

Abstract
Unilamellar vesicles or “liposomes” are commonly used as simple cell models and as drug delivery vehicles. A major limitation of unilamellar liposomes in these applications has been premature contents release in physiological environments. This premature release is likely due to enzyme degradation or protein insertion into the liposome membrane, which significantly increases the bilayer permeability. Encapsulating unilamellar liposomes within a second bilayer to form multicompartment “vesosomes” extends contents retention by two orders of magnitude by preventing enzymes and/or proteins from reaching the interior bilayers. The multicompartment structure of the vesosome can also allow for independent optimization of the interior compartments and exterior bilayer; however, just the bilayer within a bilayer structure of the vesosome is sufficient to increase drug retention from minutes to hours. The vesosome is a better mimic of eukaryotic cell structure and demonstrates the benefits of multiple internal bilayer enclosed compartments.
Keywords: liposomes, vesicles, serum, phospholipase, bilayers
Conventional unilamellar vesicles used in drug delivery and as cell models have a single compartment delimited by a single bilayer membrane, which mimics the single compartment structure of the prokaryote cell 1. This bilayer defines the interior space, regulates release of the vesicle contents, and protects the vesicle contents from degradation 2. Essentially all of the literature on vesicles and liposomes involves modifying the chemical and physical properties of this single bilayer to optimize these tasks 1, 3–7. Despite 30 years of extensive research, only doxorubicin (or the chemically similar daunorubicin) for chemotherapy 8 and amphotericin B in anti-fungal preparations are currently clinically available in liposome formulations 1. Many other drugs, such as the antibiotic ciprofloxacin and the chemotherapy agents vinorelbine, vinblastine and vincristine, leak out too rapidly from liposomes in a physiological environment 9–15, which severely limits the efficacy of unilamellar vesicles as drug carriers. While coating the exterior of vesicles with lipids functionalized with polyethylene glycol (PEG) shields against immune recognition 1, 16, and extends in vivo circulation times to 24 – 48 hours, PEG-lipids actually increase permeability and do not enhance drug retention 17, 18.
Maximizing vesicle contents retention has lead to a variety of choices of lipid headgroup, saturation of the lipid alkyl chains, and the addition of cholesterol. Many of these modifications decrease bilayer permeability by orders of magnitude in saline; however, the differences in contents retention with bilayer composition are less pronounced in serum and other relevant physiological environments 9, 10, 13–16, 19. This suggests that vesicle contents release is due to degradation of the liposome bilayer by lipases, complement, or other enzymatic processes, rather than by simple permeation 1, 16.
Slowing drug release is essential because extending the time of vesicles in the circulation results in accumulation at tumors or inflammation sites due to the mechanism known as the enhanced permeability and retention (EPR) effect 1. Rapidly growing or remodeling blood vessels near tumors, unlike those in most normal tissues, have gaps as large as 600 – 800 nm between adjacent endothelial cells through which drug carriers can extravasate 1. In addition, because tumors have impaired lymphatic drainage, the carriers concentrate in the tumor, and large increases in tumor drug concentrations could be achieved if the drug is successfully retained in the carrier 1. The benefits of the 24 – 48 hour circulation time of liposomes coated with PEG lipids1, 16 are lost if the liposome contents leak out over shorter periods of time.
A novel solution to slowing drug release is to encapsulate unilamellar liposomes within another liposome, similar to the structure of the eukaryote cell. Eukaryotes developed this structure as a successful alternative to optimizing the chemistry and physics of a single bilayer. Multiple, nested compartments, each with a distinct bilayer membrane, has proven to be a successful way to separate different cellular functions while protecting important internal contents from the environment. Mimicking these nested bilayer compartments led to the development of a multicompartment structure of unilamellar vesicles trapped within an exterior membrane called the “vesosome” (Fig. 1) 20–22. The inner compartments of the vesosome can encapsulate multiple drugs (to deliver drug cocktails 23) or have different bilayer compositions to optimize release. In addition, while it is difficult to encapsulate anything larger than molecular solutions by conventional vesicle self-assembly, the vesosome construction process lends itself to trapping colloidal particles and biological macromolecules 20–22, 24. We show here that the nested bilayer compartments of the vesosome provide a degree of freedom for optimization not possible with a single compartment and are a more realistic approximation of higher order biological organization. The vesosome extends drug retention during exposure to phospholipase A2 by orders of magnitude compared to unilamellar vesicles of the same composition. The exterior membrane provides a physical barrier against the lipase, which prevents the lipase from degrading the interior compartments. The exterior membrane also protects the interior vesicles from binding to streptavidin when the interior lipid bilayers are decorated with biotinated lipids. We extend these results to the more complex environment of serum to show that model drug release is slowed by two orders of magnitude compared to unilamellar vesicles. The physical barrier of the external membrane is extremely effective in protecting the interior compartments from degradation, thereby enhancing drug retention in physiological environments.
Figure 1.
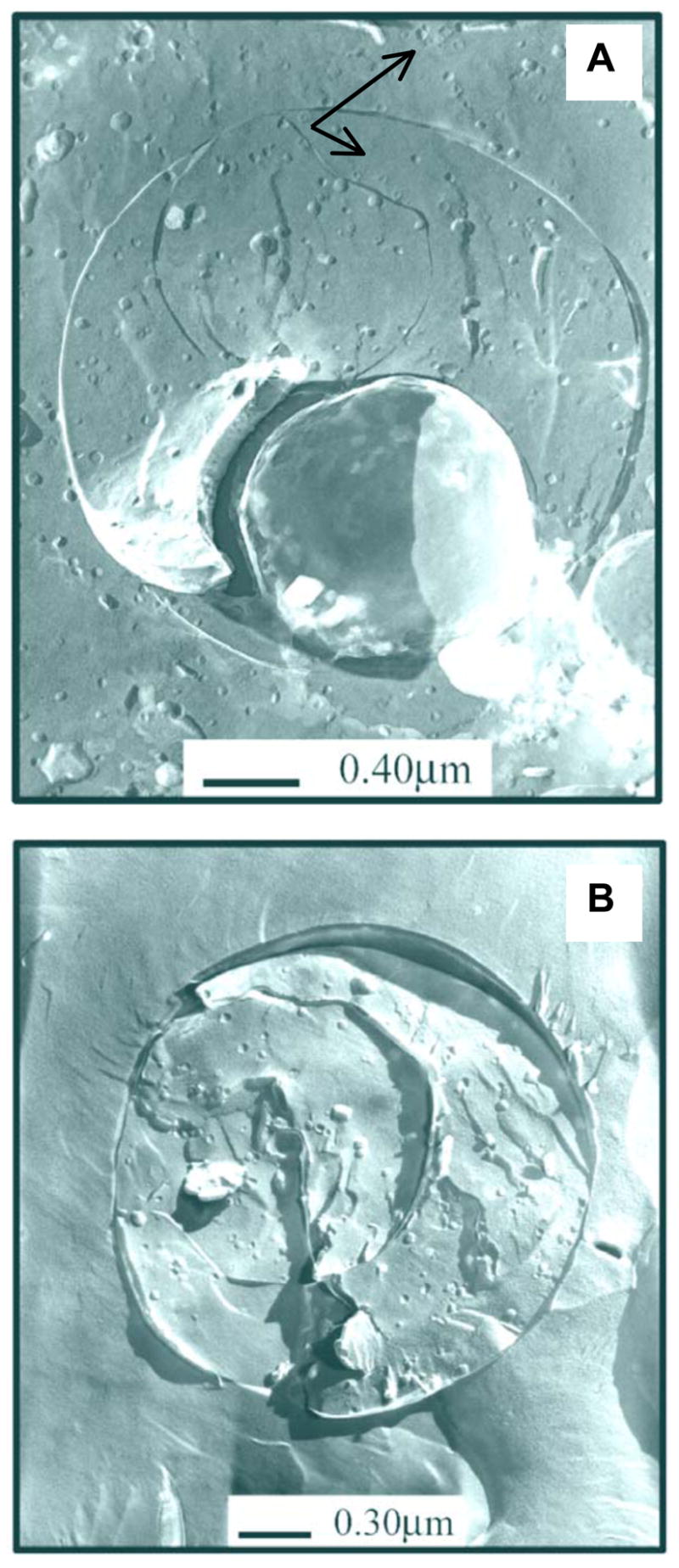
(a) Freeze-fracture TEM images of as-formed vesosome. Vesosomes consist of a single or multiple membrane shell surrounding an aqueous core that contains small, here 50 nm, unilamellar vesicles as internal compartments. The interior density of 50 nm vesicles (arrows) is the same as that outside the vesosome. (b) After one sedimentation step and two negative selections with streptavidin coated magnetic beads, there are no small vesicles outside the vesosome, but the interior density of the vesicles is the same as in (a). Freeze-fracture samples were prepared by standard techniques 32–36.
Results
To make vesosomes (or to encapsulate other colloidal suspensions), it is necessary to encapsulate vesicles efficiently, without damage, within a different membrane. In practical terms, open bilayer sheets must be formed which can then be closed without compromising the drug-carrying lipid vesicles to be encapsulated. For this purpose, we take advantage of the reversible, ethanol-induced interdigitation of saturated phosphatidylcholine bilayers. Below the main transition temperature, Tm, a number of saturated phospholipids form an LβI phase on addition of ethanol in which the all-trans acyl chains of the opposing monolayers interdigitate 21, 24, 25. This interdigitation increases the membrane rigidity, and the increased curvature energy leads to the formation of bilayer sheets, 21, 24 rather than closed vesicles. Heating above Tm causes the acyl chains to melt and the membrane reverts to a disordered, Lα bilayer phase. The reduced bending rigidity of the Lα phase allows the bilayers to close; in the process, small vesicles, colloidal particles, essentially anything of nanometer dimensions in the original suspension can be encapsulated 21 as shown in Fig. 1.
The fraction of biotinated vesicles encapsulated in vesosomes was determined by incubating the vesosomes with BODIPY-labeled avidin. The green fluorescence intensity of the BODIPY-labeled avidin (λex = 504nm, λem = 512nm) is linearly proportional to the number of biotin-lipids bound; this allows the direct quantification of the biotin-lipids accessible to the avidin on the unencapsulated vesicles 26, 27. The total amount of lipid in the vesicles in the suspension was determined after micellization with reduced Triton X-100 (RTX-100), by measuring the red fluorescence of the b-BODIPY 581/591 C5-HPC incorporated in the bilayers of the vesicles (λex = 582nm, λem = 593nm) against calibration samples.
Table 1 shows that vesosome formation results in 58% of the biotinylated vesicles not binding to the fluorescently labeled avidin. These vesicles were encapsulated and effectively shielded from binding to avidin by the exterior bilayer of the vesosome. This encapsulated fraction is reasonable in that a random packing of monodisperse spheres occupies a volume fraction of 56 – 64%, depending on the details of the packing 28; polydispersity increases the encapsulated volume fraction. Encapsulation via the vesosome process is passive – the fraction of vesicles encapsulated is similar to the expected volume fraction of the random packing of polydisperse spheres (Fig. 1). Our results show that avidin, which is a 66kDa protein, cannot cross the exterior vesosome membrane; the absence of binding to 58% of the biotinylated vesicles also confirms that the interior vesicle bilayers do not fuse with the exterior vesosome membrane over the course of the experiment. Fig. 1a shows TEM images of the as-formed vesosome suspension; a roughly equal density of small vesicles is trapped within the vesosome as outside the vesosome. The TEM images also show that the interior vesicles are distinct and do not fuse with the exterior membrane 21, 24, 29.
Table 1.
Vesicle encapsulation fraction for different vesicle concentrations in the original suspension. 0.5 ml of DPPC 50 nm vesicles (labeled with 0.02mol% b-BODIPY 581/591 C5-HPC, 0.1mole% DHPE Biotin-X) at different concentrations were mixed with 25mg of interdigitated DPPC bilayers and heated at 46°C for 20 minutes to form vesosomes. Separation of unencapsulated vesicles from the vesosomes was carried out by one sedimentation step, refined by two BioMag negative selections in which the biotinylated vesicles were bound to streptavidin coated magnetic particles and physically removed from the suspension with an external magnet.
| Vesicle concentration | Raw Vesosomes | 1 Sedimentation | 1 Sedimentation + 2 Biomag |
|---|---|---|---|
| 100mg/mL | 53.1% ± 7.1% | 89.0% ± 3.8% | 99.0% ± 0.6% |
| 50mg/mL | 62.2% ± 3.2% | 87.5% ± 1.8% | 99.0% ± 0.5% |
While a mixed population of vesicles and vesosomes might be useful in certain applications, to distinguish release properties requires the quantitative separation of the unencapsulated vesicles from the vesosome suspension. This (Table 1) is achieved by diluting the vesosome suspension, typically 1mL in volume, in 125mL buffer, and allowing the vesosomes to sediment overnight, followed by collection of the sedimented material. The method takes advantage of the size difference between the vesosomes, which have an average diameter of about a micron, and the unencapsulated 50 nm vesicles. After a single sedimentation step, the encapsulated fraction increases to 88% (Table 1).
If required, the separation can be refined by a second sedimentation step or a negative selection procedure that also takes advantage of the barrier properties of the vesosome exterior membrane. Mixing the once-refined vesosome suspension with streptavidin-coated magnetic particles causes any unencapsulated, biotinylated vesicles to bind to the magnetic particles. The magnetic particles are then physically separated from the suspension with an external magnet, leaving behind the purified vesosome suspension for collection. After two negative selection steps, no fluorescence increase relative to the biotin-free control was observed on addition of BODIPY-labeled avidin; hence, there were no detectable unencapsulated vesicles in the purified vesosome solution. Freeze-fracture TEM images (Fig. 1b) confirmed the absence of unencapsulated vesicles in the suspension 29, while showing that the vesosomes retained the interior vesicles.
Vesosome and Vesicle Contents Release
The external membrane that protects the biotinylated lipids on the interior compartments from binding to the streptavidin on the magnetic particles also protects the interior contents of the vesosomes from active degradation by lipases. To quantify this protective effect, carboxyfluorescein release from unilamellar vesicles and from identical vesicles encapsulated within a vesosome of the same membrane composition are compared in Fig 2. DPPC vesicles were formed by extrusion in 20mM CF and the external CF was quantitatively removed by size exclusion chromatography. A fraction of these CF containing vesicles were then encapsulated within a DPPC membrane to form vesosomes and purified by repeated sedimentation as described earlier. At the concentration initially present in the vesicles, CF fluorescence is quenched; however any released CF is diluted in the external solution and the fluorescence is proportional to the CF solution concentration. Measuring the solution fluorescence with time gives the release profile: %release = (I(t)-I(0))/(Itot-I(0)) where I(t) is the fluorescence intensity at λex = 492nm, λem = 517nm, at time t after exposure to the lipase, Itot is the sample total fluorescence as determined after micellization by RTX-100 and I(0) is the solution fluorescence at time zero.
Figure 2.
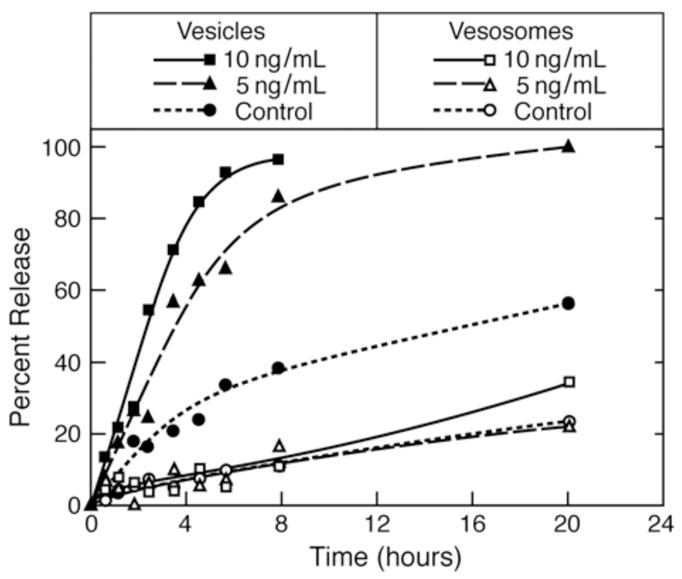
Porcine Pancreas Phospholipase A2-induced release of carboxyfluorescein (CF) from unilamellar vesicles (filled symbols) and vesosomes (open symbols for control (no PLA2), 5ng/mL and 10ng/mL PLA2 at 37°C. The unilamellar vesicles were quickly degraded by the lipase and CF was released in a dose-dependent fashion much faster than the control vesicles. Release from the vesosomes was not increased by the lipase and there was no significant difference with release from the control vesosomes. Lines are a guide to the eye.
Phospholipase A2 (Porcine pancreatic PLA2, Sigma, St. Louis, Mo) catalyzes the hydrolysis of the ester linkage of the sn-2 tail of phospholipids in the presence of calcium ions, producing lysophospholipids and fatty acids. This yields a rapid increase in membrane permeability and subsequent loss of the vesicle contents (Fig. 2) 30. Unilamellar vesicles and vesosomes samples were diluted with a calcium-containing buffer (85 mM NaCl, 50 mM TES, 10 mM CaCl2, pH 7.4) to 0.1 mg/mL total lipid. PLA2 (3ug/mL stock solution) was added to the samples at the desired concentrations, and the same volume of buffer was added to the control samples. The initial fluorescence I(0) was measured, each sample was divided into a series of 1mL aliquots which were then incubated at 37°C in a water bath to induce PLA2 enzymatic activity. At different times up to 20 hours, aliquots were collected and their fluorescence measured, producing the PLA2-induced release profiles shown in Figure 2. The control vesicle release profile gives the permeability of the DPPC vesicles at 37°C; the half-life for release is > 20 hours. PLA2 increases the CF release in a concentration-dependent manner, reducing the half-life for release to ~ 3 hours for 5 ng/ml PLA2 and to ~1.5 hour for 10 ng/ml PLA2.
The multicompartment structure of the vesosome more than doubles the half-life for release without PLA2. More importantly, PLA2 does not significantly increase CF release from vesosome. After 20 hours of exposure to PLA2 at 37°C, the vesosomes still retain the same amount of CF as the control vesosomes. This indicates that the PLA2 cannot traverse the exterior bilayer of the vesosome to degrade the internal vesicles during the time of the experiment. While PLA2 increases the permeability of the small molecule CF (molecular mass of 376 Da) from unilamellar vesicles, apparently it takes much longer for PLA2 (molecular mass of 14 kDa) itself to cross the exterior lipid bilayer of the vesosome 24. While the exterior membrane of the vesosome is likely permeablized to small molecules, it retains its ability to prevent the enzyme from interacting with the interior compartments. The compartment within a compartment structure of the vesosome is sufficient to eliminate the effects of the lipase on CF release even though the chemical composition of the interior or exterior bilayers of the vesosome are the same as that of the vesicles.
Serum is a more complex and biologically relevant medium containing a variety of proteins, enzymes and molecules that can interact with lipid membranes. Vesicles and vesosomes with 20 mM internal CF were diluted to 0.1mg/mL lipid in different mixtures of newborn calf serum with buffer. The initial fluorescence I(0), and total fluorescence Itot were determined and the remainder of each sample was divided into a series of 1mL aliquots which were incubated at 37°C in a water bath. There was more variation between different experiments in serum than PLA2; different batches of serum were more or less active in enhancing CF release. The data presented are representative experimental runs; in all cases, identical trends were seen between batches of serum.
Figure 3 shows the release profile from unilamellar vesicles in different serum/buffer mixtures. Serum induced faster release from the control vesicles in buffer; the release rate increased with increasing serum fraction. The total release from the vesicles at 10 hours was proportional to the fraction of serum, suggesting that there was a limiting amount of whatever serum component that enhanced lipid degradation 16. Figure 4 shows that the corresponding release rates from vesosome samples were much slower than those from unilamellar vesicles. After 12 hours, vesosomes released ~ 20% of the CF, independent of the serum/buffer ratio, while the unilamellar vesicles released 95% when exposed to 75% serum. Not only does the vesosome structure increase serum stability, but it also protects in a serum concentration independent manner, indicating that the serum components have not been able to traverse the external membrane to reach the contents of the vesosome within 12 hours.
Figure 3.

Calf serum-induced release at 37°C of carboxyfluorescein from unilamellar DPPC vesicles after mixing with various fractions of buffer. Serum induced rapid release from the vesicles compared to controls in a dose-dependent fashion. The release rate appeared to slow at longer times, suggesting that the enzymes or lipases responsible for the increased release rates were being consumed during the process. Lines are a guide to the eye.
Figure 4.
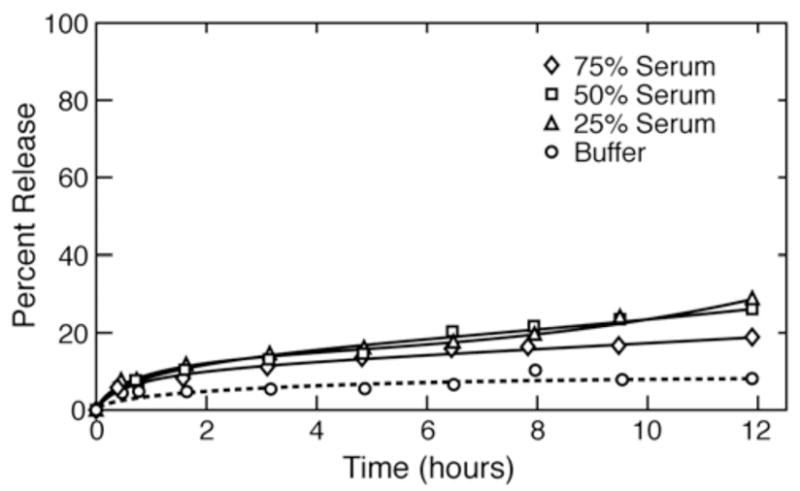
Calf serum-induced release at 37°C of carboxyfluorescein from DPPC vesosomes after mixing with various fractions of buffer. Serum did not increase the release rate from the vesosomes within the time of this experiment compared to controls. The multicompartment vesosome structure is responsible for the decrease in release as the interior vesicles and exterior bilayer of the vesosome were made from DPPC, the same lipid that was used in the vesicles in Fig. 3. Lines are a guide to the eye.
In a second series of experiments, CF release into serum/buffer mixtures was followed for an extended period (Fig. 5). The vesosome structure extended the half-life for release from vesosomes in serum to more than 50 hours, and there appears to be a delay in release for ~ 10 hours before any significant amount of CF is released. There is some variability between serum samples in promoting release from the vesosomes; however, the differences in release between vesicles and vesosomes were significant for all serum samples examined. The serum components responsible for degrading the bilayers and increasing permeability were effectively shielded from the interior compartments for this initial lag period.
Figure 5.
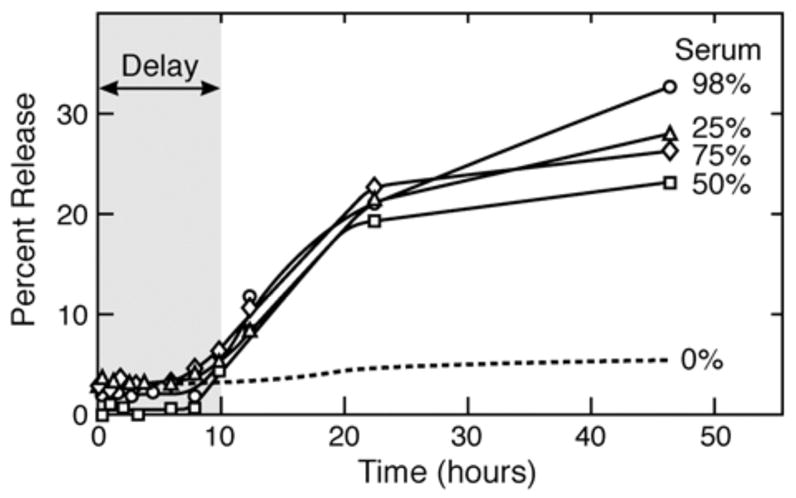
Calf serum-induced release at 37°C of CF from DPPC vesosomes followed for 48 hours. As in Fig. 4, there is an initial delay of release during which the exterior vesosome membrane effectively shields the internal compartments from degradation by serum. At later times, the rate of release increases, but is still significantly slower than that from unilamellar vesicles (Fig. 3). Over 70% of the CF is retained for 48 hours in the vesosome. Lines are a guide to the eye.
Discussion
Previous work 9, 10, 13–16, 19 has shown that modifying the composition of unilamellar vesicles has little effect on contents release in physiological environments. However, adding a second barrier to the lipases and serum components responsible for premature release significantly increases the half-life of carboxyfluorescein in PLA2 and in serum. This shows the benefits of the eukaryotic-mimic structure of the vesosome. The protection afforded the interior compartments by the exterior membrane is made obvious by (1) the inability of streptavidin or avidin to bind to the interior compartments, which we have used as a negative selection step in vesosome purification; and (2) by the absence of a permeability increase in the vesosome after exposure to phospholipase A2 and serum. The additional bilayer membrane provides a physical barrier to direct interaction with the external medium and also efficiently delays CF leakage.
We expect that varying the internal vesicle membrane composition could further optimize contents release in serum, and perhaps help identify the components of serum responsible for the membrane degradation that leads to premature drug release. For example, sphingomyelin based internal capsules would be resistant to phospholipase degradation even after the lipase degraded the external barrier membrane. This encapsulation-protection concept could easily be applied to any colloidal delivery system that suffers from early degradation-induced drug leakage. The vesosome also offers the flexibility to deliver multiple drugs within a single carrier, which has been shown to offer important advantages in chemotherapy 23. The vesosome structure also shows a potentially important advantage enjoyed by eukaryotes – the internal compartmentalization protects important organelles from interacting with a hostile environment.
Materials and Methods
DPPC was purchased from Avanti Polar Lipids (Alabaster, Al) and used as received. Porcine pancreatic PLA2 and Triton X-100 (RTX-100) were from Sigma (St. Louis, Mo). Carboxyfluorescein, BODIPY 581/591 C5-HPC, DHPE Biotin-X and fluorescently labeled avidin, (BODIPY FL) were from Invitrogen Molecular Probes (US). Bio-Mag particles labeled with streptavidin were from Qiagen (Valencia, Ca). Size exclusion chromatography was done with PD-10 columns (Amersham Biosciences) pre-packed with 8.3 ml bed volume of Sephadex G-25 between two sintered polyethylene frits of pore sizes 20 –85 microns. 50 nm diameter DPPC vesicles were prepared by standard extrusion methods using a Lipex Biomembranes Extruder (Vancouver, Canada).
Vesosomes were made by first inducing the transition from the normal (Lβ′) bilayer phase to the interdigitated (LI) bilayer phase by dropwise addition of 0.106 mL of ethanol (3 molar net ethanol concentration) to 0.5mL of a 50 mg/ml DPPC vesicle suspension, under stirring, at room temperature 31. The initially bluish vesicle suspension turns milky white and its viscosity increases significantly. After 30 minutes of stirring, the interdigitated sheets were twice washed of excess ethanol by adding 4mL of buffer, followed by centrifugation and pellet collection. The interdigitated sheets are stable to the removal of the ethanol and replacement with buffer as long as T < Tm 29, 31. The DPPC vesicles to be encapsulated were labeled with 0.02mol% of the fluorescent dye b-BODIPY 581/591 C5-HPC and 0.1mole% of the specific receptor DHPE Biotin-X. Vesosomes with two different interior vesicle concentrations were prepared by mixing 0.5mL of either 100mg/mL or 50mg/mL of the labeled vesicles with the washed interdigitated sheets. The mixture was then heated at 46°C – above the 41°C main transition temperature of DPPC - for 20 minutes under gentle stirring, driving the sheets to close around the vesicles in suspension to form the vesosomes.
Freeze-fracture samples of the vesosomes were prepared by first depositing a film of sample liquid 100 microns thick between two copper planchettes. The samples were frozen by plunging the sample into a liquid propane/liquid ethane bath cooled by liquid nitrogen. The frozen sample was transferred under liquid nitrogen to the sample block of a JEM Cryofract freeze-fracture device. After temperature (−170 °C) and pressure (< 10−7 torr) equilibration, the sample was fractured and the two resulting surfaces were replicated with 1.5 nm platinum deposited at a 45° angle, followed by 15 nm of carbon deposited normal to the surface. The copper planchettes were dissolved in a mixture of chromic acid, sulfuric acid and water, and then the replicas were washed in water and allowed to stand in ethanol several days to dissolve any remaining lipid. The replicas were collected on formvar-coated TEM grids (Ted Pella, Redding, California) 32–36. A Gatan CCD camera was used to record digital bright field images using a FEI Technai transmission electron microscope at 100 KeV.
Acknowledgments
NSF Grant CTS-0436124 and a NIH Program of Excellence in Nanotechnology Grant HL080718 supported this work: Nanotherapy for Vulnerable Plaques
Footnotes
This information is available free of charge via the internet at http://pubs.acs.org.
Supporting Information: There is no supporting information for this paper.
References
- 1.Allen TM, Cullis PR. Drug Delivery Systems: Entering the Mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 2.Lasic DD. Liposomes: From Physics to Applications. Elsevier; Amsterdam: 1993. [Google Scholar]
- 3.Discher BM, Won YY, Ege DS, Lee JM, Bates FS, Hammer D. Polymersomes: Tough Vesicles Made from Diblock Copolymers. Science. 1999;284:1143–1146. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- 4.Barauskas J, Johnsson M, Tiberg F. Self-Assembled Lipid Superstructures: Beyond Vesicles and Liposomes. Nano Letters. 2005;5:1615–1619. doi: 10.1021/nl050678i. [DOI] [PubMed] [Google Scholar]
- 5.Discher DE, Ahmed F. Polymersomes. Annu Rev Biomed Eng. 2006;8:323–341. doi: 10.1146/annurev.bioeng.8.061505.095838. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Granick S. How to Stabilize Phospholipid Liposomes (Using Nanoparticles) Nano Letters. 2006;6:694–698. doi: 10.1021/nl052455y. [DOI] [PubMed] [Google Scholar]
- 7.Zelikin AN, Becker AL, Johnston APR, Wark KL, Turatti F, Caruso F. A General Approach for DNA Encapsulation in Degradable Polymer Microcapsules. ACS Nano. 2007;1(1):63–69. doi: 10.1021/nn700063w. [DOI] [PubMed] [Google Scholar]
- 8.Abraham SA, Waterhouse DN, Mayer LD, Cullis PR, Madden TD, Bally MB. The Liposomal Formulation of Doxorubicin. Methods in Enzymology. 2005;391:71–97. doi: 10.1016/S0076-6879(05)91004-5. [DOI] [PubMed] [Google Scholar]
- 9.Maurer N, Wong KF, Hope MJ, Cullis PR. Anomalous Solubility Behavior of the Antibiotic Ciprofloxacin Encapsulated in Liposomes: A H-Nmr Study. Biochimica et Biophysica Acta. 1998;1374:9–20. doi: 10.1016/s0005-2736(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 10.Maurer-Spurej E, Wong KF, Maurer N, Fenske DB, Cullis PR. Factors Influencing Uptake and Retention of Amino-Containing Drugs in Large Unilamellar Vesicles Exhibiting Transmembrane Ph Gradients. Biochimica et Biophysica Acta. 1999;1416:1–10. doi: 10.1016/s0005-2736(98)00204-1. [DOI] [PubMed] [Google Scholar]
- 11.Bakker-Woudenberg I, ten Kate MT, Guo L, Working P, Mouton JW. Improved Efficacy of Ciprofloxacin Administered in Poyethylene Glycol-Coated Liposomes for Treatment of Klebisiella Pneumoniae Pneumonia in Rats. Antimicrobial Agents and Chemotherapy. 2001;45:1487–1492. doi: 10.1128/AAC.45.5.1487-1492.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakker-Woudenberg I, Schiffelers RM, Storm G, Becker MJ, Guo L. Long-Circulating Sterically Stabilized Liposomes in the Treatment of Infections. Methods in Enzymology. 2005;391:228–260. doi: 10.1016/S0076-6879(05)91014-8. [DOI] [PubMed] [Google Scholar]
- 13.Webb MS, Boman NL, Wiseman DJ, Saxon D, Sutton K, Wong KF, Logan P, Hope MJ. Antibacterial Efficacy against an in Vivo Salmonella Typimurium Infection Model and Pharmacokinetics of a Liposomal Ciprofloxacin Formulation. Antimicrobial Agents and Chemotherapy. 1998;42:45–52. doi: 10.1128/aac.42.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhigaltsev IV, Maurer N, Akhong QF, Leone R, Leng E, Wang J, Semple SC, Cullis PR. Liposome-Encpasulated Vincristine, Vinblastine and Vinorelbine: A Comparitive Study of Drug Loading and Retention. J Controlled Release. 2005;104:103–111. doi: 10.1016/j.jconrel.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Zhigaltsev IV, Maurer N, Edwards K, Karlsson G, Cullis PR. Formation of Drug-Arylsulfonate Complexes inside Liposomes: A Novel Approach to Improve Drug Retention. J Controlled Release. 2006;110:378–386. doi: 10.1016/j.jconrel.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 16.Auguste DT, Prud’homme RK, Ahl PL, Meers P, Kohn J. Association of Hydrophobically Modified Poly(Ethylene Glycol) with Fusogenic Liposomes. Biochimica et Biophyisca Acta. 2003;1616:184–195. doi: 10.1016/j.bbamem.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 17.Webb MS, Saxona D, Wonga F, Lima H, Wang Z, Bally M, Choi L, Cullis P, Mayer L. Comparison of Different Hydrophobic Anchors Conjugated to Poly(Ethylene Glycol): Effects on the Pharmacokinetics of Liposomal Vincristine. Biochimica et Biophyisca Acta. 1998;1372:272–282. doi: 10.1016/s0005-2736(98)00077-7. [DOI] [PubMed] [Google Scholar]
- 18.Johnsson M, Edwards K, Liposomes Disks. Spherical Micelles: Aggregate Structure in Mixtures of Gel Phase Phosphatidylcholines and Poly(Ethylene Glycol) Phospholipids. Biophysical Journal. 2003;85:3839–3847. doi: 10.1016/S0006-3495(03)74798-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Needham D, Dewhirst MW. The Development and Testing of a New Temperature-Sensitive Drug Delivery System for the Treatment of Solid Tumors. Advanced Drug Delivery Reviews. 2001;53:285–305. doi: 10.1016/s0169-409x(01)00233-2. [DOI] [PubMed] [Google Scholar]
- 20.Walker SA, Kennedy MT, Zasadzinski JA. Vesicles inside Vesicles: A Simple Method of Bilayer Encapsulation by Self-Assembly. Nature. 1997;387:61–64. doi: 10.1038/387061a0. [DOI] [PubMed] [Google Scholar]
- 21.Kisak ET, Coldren B, Zasadzinski JA. Nanocompartments Enclosing Vesicles, Colloids and Macromolecules Via Interdigitated Lipid Bilayers. Langmuir. 2002;18(1):284–288. [Google Scholar]
- 22.Evans CA, Zasadzinski JA, Encapsulating Vesicles. Colloids in Cochleate Cylinders. Langmuir. 2003;19:3109–3113. [Google Scholar]
- 23.Sengupta S, Eavarone D, Capila I, Zhao G, Watson N, Kiziltepe T, Sasisekharan R. Temporal Targeting of Tumour Cells and Neovasculature with a Nanoscale Delivery System. Nature. 2005;436:568–572. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 24.Kisak ET, Coldren B, Evans CA, Boyer C, Zasadzinski JA. The Vesosome- a Multicompartment Drug Delivery Vehicle. Current Medicinal Chemistry. 2004;11:199–219. doi: 10.2174/0929867043456197. [DOI] [PubMed] [Google Scholar]
- 25.Ahl PL, Chen L, Perkins WR, Minchey SR, Boni LT, Taraschi TF, Janoff AS. Interdigitation-Fusion: A New Method for Producing Lipid Vesicles of High Internal Volume. Biochimica et Biophysica Acta. 1994;1195:237–244. doi: 10.1016/0005-2736(94)90262-3. [DOI] [PubMed] [Google Scholar]
- 26.Emans N, Biwersi J, Verkman AS. Imaging of Endosome Fusion in Bhk Fibroblasts Based on a Novel Fluorimetric Avidin-Biotin Binding Assay. Biophysical Journal. 1995;69:716–728. doi: 10.1016/S0006-3495(95)79947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kisak E, Kennedy MT, Trommeshauser D, Zasadzinski JA. Self-Limiting Aggregation by Controlled Ligand-Receptor Stoichiometry. Langmuir. 2000;16:2825–2831. [Google Scholar]
- 28.Donev A, Cisse I, Sachs D, Variano EA, Stillinger FH, Connelly R, Torquato S, Chaikin PM. Improving the Density of Jammed Disordered Packings Using Ellipsoids. Science. 2004;303:990–993. doi: 10.1126/science.1093010. [DOI] [PubMed] [Google Scholar]
- 29.Boyer C. Ph D. University of California; Santa Barbara, Santa Barbara: 2005. Development and Characterization of a Liposome-Based Delivery Vehicle. [Google Scholar]
- 30.Jorgensen K, Davidsen J, Mouritsen OG. Biophysical Mechanisms of Phospholipase A2 Activation and Their Use in Liposome-Based Drug Delivery. FEBS Letters. 2002;531:23–27. doi: 10.1016/s0014-5793(02)03408-7. [DOI] [PubMed] [Google Scholar]
- 31.Ahl PL, Perkins WR, Interdigitation-Fusion Liposomes. Methods in Enzymology. 2003;367:80–98. doi: 10.1016/S0076-6879(03)67007-2. [DOI] [PubMed] [Google Scholar]
- 32.Zasadzinski JA, Bailey SM. Applications of Freeze-Fracture Replication to Problems in Materials and Colloid Science. J Electron Microsc Technique. 1989;13:309–334. doi: 10.1002/jemt.1060130406. [DOI] [PubMed] [Google Scholar]
- 33.Zasadzinski JA, Scriven LE, Davis HT. Liposome Structure and Defects. Philosophical Magazine A. 1985;51:287–302. [Google Scholar]
- 34.Keller SL, Boltenhagen P, Pine DJ, Zasadzinski JA. Direct Observation of Shear Induced Structures in Wormlike Micellar Solutions by Freeze-Fracture Electron Microscopy. Physical Review Letters. 1998;80:2725–2728. [Google Scholar]
- 35.Spector MS, Naranjo E, Chiruvolu S, Zasadzinski JA. Conformations of a Tethered Membrane - Crumpling in Graphitic Oxide. Phys Rev Lett. 1994;73:2867–2870. doi: 10.1103/PhysRevLett.73.2867. [DOI] [PubMed] [Google Scholar]
- 36.Spector MS, Zasadzinski JA, Sankaram MB. Topology of Multivesicular Liposomes, a Model Biliquid Foam. Langmuir. 1996;12:4704–4708. [Google Scholar]