, Ipp E. Counterregulatory hormones oscillate during steady-state hypoglycemia. Am J Physiol Endocrinol Metab 275: E821–E829, 1998. [DOI] [PubMed] [Google Scholar]
- 40.Gerich JE Lilly lecture 1988. Glucose counterregulation and its impact on diabetes mellitus. Diabetes 37: 1608–1617, 1988. [DOI] [PubMed] [Google Scholar]
- 41.Gerich JE, Charles MA, Grodsky GM. Characterization of the effects of arginine and glucose on glucagon and insulin release from the perfused rat pancreas. J Clin Invest 54: 833–841, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerich JE, Langlois M, Noacco C, Karam JH, Forsham PH. Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science 182: 171–173, 1973. [DOI] [PubMed] [Google Scholar]
- 43.Gopel SO, Kanno T, Barg S, Rorsman P. Patch-clamp characterisation of somatostatin-secreting-cells in intact mouse pancreatic islets. J Physiol 528: 497–507, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grapengiesser E, Salehi A, Quader SS, Hellman B. Glucose induces glucagon release pulses antisynchronous with insulin and sensitive to purinoceptor inhibition. Endocrinology 147: 3472–3477, 2006. [DOI] [PubMed] [Google Scholar]
- 45.Havel PJ, Ahren B. Activation of autonomic nerves and the adrenal medulla contributes to increased glucagon secretion during moderate insulin-induced hypoglycemia in women. Diabetes 46: 801–807, 1997. [DOI] [PubMed] [Google Scholar]
- 46.Havel PJ, Taborsky GJ Jr. The contribution of the autonomic nervous system to changes of glucagon and insulin secretion during hypoglycemic stress. Endocr Rev 10: 332–350, 1989. [DOI] [PubMed] [Google Scholar]
- 47.Hayashi M, Yamada H, Uehara S, Morimoto R, Muroyama A, Yatsushiro S, Takeda J, Yamamoto A, Moriyama Y. Secretory granule-mediated co-secretion of l-glutamate and glucagon triggers glutamatergic signal transmission in islets of Langerhans. J Biol Chem 278: 1966–1974, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Heimberg H, De Vos A, Moens K, Quartier E, Bouwens L, Pipeleers D, Van Schaftingen E, Madsen O, Schuit F. The glucose sensor protein glucokinase is expressed in glucagon-producing alpha-cells. Proc Natl Acad Sci USA 93: 7036–7041, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hermansen K, Christensen SE, Orskov H. Characterization of somatostatin release from the pancreas: the role of potassium. Scand J Clin Lab Invest 39: 717–722, 1979. [DOI] [PubMed] [Google Scholar]
- 50.Hoffman RP, Arslanian S, Drash AL, Becker DJ. Impaired counterregulatory hormone responses to hypoglycemia in children and adolescents with new onset IDDM. J Pediatr Endocrinol 7: 235–244, 1994. [DOI] [PubMed] [Google Scholar]
- 51.Hope KM, Tran PO, Zhou H, Oseid E, Leroy E, Robertson RP. Regulation of alpha-cell function by the beta-cell in isolated human and rat islets deprived of glucose: the “switch-off” hypothesis. Diabetes 53: 1488–1495, 2004. [DOI] [PubMed] [Google Scholar]
- 52.Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 43: 1012–1019, 2000. [DOI] [PubMed] [Google Scholar]
- 53.Inagaki N, Kuromi H, Gonoi T, Okamoto Y, Ishida H, Seino Y, Kaneko T, Iwanaga T, Seino S. Expression and role of ionotropic glutamate receptors in pancreatic islet cells. FASEB J 9: 686–691, 1995. [PubMed] [Google Scholar]
- 54.Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB. Islet β-cell secretion determines glucagon release from neighboring α-cells. Nat Cell Biol 5: 330–335, 2003. [DOI] [PubMed] [Google Scholar]
- 55.Ito K, Maruyama H, Hirose H, Kido K, Koyama K, Kataoka K, Saruta T. Exogenous insulin dose-dependently suppresses glucopenia-induced glucagon secretion from perfused rat pancreas. Metabolism 44: 358–362, 1995. [DOI] [PubMed] [Google Scholar]
- 56.Jaspan JB, Lever E, Polonsky KS, Van Cauter E. In vivo pulsatility of pancreatic islet peptides. Am J Physiol Endocrinol Metab 251: E215–E226, 1986. [DOI] [PubMed] [Google Scholar]
- 57.Johnson ML, Pipes L, Veldhuis PP, Farhy LS, Nass RM, Thorner MO, Evans WS. AutoDecon: A robust numerical method for the quantification of pulsatile events. Methods Enzymol. In press. [DOI] [PMC free article] [PubMed]
- 58.Johnson ML, Straume M. Innovative quantitative neuroendocrine techniques. In: Sex-Steroid Interactions with Growth Hormone (Serona Symposia). New York: Springer, 1999, p. 318–326.
- 59.Johnson ML, Virostko A, Veldhuis JD, Evans WS. Deconvolution analysis as a hormone pulse-detection algorithm. Methods Enzymol 384: 40–53, 2004. [DOI] [PubMed] [Google Scholar]
- 60.Kawai K, Unger RH. Inhibition of glucagon secretion by exogenous glucagon in the isolated, perfused dog pancreas. Diabetes 31: 512–515, 1982. [DOI] [PubMed] [Google Scholar]
- 61.Kieffer TJ, Heller RS, Unson CG, Weir GC, Habener JF. Distribution of glucagon receptors on hormone-specific endocrine cells of rat pancreatic islets. Endocrinology 137: 5119–5125, 1996. [DOI] [PubMed] [Google Scholar]
- 62.Klaff LJ, Taborsky GJ Jr. Pancreatic somatostatin is a mediator of glucagon inhibition by hyperglycemia. Diabetes 36: 592–596, 1987. [DOI] [PubMed] [Google Scholar]
- 63.Kleinman R, Gingerich R, Wong H, Walsh J, Lloyd K, Ohning G, De Giorgio R, Sternini C, Brunicardi FC. Use of the Fab fragment for immunoneutralization of somatostatin in the isolated perfused human pancreas. Am J Surg 167: 114–119, 1994. [DOI] [PubMed] [Google Scholar]
- 64.Kleinman R, Gingerich R, Ohning G, Wong H, Olthoff K, Walsh J, Brunicardi FC. The influence of somatostatin on glucagon and pancreatic polypeptide secretion in the isolated perfused human pancreas. Int J Pancreatol 18: 51–57, 1995. [DOI] [PubMed] [Google Scholar]
- 65.Kovatchev BP, Farhy LS, Cox DJ, Straume M, Yankov VI, Gonder-Frederick LA, Clarke WL. Modeling insulin-glucose dynamics during insulin-induced hypoglycemia. Evaluation of glucose counterregulation. J Theor Med 1: 313–323, 1999. [Google Scholar]
- 66.Leblanc H, Anderson JR, Sigel MB, Yen SS. Inhibitory action of somatostatin on pancreatic alpha and beta cell function. J Clin Endocrinol Metab 40: 568–572, 1975. [DOI] [PubMed] [Google Scholar]
- 67.Ludvigsen E, Olsson R, Stridsberg M, Janson ET, Sandler S. Expression and distribution of somatostatin receptor subtypes in the pancreatic islets of mice and rats. J Histochem Cytochem 52: 391–400, 2004. [DOI] [PubMed] [Google Scholar]
- 68.Matthews DR, Hermansen K, Connolly AA, Gray D, Schmitz O, Clark A, Orskov H, Turner RC. Greater in vivo than in vitro pulsatility of insulin secretion with synchronized insulin and somatostatin secretory pulses. Endocrinology 120: 2272–2278, 1987. [DOI] [PubMed] [Google Scholar]
- 69.Maruyama H, Hisatomi A, Orci L, Grodsky GM, Unger RH. Insulin within islets is a physiologic glucagon release inhibitor. J Clin Invest 74: 2296–2299, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meier JJ, Kjems LL, Veldhuis JD, Lefebvre P, Butler PC. Postprandial suppression of glucagon secretion depends on intact pulsatile insulin secretion: further evidence for the intraislet insulin hypothesis. Diabetes 55: 1051–1056, 2006. [DOI] [PubMed] [Google Scholar]
- 71.Oshima I, Hirota M, Ohboshi C, Shima K. Comparison of half-disappearance times, distribution volumes and metabolic clearance rates of exogenous glucagon-like peptide 1 and glucagon in rats. Regul Pept 21: 85–93, 1988. [DOI] [PubMed] [Google Scholar]
- 72.Portela-Gomes GM, Stridsberg M, Grimelius L, Oberg K, Janson ET. Expression of the five different somatostatin receptor subtypes in endocrine cells of the pancreas. Appl Immunohistochem Mol Morphol 8: 126–132, 2000. [DOI] [PubMed] [Google Scholar]
- 73.Ravier MA, Rutter GA. Glucose or insulin, but not zinc ions, inhibit glucagon secretion from mouse pancreatic alpha cells. Diabetes 54: 1789–1797, 2005. [DOI] [PubMed] [Google Scholar]
- 74.Rorsman P, Berggren PO, Bokvist K, Ericson H, Mohler H, Ostenson CG, Smith PA. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 341: 233–236, 1989. [DOI] [PubMed] [Google Scholar]
- 75.Rorsman P, Hellman B. Voltage-activated currents in guinea pig pancreatic alpha 2 cells. Evidence for Ca2+-dependent action potentials. J Gen Physiol 91: 223–242, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salehi A, Quader SS, Grapengiesser E, Hellman B. Pulses of somatostatin release are slightly delayed compared with insulin and antisynchronous to glucagon. Regul Pept 144: 43–49, 2007. [DOI] [PubMed] [Google Scholar]
- 77.Samols E, Stagner JI. Intra-islet regulation. Am J Med 85: 31–35, 1988. [DOI] [PubMed] [Google Scholar]
- 78.Samols E, Stagner JI. Islet somatostatin—microvascular, paracrine, and pulsatile regulation. Metabolism 39, Suppl 2: 55–60, 1990. [DOI] [PubMed] [Google Scholar]
- 79.Schuit FC, Derde MP, Pipeleers DG. Sensitivity of rat pancreatic A and B cells to somatostatin. Diabetologia 32: 207–212, 1989. [DOI] [PubMed] [Google Scholar]
- 80.Schuit FC, Huypens P, Heimberg H, Pipeleers DG. Glucose sensing in pancreatic beta-cells: a model for the study of other glucose-regulated cells in gut, pancreas, and hypothalamus. Diabetes 50: 1–11, 2001. [DOI] [PubMed] [Google Scholar]
- 81.Segel SA, Paramore DS, Cryer PE. Hypoglycemia-associated autonomic failure in advanced type 2 diabetes. Diabetes 51: 724–733, 2002. [DOI] [PubMed] [Google Scholar]
- 82.Singer-Granick C, Hoffman RP, Kerensky K, Drash AL, Becker DJ. Glucagon responses to hypoglycemia in children and adolescents with IDDM. Diabetes Care 11: 643–649. 1988. [DOI] [PubMed] [Google Scholar]
- 83.Stagner JI, Samols E, Marks V. The anterograde and retrograde infusion of glucagon antibodies suggests that A cells are vascularly perfused before D cells within the rat islet. Diabetologia 32: 203–206, 1989. [DOI] [PubMed] [Google Scholar]
- 84.Stagner JI, Samols E, Bonner-Weir S. Beta—-alpha—-delta pancreatic islet cellular perfusion in dogs. Diabetes 37: 1715–1721, 1988. [DOI] [PubMed] [Google Scholar]
- 85.Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin inhibits insulin and glucagon secretion via two receptors subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology 141: 111–117, 2000. [DOI] [PubMed] [Google Scholar]
- 86.Sumida Y, Shima T, Shirayama K, Misaki M, Miyaji K. Effects of hexoses and their derivatives on glucagon secretion from isolated perfused rat pancreas. Horm Metab Res 26: 222–225, 1994. [DOI] [PubMed] [Google Scholar]
- 87.Suzuki M, Fujikura K, Kotake K, Inagaki N, Seino K, Takata S. Immuno-localization of sulphonylurea receptor 1 in rat pancreas. Diabetologia 42: 1204–1211, 1999. [DOI] [PubMed] [Google Scholar]
- 88.Taborsky GJ Jr, Ahren B, Havel PJ. Autonomic mediation of glucagon secretion during hypoglycemia: implications for impaired alpha-cell responses in type 1 diabetes. Diabetes 47: 995–1005, 1998. [DOI] [PubMed] [Google Scholar]
- 89.Tapia-Arancibia L, Astier H. Glutamate stimulates somatostatin release from diencephalic neurons in primary culture. Endocrinology 123: 2360–2366, 1988. [DOI] [PubMed] [Google Scholar]
- 90.Tirone TA, Norman MA, Moldovan S, DeMayo FJ, Wang XP, Brunicardi FC. Pancreatic somatostatin inhibits insulin secretion via SSTR-5 in the isolated perfused mouse pancreas model. Pancreas 26: e67–e73, 2003. [DOI] [PubMed] [Google Scholar]
- 91.Uehara S, Muroyama A, Echigo N, Morimoto R, Otsuka M, Yatsushiro S, Moriyama Y. Metabotropic glutamate receptor type 4 is involved in autoinhibitory cascade for glucagon secretion by alpha-cells of islet of Langerhans. Diabetes 53: 998–1006, 2004. [DOI] [PubMed] [Google Scholar]
- 92.Utsumi M, Makimura H, Ishihara K, Morita S, Baba S. Determination of immunoreactive somatostatin in rat plasma and responses to arginine, glucose and glucagon infusion. Diabetologia 17: 319–323, 1979. [DOI] [PubMed] [Google Scholar]
- 93.Van Schravendijk CF, Foriers A, Van den Brande JL, Pipeleers DG. Evidence for the presence of type I insulin-like growth factor receptors on rat pancreatic A and B cells. Endocrinology 121: 1784–1788, 1987. [DOI] [PubMed] [Google Scholar]
- 94.Wang JL, McDaniel ML. Secretagogue-induced oscillations of cytoplasmic Ca2+ in single beta and alpha-cells obtained from pancreatic islets by fluorescence-activated cell sorting. Biochem Biophys Res Commun 166: 813–818, 1990. [DOI] [PubMed] [Google Scholar]
- 95.Weir GC, Leahy JL, Barras E, Braunstein LP. Characteristics of insulin and glucagon release from the perfused pancreas, intact isolated islets, and dispersed islet cells. Horm Res 24: 62–72, 1986. [DOI] [PubMed] [Google Scholar]
- 96.Wendt A, Birnir B, Buschard K, Gromada J, Salehi A, Sewing S, Rorsman P, Braun M. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes 53: 1038–1045, 2004. [DOI] [PubMed] [Google Scholar]
- 97.Xu E, Kumar M, Zhang Y, Ju W, Obata T, Zhang N, Liu S, Wendt A, Deng S, Ebina Y, Wheeler MB, Braun M, Wang Q. Intraislet insulin suppresses glucagon release via GABA-GABAA receptor system. Cell Metab 3: 47–58, 2006. [DOI] [PubMed] [Google Scholar]
- 98.Zhou H, Tran PO, Yang S, Zhang T, LeRoy E, Oseid E, Robertson RP. Regulation of alpha-cell function by the beta-cell during hypoglycemia in Wistar rats: the “switch-off” hypothesis. Diabetes 53: 1482–1487, 2004. [DOI] [PubMed] [Google Scholar]
- 99.Zhou H, Zhang T, Oseid E, Harmon J, Tonooka N, Robertson RP. Reversal of defective glucagon responses to hypoglycemia in insulin-dependent autoimmune diabetic BB rats. Endocrinology 148: 2863–2869, 2007. [DOI] [PubMed] [Google Scholar]
- 100.Zhou H, Zhang T, Harmon JS, Bryan J, Robertson RP. Zinc, not insulin, regulates the rat α-cell response to hypoglycemia in vivo. Diabetes 56: 1107–1112, 2007. [DOI] [PubMed] [Google Scholar]