

Abstract
8-Vinyl-2’-deoxyadenosine (8vdA) is a fluorophore with a quantum yield comparable to 2-aminopurine nucleoside. 8vdA was incorporated into a 10-mer stem-tetraloop RNA (8vdA-10) structure to characterize the properties of the base, 8-vinyladenine (8-vA), with respect to adenine as a substrate or inhibitor for ribosome inactivating proteins. Ricin Toxin A-chain (RTA) and Pokeweed Antiviral Protein (PAP) catalyze the release of adenine from a specific adenosine on a stem-tetraloop (GAGA) sequence at the elongation factor (eEF2) binding site of the 28S subunit of eukaryotic ribosomes, thereby arresting translation. RTA does not catalyze 8-vinyladenine release from 8vdA-10. Molecular dynamics simulations implicate a role for Arg 180 in oxacarbenium ion destabilization and lack of catalysis. However, 8vdA-10 is an active site analog and inhibits RTA with a Ki value of 2.4 μM. Adenine is also released from the second adenosine in the modified tetraloop demonstrating an alternative mode for the binding of this motif in the RTA active site. The 8vdA analog defines the specificities of RTA for the two adenylate depurination sites in an RNA substrate with a GAGA tetraloop. The rate of non-enzymatic acid-catalyzed solvolysis of 8-vinyladenine from the stem-loop RNA is described. Unlike RTA, PAP catalyzes the slow release of 8-vinyladenine from 8vdA-10. The isolation of 8-vA and its physicochemical characterization is described.
Introduction
Ricin is a protein found in castor beans and it inhibits protein synthesis in eukaryotes by depurination of ribosomes at a site where elongation factors bind during translation (1-4). The B-chain of this heterodimeric protein is a lectin that directs the catalytic A-chain to the cell surface. Disulfide bond cleavage releases the A-chain which is then internalized via endocytosis (5). Once inside the cell, retrograde transport shuttles the protein to the endoplasmic reticulum (6). The A-chain is then translocated into the cytosol wherein it binds to a GAGA nucleotide sequence on the sarcin-ricin tetraloop (SRL) of the 28S ribosomal RNA and specifically depurinates the first adenosine (A4324 on the rat ribosome; GA4324GA in the 5’→3’ direction) (Figure 1). The kcat for RTA activity on rat liver ribosomes is ~1800 min-1 and a single molecule can kill a eukaryotic cell (7). A dose of ~1-2 μg/kg is lethal for dogs. The potency of RTA has been exploited in the assassination of Georgi Markov (8) and an attempt on Alexander Solzhenitsyn’s life (9) being the most infamous cases. With a typical content of 105-107 ribosomes, depurination in a rat hepatocyte would be complete in ~40 hrs which is consistent with the rate of fatal ricin poisoning. RTA more recently has also been used as a bioterrorism agent. Inhalation of aerosolized ricin causes rapid and irreversible pulmonary dysfunction. Reliable means of detection (10) and the development of antidotes (11) are important areas of research for this toxin.
Figure 1.
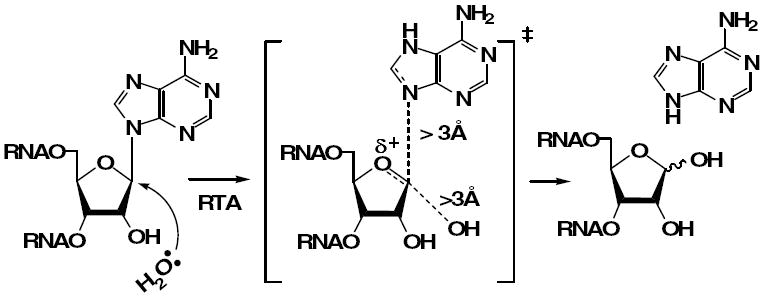
The hydrolytic cleavage of an adenosine moiety catalyzed by RTA proceeds through an oxacarbenium ion transition state.
The activity of RTA on small 10-14mer stem-tetraloop RNA substrates at acidic pH has been demonstrated (12) and the Kcat for the hydrolysis of 2’-deoxyadenosine in a hybrid GdAGA tetraloop was determined to be ~16 fold faster than that for an adenosine tetraloop (13). Kinetic isotope effects (KIEs) on the hydrolysis of an appropriately radiolabeled 10-mer RNA substrate indicate that the reaction proceeds through a dissociative transition state (TS) (14). TS formation at the RTA active site involves leaving group activation, oxacarbenium ion formation and generation of the insipient water nucleophile. Leaving group activation is the major driving force for catalysis. We hypothesized that the incorporation of 8-vinyl-2’-deoxyadenosine into an RNA 10-mer structure could provide a useful fluorescent substrate or an inhibitor of RTA. It is likely that the vinyl moiety, with its ability to delocalize electrons, makes 8-vA a better leaving group than adenine. If 8-vA was released, the reaction would become amenable to direct spectroscopic characterization and thus yield a continuous assay system. If not a substrate, an RNA construct with 8-vA could inhibit RTA, and provide useful information in the design of inhibitors. The following sections describe the incorporation of the vinyl nucleoside into RNA, the binding of the modified stem-loop to RTA and a molecular dynamics interpretation of the interaction.
Experimental Section
Materials and General Experimental
RTA was purchased from Sigma. Pokeweed Antiviral Protein was purchased from Worthington Biochemicals. Calf intestinal alkaline phosphatase was from Promega (Madison, WI), and phosphodiesterase I was from Sigma Chemical Co. (St. Louis, MO). RNase inhibitors and RNase-free DNase were from Ambion (Austin, TX). Nucleoside phosphoramidites and other reagents for oligoribonucleotide synthesis were purchased from Glen Research (Sterling, VA), except for phosphoramidites attached directly to CPG solid supports, which were obtained from ChemGene Corp. (Ashland, MA). All other chemicals were purchased from Aldrich Chemical Co. (Milwaukee, WI) and were of the highest purity available. These reagents were used without further purification. Purification of reaction intermediates of the phosphoramidite synthetic pathway was completed by flash column chromatography using Merck silica gel 60 (230-400 mesh). Purification by HPLC was performed on a Waters 626 pump with either a 996 photodiode array detector or a 487 dual wavelength detector and using the Millennium software package. RNA concentrations were determined by UV-Vis measurements using a Cary 100 or 300 diode array spectrophotometer from Varian.
Synthesis of the 8-vinyl-2’-deoxyadenosine phosphoramidite
The synthesis of the vinyl phosphoramidite was carried out as reported by Mély et al. (15). This phosphoramidite can be purchased from Berry and Associates, Inc. However, at the beginning of our work and through the completion of the synthesis, this product was not commercially available.
Synthesis and Purification of 8vdA-10
The oligonucleotide was chemically synthesized using a phosphoramidite methodology on an Expedite 8900 Nucleic Acid Synthesis System from Perseptive Biosystems. Syntheses were carried out on a 1 μmol scale with coupling times extended to 15 min, in the trityl-off mode. The coupling times for guanosine and 8-vinyl-2’-deoxyadenosine were extended to 40 min. Deprotection of the products was accomplished using a concentrated NH4OH/ethanol mixture (3:1, v/v) and triethylamine trihydrofluoride as described by Chen et al. (12) All products were purified by HPLC using a Waters XTerra Prep MS C18 column (7.8 mm × 50 mm). These columns are specially designed for purification of oligonucleotides in the trityl-off mode. An ion pairing mobile phase of 50 mM triethylammonium carbonate (pH 6.4) with a gradient from 3 to 80% of 50% methanol was employed for elution. The homogeneity of the product was confirmed by ion exchange chromatography on a Protein-Pak Q-15 HR DEAE column eluting with 50 mM ammonium acetate (pH 5.0) containing 15% methanol and 1.0 M LiCl. The nucleotide composition was confirmed by enzymatic digestion of a 50 μL aliquot of a 2.5 μM solution of the oligonucleotide with 1 unit each of calf intestinal alkaline phosphatase and snake venom phosphodiesterase I overnight at 37 °C, and quantitation of the released nucleosides relative to known standards by HPLC on a Waters Delta-Pak C18 analytical column (3.9 mm × 300 mm) eluted with 50 mM ammonium acetate (pH 5.0) containing 5% methanol. Nucleoside and deoxynucleoside standards were used to determine the relative amounts of each component in the digest. Product formation was further confirmed by MALDI-TOF mass spectrometry analysis on an Applied Biosystems 4115 Voyager system. Masses were acquired in the 500-5000 Da range.
Synthesis and Purification of 8-Vinyladenine
8-Vinyladenine was isolated from an intermediate of the phosphoramidite synthesis pathway. Thus, a solution of 25 mgs of 5’-dimethoxytrityl-6-N,N-dimethylformamidino-8-vinyl-2’-deoxyadenosine in 10 ml of 15% dichloroacetic acid in dichloromethane was allowed to stir at room temperature for 3 hrs. The solvent was then removed in-vacuo and the residue was dissolved in 5 ml of a 1:1 solution of triethylamine in methanol and stirred at room temperature for 24 hrs. The solvent was then removed in-vacuo and the crude material was purified by HPLC on a Waters C18 Deltapak analytical column (3 mm × 300 mm) eluting with 50mM ammonium acetate (pH 5.0) and a gradient of 3 to 80% of 50% methanol over 40 minutes. The collected fractions were pooled, concentrated and finally desalted using a C18 zip-tip prior to ESI mass analysis in the negative mode. 1H NMR (CD3OD, 300 MHz): δ 5.9 (d, J=11.3 Hz, 1H, cis-vinyl), 6.45 (d, J=17.7 Hz, 1H, trans-vinyl), 6.95 (dd, J=11.3 Hz, J=17.7 Hz, 1H, vinyl), 8.35 (s, 1H, C2-H). Calculated Mass: 161.07; Observed Mass: 160.0.
Synthesis and Purification of 8-Vinyladenosine
The synthesis of 8-vinyladenosine was carried out as reported by Manfredini et al. (16).
Kinetic experiments
Reaction rates with RTA were determined in 10 mM potassium citrate buffer (pH 4.0) containing 1 mM EDTA. The total reaction volume was 100 μL. Reactions were started by the addition of RTA at concentrations of 0.5-1 μM. After incubation of the reaction vials at 37°C for the allotted time, the reactions were quenched by inactivating the enzyme with 500 mM potassium phosphate buffer (pH 8.3, 100 μL of a 1 M solution). The samples were then injected onto a reversed-phase C18 Waters Delta-Pak guard and analytical column (3.9 mm × 300 mm) with isocratic elution in 50 mM ammonium acetate (pH 5.0) containing 5% methanol, at a flow rate of 1 mL/min. The enzyme protein is retained on the guard column under these conditions. The extent of hydrolysis of RNA by RTA or by PAP was measured by quantitating the released adenine by monitoring the peak at 260 nm and using a comparison with standards treated with the same protocol. All substrates used and 8vdA-10 were heated to 80°C for 1 min, cooled on ice, and incubated at 37°C for 15 min prior to their addition to the assay mix to reduce any variability in the turnover rate that might result from conformational heterogeneity (hairpins vs other forms) in solution. In initial rate experiments, the extent of substrate hydrolysis was less than 15%. Product formation was shown to conform to initial rate conditions for the duration of the assay. Substrate reaction rates were fitted to the Michealis-Menten equation. For 8vdA-10, the binding constant, Kd was assumed to be equal to the competitive dissociation constant (Ki). The latter was determined by fitting the initial rates to the equation for competitive inhibition, v = kcat[S]/[[S] + Km(1 + I/Ki)], where v is the initial reaction rate, [S] is the substrate concentration, Km is the Michaelis constant for the substrate (2.9 μM for the competitive substrate, A-10, under the assay conditions), I is the concentration of 8vdA-10, and kcat is the rate of catalytic turnover at substrate saturation. The concentration of 7 μM used for A-10 in this assay is ~ 2.5 times above its Km, and is a convenient value for competitive inhibitor analysis (17). All reactions with PAP were carried out either in 25 mM HEPES or 25 mM Tris/HCl buffer of pH 7.8 at 25 °C.
Steady State Fluorescence Measurements
Steady-state fluorescence emission spectra of the 8vA-containing oligonucleotide samples were measured on a SPEX Fluoromax spectrofluorometer using a 3 mm square cuvette. The emission spectra were recorded over the wavelength range 315-450 nm with an excitation wavelength of 305 or 310 nm. The spectral bandpass was 5 nm for all emission spectra. The excitation spectra were recorded over the wavelength range 240-315 nm by monitoring the emission 380 nm. Inner filter effects were corrected for the sample concentrations that showed a maximal absorbance at the excitation wavelength of >0.1. The concentrations of oligonucleotides used were typically in the range of 2.5-5 μM. The concentrations of 2-aminopurine, 8-vinyladenine and tryptophan used for measurement of quantum yield were <1 μM and did not require any correction. Quantum yield values (18) were calculated using the equation: ϕvA = ϕref(IvA/Iref)/(10(ODvA-ODref)/2); where ϕvA and ϕref are the quantum yields of 8-vinyladenine and the reference sample respectively, I refers to the integrated emission intensities and OD to the optical densities at the excitation wavelength of 305 nm.
Determination of the pKa of N9 in 8-Vinyladenine
1D 1H NMR experiments were performed at 25 °C on a Bruker DRX 300 MHz NMR spectrometer equipped with pulsed field gradients. 3-(Trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt was used as the internal standard to which the 1H chemical shifts were referenced. A 1 mM solution of 8-vinyladenine in 0.5 ml of D2O containing 10% CD3OD as a solubility aid, was used in the study. Titrations were performed by adding small aliquots of 0.1 M NaOD or DCl. The pKa value for the N9 of the 8-vinyladenine was determined by following the pH-dependent chemical shift of the CH=CH2 proton in the vinyl moiety. The apparent electrode readings were not corrected for deuterium isotope effects. The titration data was biphasic, with the chemical shift of the proton in the vinylic system conclusively reporting on the N9 pKa, whereas the C2 proton reports a pKa that may correspond to protonation of either N1 or N3. The data was fitted by nonlinear regression analysis to the following equation: δppm = δ1 + ((Δδ2*10(pka1-pH))/(1+10(pKa1-pH)))+((Δδ3*10(pka2-pH))/(1+10(pka2-pH))); where δppm and δ1 are the observed chemical shift at a particular pH and the chemical shift of the unprotonated form, respectively. Δδ2 and Δδ3 represent differences in shifts from the chemical shifts at the low and high limiting pH values respectively.
Stochastic deformable boundary (SDB) dynamics
The starting structure for the simulations described here was based on the crystal structure of RTA in complex with the dinucleotide, adenyl-3’,5’-guanosine (ApG: PDB Id:1APG). (19) Due to the lack of a crystallographic water molecule in the X-ray structure, an important catalytic water molecule was placed into the active site explicitly. Robertus et al. have reported the crystal structure of apo-ricin A and B-chain at 2.5 Å resolution (PDB Id: 2AAI) (20) and identified one water molecule (WAT 323) within the active site pocket as the catalytic water forming strong interactions with Glu177 and Arg180. Therefore, we modeled WAT323 into the active site of the RTA/ApG complex. We also modelled 8-vinyladenyl-3’,5’-guanosine (vApG) in complex with RTA/vApG using the RTA/ApG complex structure by substituting the C8 position of adenine ring with vinyl group. 2’-deoxyadenyl-3’,5’-guanosine (dApG) and the vinyl counterpart, 2’-deoxy-8-vinyladenyl-3’,5’-guanosine (vdApG) were also modelled in similar fashion.
To investigate the local structural and interaction characteristics of dinucleotide binding to the active site of RTA, the stochastic boundary approach (21) was applied and all four simulations (RTA/ApG; RTA/vApG; RTA/dApG and RTA/vdApG) were carried out with the CHARMM program. (22) Hydrogen atoms were built using the ‘hbuild’ module in CHARMM. The simulations were run with the truncated protein SDB simulations. The simulation system contained a spherical selection of residues centered around the C1’ atom of adenine dinucleotide. All protein residues with at least one atom within 18 Å of the center were selected. Protein residues outside the spherical zone were held fixed throughout the entire SDB simulation. The systems were then solvated by the superimposing a 23-Å-radius sphere of water (Jorgensen’s TIP3P model) (23) and deleting any added water molecule whose oxygen atom was within 2.8 Å distance of another non-hydrogen atom. One chloride ion was added to neutralize the system. The structure of the model set up for SDB simulation is shown in Figure S1 in supporting information. Water molecules were minimized by applying a harmonic restraint of 10 kcal mol-1 Å2 with 500 steps of steepest descent (SD) minimization. All atoms within the spherical 18-Å-radius were then minimized with 500 steps each of steepest descent (SD) and adopted basis Newton Raphson (ABNR) minimization.
All atoms of the system were treated molecular mechanically, using the CHARMM22 all-atoms force field. Throughout, a 5-Å buffer zone was defined as all atoms further than 18 Å away from the center of the sphere, in which the non-solvent heavy atoms were harmonically restrained to their crystal coordinates with the force constants. During the dynamics simulations, Langevin dynamics were applied to the buffer region, using friction coefficients of 250 ps-1 for non-hydrogen protein atoms and 62 ps-1 on water oxygen atoms. (24) A deformable boundary potential was applied on the water oxygens. Simulations were run with a time step of 1 fs and the SHAKE algorithm (25) with a tolerance of 10-8 was used to constrain all bonds involving hydrogen atoms. Nonbonded interaction energies and forces were smoothly shifted to zero at a cutoff of 12.0 Å by the atom-based truncation method with a force-shifting function. The nonbonded pair list was generated for atoms within a 12 Å cutoff and the nonbonded list was updated heuristically. The force shift option causing the interaction energies and the force to vanish smoothly at the distance of 10 Å was used. The relative dielectric constant was set to 1.0. Simulation systems were heated from 100 to 300 K over 20 ps. The temperature was progressively increased and the systems were then equilibrated for 80 ps at 300 K and continued for the production run for a further 300 ps. Trajectories were saved every 10 fs.
Results and Discussion
Incorporation of 8-vinyl-2’-deoxyadenosine (8vdA) into RNA
Mély et al. have reported the synthesis of 8-vinyl-2’-deoxyadenosine (8vdA) and its phosphoramidite and subsequent incorporation into DNA (15). We followed their synthetic route but aimed to incorporate the novel nucleoside phosphoramidite into a small 10-mer stem-tetraloop RNA, 5’-CGCG(8vdA)GAGCG-3’ (8vdA-10). It should be noted, however, that the modified nucleoside is flanked by two guanosines in this sequence and that Mẻly et al. were only able to incorporate this residue into DNA when the flanking residues were either A, T or C. When the flanking residue was G, a compound of mass ΔM = +151 was obtained which was consistent with the formation of a guanine adduct. This presumably results from a 1,4 type Michael addition of the N7 of the 2’-deoxyguanosine phosphoramidate on the α,β-unsaturated vinyladenine system. In our approach, every residue other than the vinyl nucleoside was a ribonucleoside, thus the greater inherent acid stability of the guanosine phosphoramidate during RNA synthesis was proposed to decrease the extent of Michael addition. Another concern was the nucleophilic addition of ammonia to the vinyl moiety during deprotection of the DNA from the solid support. This was reduced by allowing the deprotection reaction for 4 hrs at room temperature instead of the standard 16-18 hrs. This problem was also encountered in our initial synthesis but was circumvented by using a 3’-end nucleoside that was derivatized on an oxalyl-controlled pore glass (CPG) solid support (26) instead of the standard succinyl-CPG. The former has the advantage of being labile to ammonia-free cleavage with basic reagents like triethylamine which are not nucleophilic. Further, we used rapid base labile protecting groups on the C, G and A residues. 8-vinyl-2’-deoxyadenosine is both acid labile (hydrolysis of the glycosidic bond) and base susceptible (addition to the vinyl moiety) and thus care is needed in its exposure time to potentially degradative reagents. A solution of 0.5 M iodine in pyridine is the standard reagent used in DNA synthesis to oxidize the phosphite to the phosphate. In our synthesis of RNA, we replaced this with a solution of 2 M tert-butyl hydroperoxide (TBHP) in toluene/dichloromethane (27) to avoid exposure of the vinyl moiety to pyridine. We also used 3% dichloroacetic acid in dichloromethane, a mild acid, to effect the removal of the 5’-dimethoxytrityl groups. The synthesis of 8vdA-10 was carried out on a 3 μmole scale with a yield of ~ 20%. MALDI mass spectrometry and enzymatic digestion with snake venom phosphodiesterase and alkaline phosphatase confirmed the incorporation of the vinyl nucleoside into the RNA and the composition of the 10-mer species. This is the first report of the incorporation of a vinyl nucleoside into an RNA scaffold.
Fluorescence of 8-vinyl-2’-deoxyadenosine within the stem-loop RNA, 8vdA-10
The emission spectrum of 8vdA in the oligonucleotide, 8vdA-10 has a maximum at 388 nm. The quantum yield of the oligonucleotide was determined relative to 2-aminopurine (28) and compared with similarly determined quantum yields of 8-vinyl-2’-deoxyadenosine, 8-vinyladenosine and 8-vinyladenine. The quantum yield of 8vdA-10 was found to be quenched 270 fold relative to that of the vinyl deoxyribonucleoside (Figure 2A, 2B and Table 1). The spectroscopic behavior of 8vdA in the hairpin RNA is governed by intra-tetraloop conformational factors. It is known that the ‘GAGA’ tetraloop adopts a structure in solution such that the three purine rings on the 3’-side (ie. A, G and A) are stacked upon each other (Figure 2D) (29, 30). A similar conformation is likely for the ‘G8vdAGA’ tetraloop as well and along with the two flanking guanosines explains the dramatic quenching of the vinyl nucleoside fluorescence in the RNA. Further, the emission maximum of the free base, 8-vinyladenine, was found to be at 378 nm (the synthesis of 8-vA is described in a later section) and that of the vinyl nucleosides (both ribo and deoxy) at 384 nm. The 4 nm red shift of fluorescence of the 8-vinyladenine moiety in RNA suggests that it is solvent exposed. An examination of the tetraloop structure reveals that the turn in the phosphate backbone between the first guanosine of the loop and the second adenosine (or 8vdA) disposes these residues in spatially different orientations thus providing solvent accessibility to the first purine of the triple base stack. A temperature dependence of the fluorescence intensity of 8vdA-10 was also carried out (Figure 2C). A two fold increase in intensity was observed when the temperature was raised from 22 °C to 50 °C. The emission maximum was also found to be blue shifted by 6 nm. The tetraloop is clearly more dynamic at higher temperatures resulting in an unstacking of the 8-vinyadenine moiety. The disruption of the quenching/stacking effect of the adjacent guanosines at higher temperatures results in the observed changes in fluorescence intensities. At temperatures greater than 60 °C, the 8vdA-10 starts undergoing intrinsic chemical hydrolysis and releases 8-vinyladenine resulting in a dramatic increase of fluorescence intensity.
Figure 2.
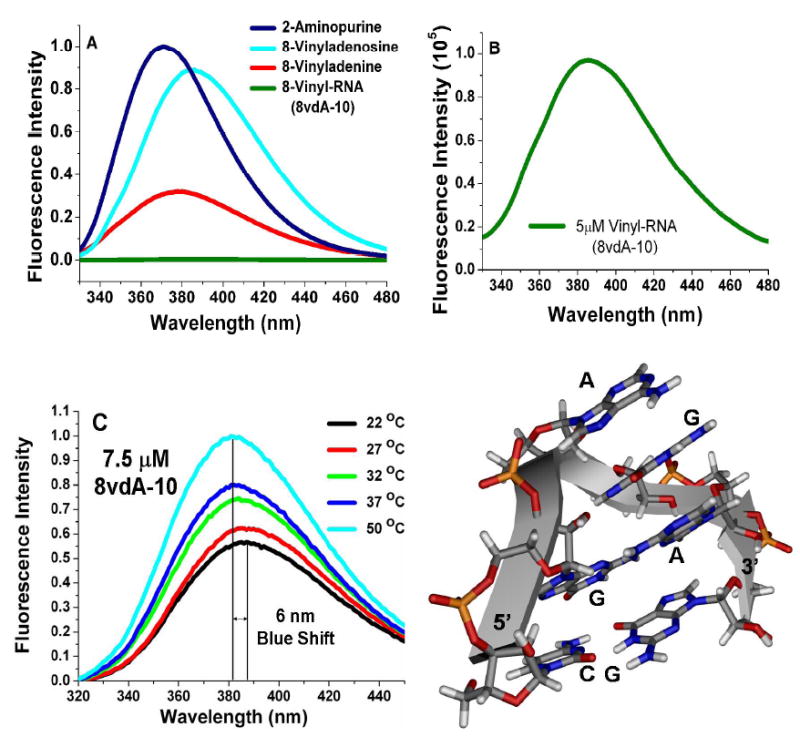
A) Normalized fluorescence intensities of equimolar concentrations of 2-AP, 8vAde, 8vdA and 8vdA-10 in water at pH 7.0 and 25 °C B) Absolute fluorescence intensity of a 5 μM solution of 8vdA-10 in 10 mM phosphate/0.1mM EDTA buffer, pH 7.1 and 25 °C. C) Normalized fluorescence intensities of a 7.5 μM solution of 8vdA-10 in 10 mM citrate/0.1mM EDTA buffer, pH 4.0 and D) The structure of the GAGA tetraloop (PDB ID: 1ZIG). 29 The tetraloop residues are shown as stick models. Residues A, G and A on the 3’ -side of the loop form a triple purine stack. The first adenosine on the 5’-side is depurinated by RTA.
Table 1.
Relative quantum yields of the 8-vinyl nucleosides, the base and the RNA 10-mer oligonucleotide sequence. 8-vinyladenosine was synthesized using the procedure reported by Manfredini et al. (16).
Quenching mechanism for 8-vA stacked with A, T and C has been studied theoretically in dimer and trimer species and compared with the observed quenching of its quantum yield when incorporated into oligonucleotides bearing A, T or C adjacent to it in the sequence (31). However, the quenching mechanism for 8-vA flanked with G was not studied since an oligonucleotide sequence with a G adjacent to 8-vA could not be synthesized. Our successful synthesis of 8vdA-10 with the vinyl nucleoside adjacent to two guanosines and the characterization of its spectral properties in the RNA provides a good experimental oligonucleotide model for comparison with a theoretical analysis of the 8-vA|G quenching mechanism.
Substrate Activity of 8vdA -10 with RTA
8vdA-10 was tested as a substrate for RTA at pH 4.0 and 37 °C with assay conditions that have been reported previously for stem-loop substrates of RTA (10,11). The reaction was monitored by HPLC using dual wavelength detection at 260 nm and 305 nm. (The λmax of 8vdA is 295 nm). The initial conditions followed the reaction for 1 hr at 37 °C. No release of 8-vinyladenine was observed. However, HPLC showed the release of adenine under the prolonged conditions (~ 12 hrs). A new peak at 305 nm was also observed under these conditions and was assumed to be that of 8-vinyladenine (Figure 3). Control incubations at pH 4.0 without RTA indicated no release of adenine. However, both control and RTA samples released similar amounts of 8-vinyladenine on prolonged incubations indicating a slow non-enzymatic solvolysis of 8-vinyladenine. Although 8vdA-10 is not a substrate for RTA at the primary depurination site, enzymatic release of adenine from the second site confirms that it binds at the catalytic site. Further, control experiments (10 mM HEPES buffer) at pH 7.0 do not release either adenine or 8-vA on prolonged incubation. MALDI analysis also established masses corresponding to unreacted RNA, deadenylated RNA and the devinylated RNA (Figure 3).
Figure 3.
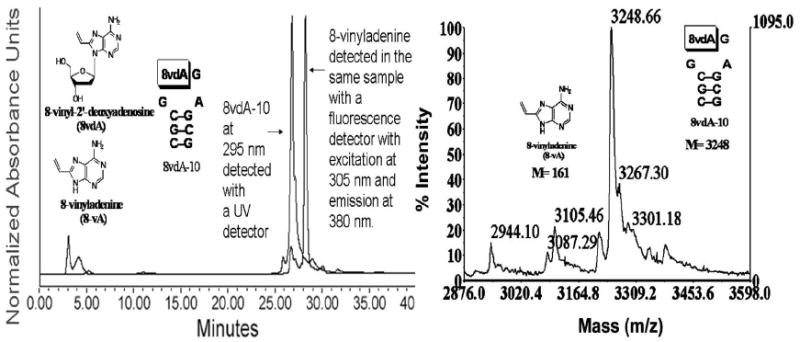
Left Panel: HPLC profile of 8vdA-10 upon incubation at 37 °C for 12 hours in 10 mM citrate buffer, pH 4.0. The peak at 27 minutes corresponds to the RNA (8vdA-10) and the peak at 29 minutes corresponds to 8-vinyladenine. The latter was undetectable at 260 nm. However, it can be detected either at 305 nm (where the sensitivity of detection is ~ 40 pmoles) or by a fluorescence detector set at an excitation wavelength of 305 nm and emission of 380 nm. Right Panel: MALDI spectrum of 8vdA-10 upon incubation at 37 °C for 12 hours in 10 mM citrate buffer, pH 4.0 shows the loss of 8-vinyladenine (M-161; the peak with mass 3105 is the [M+ + NH4+] peak).
RTA catalyzes the specific depurination of the first adenosine in synthetic stem-loop RNA and DNA molecules containing a GAGA tetraloop motif. Chen et al. showed that the kcat for the depurination of an all DNA stem-tetraloop substrate, dA-10 at this site was ~ 0.38 min-1 and the Km for its binding was ~ 2.6 μM (12). Prolonged incubation of dA-10 with RTA caused the release of a second equivalent of adenine as a result of both 2’-deoxyadenosine residues in the DNA tetraloop being hydrolyzed by RTA. The kinetic constants of depurination at the second site were determined using a DNA 10-mer, dR-10, where an abasic residue, 1,2-dideoxy-D-ribose, was substituted for the first dA in the d(GAGA) tetraloop (32). The kcat for depurination at the second site (k1) was ~ 0.46 min-1 and the Km was ~160 μM. Chen et al. determined the specificity constant, kcat/Km, for depurination at the second site to be ~60-fold lower than at the first. However, this experiment does not provide a rate for depurination at the second site while the first remains intact. In addition, the structure of the tetraloop with an abasic site (‘GabGA’) is different from an intact GAGA tetraloop. Thus, Chen’s specificity constant comparison for the two adenosines is an approximation.
Adenine release from 8vdA-10 could occur before or after the oligonucleotide is depurinated at the vinyl site. If the 8vdA-10 binds to RTA and only deadenylation takes place, an accurate rate of depurination from the second site could be obtained. And if the binding constant of 8vdA-10 is similar to A-10, the second site depurination rate could be directly compared with the rate of adenine release from the first site of A-10, thus providing a true discrimination of specificities for the two sites. Scheme 1 shows the fate of 8vdA-10 upon binding to RTA. Literature suggests that RTA can cause both deadenylation of the first adenosine in a GAGA tetraloop and non-specific deadenylation and deguanylation from other sites in stem-loop RNA (33). This study uses synthetic 28-mer stem-loop RNA and 5S rRNA as substrates and reports deadenylation and deguanylation on the basis of denaturing gels. In our work, we determine the rates of depurination at individual sites.
Scheme 1.
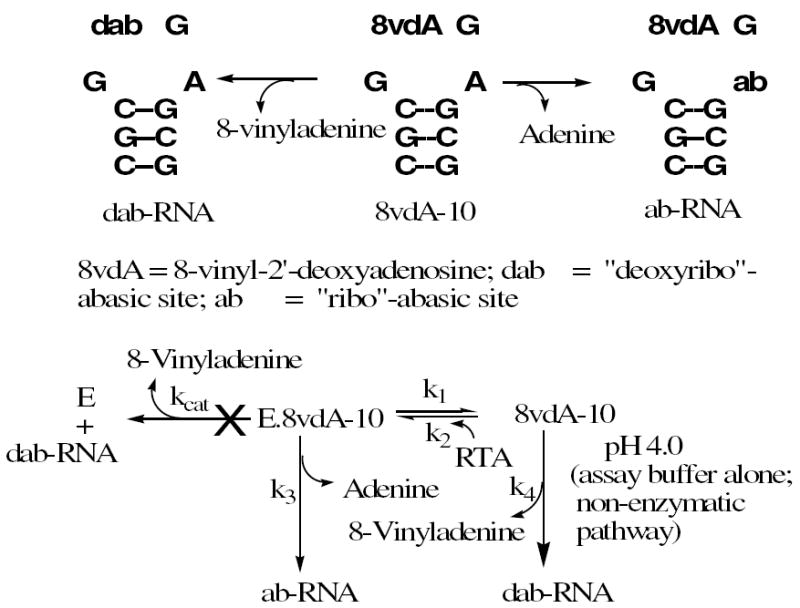
The fate of the stem-loop RNA, 8vdA-10 under enzymatic and non-enzymatic conditions. 8vdA-10 binds to RTA (Ki = k2/k1) but is not hydrolyzed. Instead, adenine is released from the second site in the tetraloop (k3). However a competing non-enzymatic process in the pH 4.0 assay buffer results in depurination (k4).
Binding of 8vdA-10 to RTA: Competitive Inhibition and Fluorescence Studies
The dissociation constant of 8vdA-10 for RTA, Ki, was determined by using a 10-mer stem-loop RNA, A-10, as a competitive substrate. Under assay conditions where RTA satisfies initial rate conditions on A-10 and does not release either adenine or 8-vinyladenine (8-vA) from 8vdA-10, the competitive assay gives a Ki (Figure 4A) of 2.4 ± 0.34 μM for 8vdA-10, a value close to the Km for A-10 (2.9 μM). Thus, 8vdA-10 binds as well as the A-10 substrate. However, despite its good binding, 8vdA is not hydrolyzed.
Figure 4.
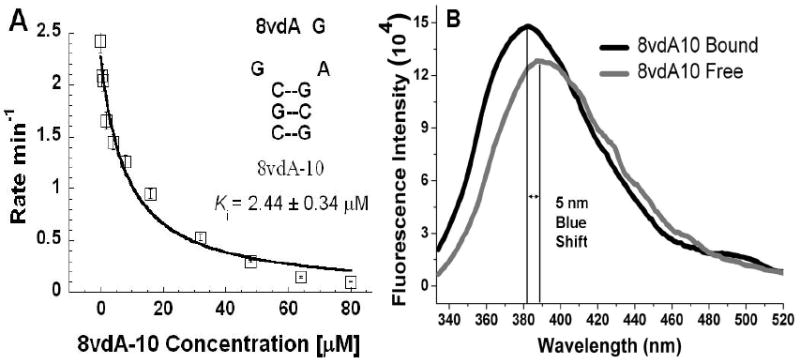
A) The binding constant for 8vdA-10 to RTA, Ki, was determined from fits of initial rate data for the competitive substrate, A-10 at a given concentration of 8vdA-10 to the equation v = kcat[S]/[[S] + Km(1 + I/Ki)]. B) Fluorescence intensities of the free and bound form of 8vdA-10. Binding of the RNA to RTA causes a 5 nm blue shift in the emission maximum.
Fluorescence spectroscopy was also employed for a direct determination of the binding constant of 8vdA-10 and to explore the base-flipping mechanism. If 8vdA-10 were to adopt a hairpin tetraloop structure with a triple purine stack and if RTA were to alter the torsion angle of the glycosidic bond of the vinyl nucleoside, it would disrupt this stack, make the vinyladenine more solvent exposed and increase the fluorescence of the “flipped-out” bound species through disruption of the quenching effect of the adjacent guanosines. However, a titration of increasing concentrations of RTA with a fixed concentration of 8vdA-10 only caused an increase of about 15% in fluorescence intensity at saturation (Figure 4B) making it difficult to determine the Kd of 8vdA-10 directly from the fluorescence experiment. The fluorescence of the bound species was blue shifted by about 5 nm suggesting an unstacking of the 8-vinyladenine (similar to the blue shift in the temperature dependence experiment discussed earlier). However, the small increase in fluorescence suggests that the flipped out base becomes stacked between the two active site tyrosines (Tyr 80 and Tyr 123) which then efficiently quench its fluorescence. Thus, the overall change in fluorescence intensity upon binding is minimal since the vinyladenine moves from one stacked environment in the tetraloop to another in the hydrophobic RTA active site.
Binding of the vinyladenine moiety in the RTA active site: Molecular Dynamics Simulations
Attempts in crystallizing either 8-vA or 8vAde with RTA were unsuccessful. Modeling of 8-vA in the crystal structure of RTA bound to adenine (Figure 5) indicates potential steric clashes with the α and β carbons of Asn 122, the aromatic ring of Tyr 123 and the carbonyl oxygen of Gly 121. It has been suggested that RTA might use a base-flipping mechanism to dock its target adenosine in the active site before depurination (34). In the uncomplexed structure of the Sarcin Ricin Loop (SRL) RNA, the glycosidic bond of the susceptible adenosine, A4324, is anti and the adenine base is involved in a triple purine stack with the bases of the adjacent guanosine and adenosine. However, the crystal structure of ricin bound to a substrate analog, formycin monophosphate (FMP), shows that the glycosidic bond is syn and the base stacks between two tyrosines, Tyr 80 and Tyr 123 (19). It is proposed that RTA recognizes the RNA with the susceptible adenosine in the anti conformation, flips it to the syn and finally catalyzes the hydrolytic reaction. Mély et al. carried out a NOESY study and determined that both syn and anti conformations were present for the base in 8-vinyl-2’-deoxyadenosine (15). It is possible in the context of an RTA bound structure that a C8 substituent forces the adenine ring to adopt an orientation (syn or anti) where steric factors preclude catalysis. Although competitive inhibition of A-10 hydrolysis by 8vdA-10 and the release of adenine from its second site both establish that this stem-loop RNA is an active site analog, we carried out molecular dynamics simulations to understand the surprising lack of catalysis.
Figure 5.
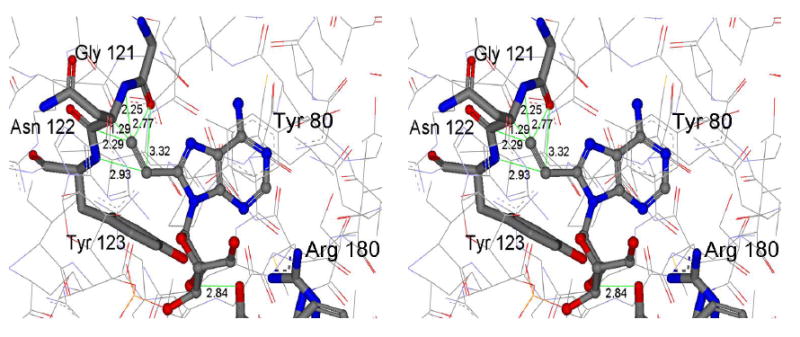
Stereoview of a docking model of 8-vinyladenine interactions in the active site of RTA created by superimposition on the coordinates of adenine in the crystal structure of RTA bound to the dinucleotide adenyl-3’,5’-guanosine (PDB ID: 1APG). The vinyl moiety experiences steric clashes with the carbonyl oxygen of Gly 121 , the backbone amide and the α and β carbons of Asn 122 and the backbone amide and the aromatic ring of Tyr 123.
The X-ray structure of RTA bound to the dinucleotide adenyl-3’,5’-guanosine, ApG (PDB Id: 1APG) (19) was used as the starting model structure for the MD simulations. Two independent simulations were run, one with ApG and the other with the dinucleotide, vApG where the adenosine is replaced by 8-vinyladenosine. The conformational binding mode for the adenine and ribose rings of adenosine is known from the X-ray structure. A comparative analysis of the simulated RTA- ApG and RTA-vApG complexes was carried out to determine the origin of the differences in catalysis between adenosine and 8-vinyladenosine. MD simulations with ApG have been reported in the literature and our ApG simulation model was in good accord with those results (35). The simulation structures averaged over 300 ps show dramatic differences between the bound orientations of adenine (in ApG) and 8-vA (in vApG) in the hydrophobic pocket of RTA (Figure 6). The two invariant residues lining this pocket are Tyr 80 in a β-strand and Tyr 123 in an α-helix. Their spatial disposition is coupled with the structural changes that accompany base binding. In particular, the backbone and side-chain dihedral angles of Tyr 80 undergo transitions that accompany the change in the dihedral angle of the C-N glycosidic bond in adenosine and 8-vinyladenosine. This dihedral angle governs the orientation of base binding in the active site. As shown in Figure 5, the hydrophobic pocket of RTA is tight such that the introduction of the vinyl substituent on the C8 of adenosine would result in several steric clashes if it was bound in the same orientation as the adenine ring in the X-ray structure, 1APG. The 300 ps MD simulation of vApG shows that these steric factors indeed play a major role since the glycosidic bond torsion in vinyladenosine rotates by 33° compared to the adenosine. This allows the vinyl moiety to be accommodated such that steric clashes with residues Gly 121, Asn 122 and Tyr 123 are avoided (See Figure 6). The dihedral angles that orient the aromatic rings in the two tyrosines change significantly to accommodate the vinyladenine in the most favorable and least sterically hindered orientation. The side-chain dihedral angle of Tyr 123 rotates from 150° in ApG to 82° in vApG and that of Tyr 80 changes from 82° to -177° respectively. It is clear from the MD simulation that both the 8-vinyladenine and the catalytic site undergo conformational changes from both the crystal structure and the simulation structure of the RTA-ApG complex. Apart from the differences in stacking, the electrostatic interaction of Arg 180 with N3 of the base is preserved in both adenosine and 8-vinyladenosine. However, for 8-vinyladenine, the contacts of N6 with Gly 121 and Val 81 and of the N1 with Val 81 are also weakened suggesting that stacking is the dominant force for binding. It must be noted however that the simulation structure of ApG also shows several changes in the active site compared to its crystal structure. Nevertheless, these are not as dramatic as seen for accommodation of the vinyladenine. The observed differences between the simulation structure and the X-ray structure are a result of the optimization of non-bonded interactions (electrostatic and van der Waals). In addition, as pointed out by Olson, ApG is a weak binding ligand such that multiple binding modes are possible (35).
Figure 6.
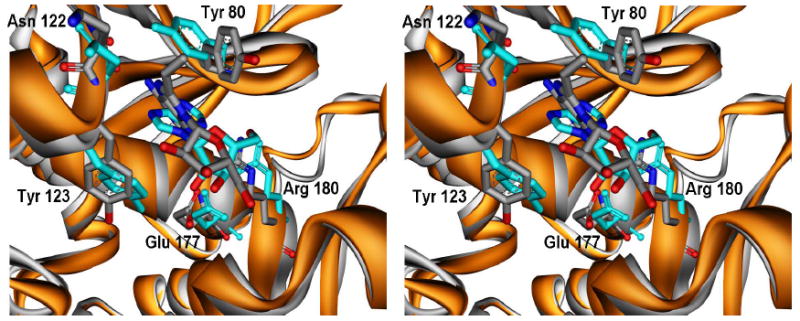
Stereoview of the superimposition of 300 ps simulated structures of RTA-ApG (cyan with ligand heteroatoms colored by atom type) and RTA-vApG (shown in grey with heteroatoms colored by atom type). The C-N glycosidic bond dihedral rotates by ~33° for the vinyladenosine which is accompanied by changes in side chain dihedral angles of Tyr 80 and Tyr 123. The 8-vinyladenine base avoids steric clashes with Asn 122 and Tyr 123 (compare with Figure 5).
Having established an alternate binding mode for the 8-vinyladenine, an analysis of the interactions around the ribose ring was carried out. This analysis was compared with that of the adenosine ribosyl in the RTA-ApG complex. The ribose pucker configuration is 2’-endo for both adenosine and 8-vinyladenosine. However, the orientation of the ribosyl ring in the latter is dramatically different. In the ApG complex, the OE1 atom of Glu 177 is 3.7 Å away from the 2’-hydroxyl and 4.6 Å from the C1’ of the ribose. In the vApG complex, these distances are 7.4 Å and 6.3 Å respectively. Glu 177 is a catalytic residue in the RTA active site that has been implicated in a) an oxacarbenium ion stabilizing role at the transition state and b) a general base role in abstracting a proton from a nucleophilic water molecule in the active site (36). Glu 208 is another catalytic residue that has been implicated in catalysis (36). A weakening of ribosyl interactions with these two residues may lead to a loss of oxacarbenium ion stabilization and thus lack of catalytic efficiency. However, literature reports an E177A mutant that only lost catalytic efficiency by 20-fold over the wild type RTA. In addition the 2’-3H KIE for hydrolysis of adenosine in a model 10-mer stem-loop RNA is reported to be 1.012 (14). It has been shown that polarization of 2’-hydroxyl group leads to a higher value of the 2’-3H KIE by a lengthening of the C2’-H2’ bond which in turn allows substantive hyperconjugative overlap between the σC2’-H2’ bond and the empty p-orbital on the anomeric carbon (C1’) of the oxacarbenium ion (37). Since this value is small in case of the RNA hydrolysis it is likely that Glu 177 in RTA does not operate via this mechanism. Instead it may directly stabilize the oxacarbenium ion by electrostatic interaction. Based on these experimental results, it was thought that disruption of interactions with Glu 177 would lead to a loss of catalytic efficiency but not complete abolishment of activity. However, the latter is observed in the case of the vinyl RNA, 8vdA-10.
A comparative analysis of distances and interaction energies of the RTA-ApG and RTA-vApG complexes then led to another interesting observation (See Tables 2 and 3 and Figure 7). The Arg 180 nitrogens, NH1 and NH2 are ~5-6 Å away from the ribose ring oxygen in case of both the X-ray and simulated structures of ApG. However, these distances close in to 3.4 and 3.1 Å respectively in case of the simulated structure of the vinyladenyl compound, vApG. The analysis of interaction energies shows that the total energy for the ribose-Arg 180 interaction is ~ 4 kcal/mol lower for vApG compared to ApG suggesting a clear electrostatic stabilization of the ribosyl conformation in the vinyl dinucleotide.
Table 2.
Comparative interaction distances (Å) for two sets of simulations. The most significant differences (1-2 Å) between adenyl and 8-vinyladenyl dinucleotides come from the interaction of Arg 180 with the sugar ring oxygen. In case of the vinyl nucleosides, the guanidinium nitrogens of Arg 180 close in on the ribosyl O4’ by 2 Å in ribo-simulations and 0.6-1.1 Å in the deoxyribo-simulations, suggesting stronger sugar-Arg 180 interactions.
| Simulation Distances (Å)
|
||||||
|---|---|---|---|---|---|---|
| Adenosine
|
2’-Deoxyadenosine
|
|||||
| Ricin | X-ray | RTA/ApG | RTA/vApG | RTA/dApG | RTA/vdApG | |
| R180:NH2 | Ade:O4’ | 5.4 | 5.2 ± 0.4 | 3.1 ± 0.2 | 3.5 ± 0.2 | 2.9 ± 0.2 |
| Ade:N3 | 3.7 | 4.0 ± 0.8 | 3.2 ± 0.3 | 3.0 ± 0.1 | 3.1 ± 0.2 | |
| R180:NH1 | Ade:N3 | 3.0 | 3.7 ± 0.6 | 3.0 ± 0.2 | 3.5 ± 0.4 | 3.4 ± 0.4 |
| Ade:O4’ | 5.0 | 5.2 ± 0.2 | 3.4 ± 0.2 | 4.2 ± 0.2 | 3.1 ± 0.2 | |
| E177:OE1 | Ade:O2’ | 3.5 | 3.7 ± 0.8 | 7.3 ± 0.4 | - | - |
| Ade:O4’ | 6.6 | 6.3 ± 0.7 | 6.1 ± 0.5 | 6.7 ± 0.4 | 5.6 ± 0.7 | |
| E177:OE2 | Ade:O2’ | 2.8 | 3.6 ± 0.8 | 7.4 ± 0.6 | - | - |
| Ade:O4’ | 5.7 | 6.2 ± 0.7 | 5.7 ± 0.3 | 6.3 ± 0.4 | 5.6 ± 0.7 | |
| Wat323:O | Ade:C1’ | 4.1 | 5.1 ± 0.4 | 4.7 ± 0.7 | 4.5 ± 0.3 | 4.5 ± 0.6 |
| Ade:O2’ | 2.5 | 3.6 ± 0.5 | 5.1 ± 0.9 | - | - | |
| E177:OE2 | R180:NH1 | 2.2 | 2.8 ± 0.2 | 2.7 ± 0.1 | 2.8 ± 0.2 | 3.0 ± 0.2 |
| E177:OE1 | R180:NH2 | 2.3 | 5.1 ± 0.5 | 5.7 ± 0.6 | 5.0 ± 0.3 | 5.0 ± 0.2 |
Table 3.
Comparative interaction energies (kcal mol-1) for two sets of simulations. The most significant energetic differences (upto 4 kcal mol-1) between adenyl and 8-vinyladenyl dinucleotides come from the interaction of Arg 180 with the sugar ring oxygen. The isolated energetics between the guanidinium nitrogens and sugar O4’ are also highlighted.
| Interacting Residues | Interaction Energies (kcal/mol)
|
||||||
|---|---|---|---|---|---|---|---|
| ApG
|
vApG
|
||||||
| vdw | elec | total | vdw | elec | total | ||
| Adenine | R180 | -0.9 | -12.3 | -13.2 | 0.1 | -13.5 | -13.4 |
| Y123 | -2.9 | -0.2 | -3.1 | -3.7 | -1.0 | -4.7 | |
| Y80 | -4.0 | -2.2 | -6.2 | -6.2 | -1.9 | -8.1 | |
| Ribose | R180 | -1.3 | -2.2 | -3.5 | -1.4 | -6.0 | -7.4 |
| E177 | 1.1 | -14.2 | -13.1 | -0.4 | -1.3 | -1.7 | |
| Ribose:O4’ | R180:NCN | -0.2 | -8.8 | -9.0 | -0.1 | -18.9 | -19.0 |
|
| |||||||
| Interacting Residues | dApG
|
vdApG
|
|||||
| vdw | elec | total | vdw | elec | total | ||
|
| |||||||
| Adenine | R180 | -0.02 | -12.3 | -12.3 | -0.1 | -12.9 | -13.0 |
| Y123 | -1.8 | -1.9 | -3.7 | -2.7 | -0.2 | -2.9 | |
| Y80 | -4.7 | -1.5 | -6.2 | -6.0 | -1.8 | -7.8 | |
| Ribose | R180 | -1.8 | -4.5 | -6.3 | -1.5 | -8.8 | -10.3 |
| E177 | -0.1 | -0.1 | -0.2 | -0.2 | -0.2 | -0.4 | |
| Ribose:O4’ | R180:NCN | -0.3 | -13.7 | -14.0 | -0.1 | -19.5 | -19.6 |
Figure 7.
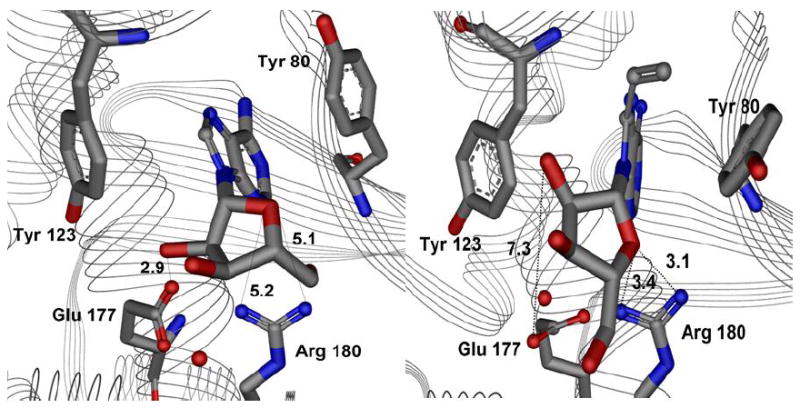
Active site interaction distances (Å) for 300 ps simulation structures of ApG (left panel) and the vinyl dinucleotide, vApG (right). Sterics reorganize the 8-vinyladenine base binding pocket and also reorient the ribosyl group. The guanidinium nitrogens of Arg 180 in the RTA- vApG complex close in on the ribosyl O4’ by 1.8 Å for NH1 and 2.1 Å for the NH2, suggesting stronger sugar-Arg 180 interactions. The 2’-hydroxyl-Glu 177 interaction is also lost as this distance extends from 2.9 Å to 7.3 Å.
Arg 180 is a conserved residue in a family of ribosome inactivating proteins and mutations of this residue are severely detrimental to catalytic activity. A contribution of 4 kcal/mol can lead to ~1000 fold loss of catalytic activity [from ΔΔG = -RT ln (k1/k2)]. It is reasonable to assume that in case of vApG, Arg 180 plays a role in ground state stabilization (electrostatic attraction) of the ribose and/or transition state destabilization (repulsion) of the oxacarbenium ion.
Enzymes such as purine nucleoside phosphorylase (PNP) have been shown to exhibit coupled protein and substrate motion such that the purine 5’-hydroxyl and ribose ring oxygen and the nucleophile phosphate oxygen form a repulsive environment that promotes the lone pair of electrons to push from the ring oxygen to C1’ resulting in oxacarbenium ion formation (38). In the case of RTA-vApG complex, coupled motion of protein and substrate allows the vinyladenine base to bind in an altered orientation. Coupled protein and ribose motion then brings the Arg 180 in close proximity to the ribose ring oxygen. This would 1) serve to stabilize the ribose in the ground state but 2) be destabilizing for oxacarbenium ion formation and 3) result in loss of catalytic activity.
Thus, the entropic cost of organizing the 8-vinyladenine in the active site is coupled with the change in ribosyl orientation. The simulation model provides a rationale for the observation that 8vdA-10 binds to RTA but is not hydrolyzed. Although the simulation only involves a dinucleotide, similar changes in ribosyl geometry in case of the RTA bound RNA structure can lead to a loss of catalytic activity. As pointed out by Pande et al., the unique feature of the GNRA class of tetraloops is a cooperative relationship between 3′-endo⇌2′-endo and closed-loop⇌open-loop transitions (39). The 2′-endo pucker modes expand the loop backbone and allow extension of the base away from the loop. It is reasonable to assume that in the dynamic open loop conformation with the depurination site base flipped out, changes in ribosyl geometry similar to those observed in the dinucleotide structure, vApG, can also be allowed in the larger RNA structure without a significant distortion of the orientations of the RNA chain on the 5’ and 3’-sides of the vinyladenosine. This also seems plausible given the 1 Å expanded backbone of the 8vApG (5’O to 3’-P distance of 5.6 Å) compared to the ApG structure.
MD simulations were also carried out for the 2’-deoxyadenyl guanosine (dApG) and its vinyl counterpart, 8-vinyl-2’-deoxyadenyl guanine (vdApG). The binding of the vinyladenine base involves similar motion of Tyr 80 (side chain dihedral changes from 81° to 177°) and Tyr 123 (side chain dihedral changes from 92° to 85°) as was described in the ribose case. The N-C glycosidic bond torsion (O4’-C1’-N9-C4) rotates from 31° in dApG to -27° in vdApG. The lack of catalysis in the case of the 2’-deoxy-8-vinyladenosine can again be rationalized on the basis of similar observations that Arg 180 forms a strong electrostatic interaction with the deoxyribose ring oxygen in vdApG but not in dApG (energy difference ~ 4 kcal/mol). The Arg 180-deoxyribose interaction is similar to the one observed with ribose. Thus, both sets of simulations show the same trends. Another interesting observation is that the geometry of the deoxyribosyl ring is 3’-endo for vdApG whereas it is 2’-endo for dApG. The interaction distances and energies are shown in Tables 2 and 3 respectively. Details of the simulation have been included in the (supporting information Figure S2 and Table S1).
Our MD simulations point toward an interesting mechanism by which RTA maintains its specificity for hydrolyzing adenosine. The hydrophobic pocket can bind a variety of base analogs (11) and is also involved in leaving group activation but not all molecules that bind are turned over. Thus, it is likely that in the RTA-8vdA-10 structure, Arg 180 is destabilizing for oxacarbenium ion formation resulting in a non-productive complex. This scenario is reminiscent of transition state destabilization in the DNA repair enzymes by 2’-fluoro substituted nucleosides which form non-productive complexes (40). Our theory of the lack of catalysis of 8vdA-10 by RTA thus invokes both steric and electronic factors. The alteration in 8-vinyladenine binding is coupled with the reorientation of ribose/deoxyribose rings (in vApG/vdApG respectively) such that the Arg 180 interaction with sugar ring oxygen is only observed in the vinyl nucleoside simulations. Figure 8 shows a schematic model for these observations.
Figure 8.
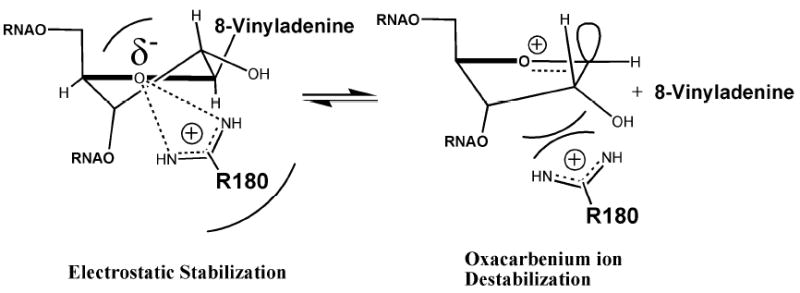
A model to explain the lack of catalysis by RTA of the 8-vinyladenyl substituted RNA, 8vdA-10. Dynamics simulations predict Arg 180 within 3 Å distance from the ribose 4’-hydroxyl. The consequence of this could be electrostatic stabilization of the ground state such that the formation of the oxacarbenium ion is destabilized and the reaction stalled.
Characterization of adenine release from the second site of the tetraloop of 8vdA-10
RTA-catalyzed adenine release for 8vdA-10 was found to be slow with a kcat of 0.023 ± 0.004 min-1, Km of 205 ± 64 μM and a kcat/Km of 2.03 ± 0.03 M-1s-1 (See Figure S3A in supporting information). Chen et al. determined the kcat/Km for the hydrolysis of stem-loop RNA 10-mer, A-10 to be 1.7×104 M-1s-1 and the kcat and Km to be ~ 4 min-1 and 4.1 μM respectively (12). A comparison of the kcat/Km of A-10 with 8vdA -10 indicates that the recognition of the second adenosine in the tetraloop by RTA is ~ 8000-fold lower than the first site.
Since the specificity of RTA for the first adenosine is much higher than the second, other depurination events follow the first one. The conformation of the tetraloop changes once the first adenosine is cleaved, resulting in altered susceptibility for the second adenosine. As described above, Chen et al. observed a kcat of ~0.46 min-1 for second site adenine release from dR-10, a substrate that was abasic in the primary site. This rate is ~ 20 fold faster than that observed for adenine release from 8vdA-10 (which is intact at the primary site).
Adenine release from the second adenosine on the tetraloop of 8vdA-10 suggests binding to RTA in two modes. One positions the 8-vA (8-vinyladenine) in the depurination site using the normal stem-loop geometry and another reverse-disposes the 8-vA and the adenine by a 180° rotation of the tetraloop with respect to the catalytic site. Stem-tetraloop structures such as 8vdA-10 are known to be thermodynamically extremely stable. However it is possible that stem-slippage could also alter loop structure. We calculated the secondary structures of the related 10-mer stem-loop molecule A-10 with the programs mFold (version 2.3; structure calculated in 1M NaCl at 25 °C) (41) and RNAshapes (42). An alternate geometry was found that results from slippage of two residues in the stem such that the second adenosine can be positioned in the RTA catalytic site for depurination. This alternate structure, TL2, (with a tetraloop, two base-pairs and a two residue overhang) is 6.1 kcal/mol higher in energy than the most stable structure with the tetraloop and 3 base-pairs, TL1 (Figure 9). However, the difference in catalytic rates of depurination at the two sites (calculated using the expression: ΔΔGTS = - RT ln(k1h/kBT)/-RT ln(k2h/kBT) where k1 and k2 are the catalytic rate constants at the two sites) is ~3.1 kcal/mol which is less than the barrier to interconversion between TL1 and TL2. Thus, the direct recognition of a slipped structure by RTA or an enzyme promoted slippage is unlikely. Disruption of base-pairs and rearrangement of secondary structure on the enzyme is likely to be more energetically demanding than in solution. A simple 180° rotation of the tetraloop more easily explains both the high Km and low turnover at the second site.
Figure 9.
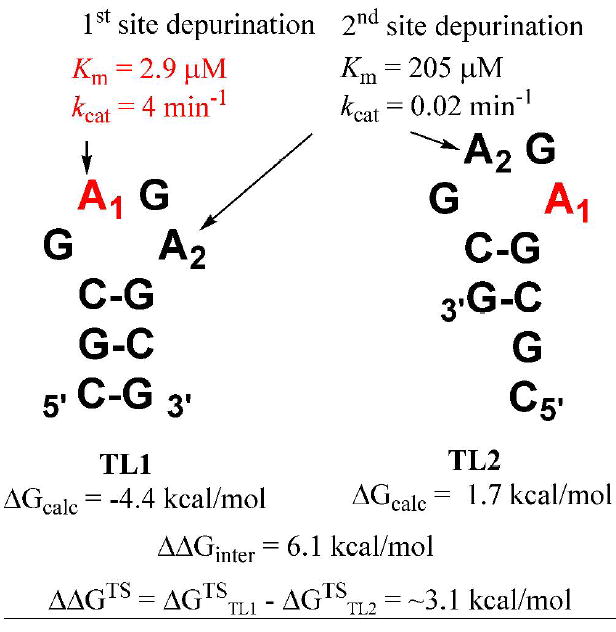
Substrate slippage model for RTA catalysis at the second adenosine site in the tetraloop. Two possible stem-tetraloop structures of A-10 calculated with the programs mFold and RNAshapes are shown. The barrier to interconversion between TL1 and TL2 in solution is higher than the difference in transition state energies for catalysis at the two sites.
Crystal structures of RTA bound either to small molecules such as adenine and formycin monophosphate (19) or simulation models of hexamer, CGAGAG (35), indicate a single site for adenine depurination. Binding of 8vdA-10 gives a Ki of 2.4 μM which is a dissociation constant with respect to A-10 (Km = 2.9 μM). This thermodynamic constant represents similar binding of 8vdA-10 and A-10. However, in catalysis at the second site, the Km of the vinyl RNA is 205 μM. Slow substrates of RTA are known to be in thermodynamic equilibrium: thus, Km is also a dissociation constant. Therefore, adenine depurination of 8vdA-10 likely involves binding of the stem-loop in a 3’→5’ orientation opposite to that of A-10. Kinetic data with 8vdA-10 indicates that the catalytic specificity (kcat/Km) for the first adenosine in a 10-mer stem-tetraloop substrate is almost three orders of magnitude more favorable than the second site.
It is not clear if second site deadenylation is physiologically relevant. However, our observations are relevant to inhibitor design in that it may be desirable to include non-hydrolysable adenine isosteres at two sites in the tetraloop. Depurination reactions can occur in the same active site as a result of recognition of multiple conformations of the tetraloop. Catalytic rate efficiency at the two sites can be tested more directly on shorter cyclic versions of tetraloops which lack stems. Preliminary results comparing the Ki of a cyclic tetraloop analog with the Km for its second site deadenylation suggest that the binding affinity of RTA for the second site adenosine is ~ 15-fold lower than the first (43). This difference is ~100-fold in case of the stem-loop substrate, 8vdA-10 and suggests that the stem plays a role in the directionality of binding (5’→3’ versus 3’→5’).
8-Vinyladenine: Synthesis and physicochemical characterization
Characterization of 8-vinyladenine has not been previously reported (44). A standard of 8-vinyladenine was synthesized for accurate quantitation of the hydrolytic rate of this novel nucleobase from the parent RNA. The precursor, “1”, was treated with triethylamine in methanol (24 hrs at rt) to remove the formamidine followed by 15% dichloroacetic acid in dichloromethane for 3 hrs to remove the 5’-dimethoxytrityl group and cleave the glycosidic bond in one step (see inset in Figure 10). The product was purified on a semi-preparative C18 reverse phase column. The identity of the product was confirmed by 1H NMR and ESI mass analysis in the negative mode.
Figure 10.
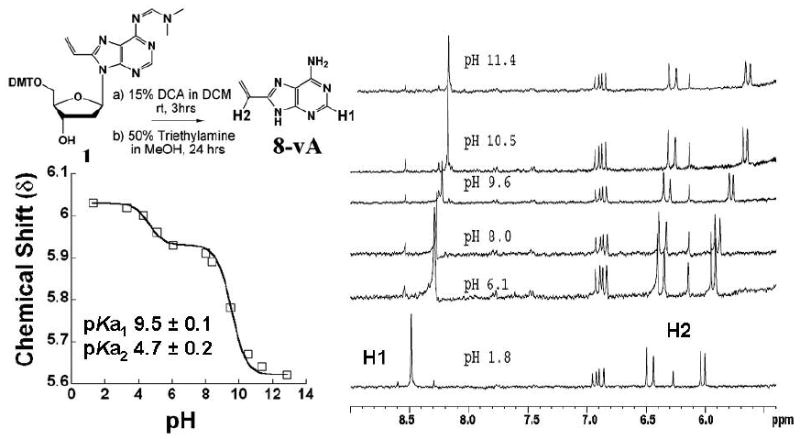
Determination of the pKa of N9 in 8-vinyladenine by 1H NMR. Data fitting is described in the text. The chemical shifts of the vinylic proton, H2 (doublet), in the 6 ppm region and that of the ring proton, H1 (singlet), in the 8.5 ppm region report on two protonation events, hence the biphasic curve. The inset shows the synthesis of 8-vinyladenine (8vA). Prolonged treatment of the dimethoxytritylated precursor nucleoside, 1, under acidic conditions effected the removal of the DMT protecting group as well as cleavage of the glycosidic bond. The N6 protection was removed under basic conditions and the final compound was purified by reverse phase HPLC.
Absorption spectra of known quantities of 8-vinyladenine were recorded to determine its extinction coefficient. The λmax was 275 nm and the absorption profile was broad with a spectral maximum between 270-290 nm. The millimolar extinction coefficient, ε, was determined to be 8.33. 8-vA was also characterized as a fluorophore (see Figure 2 and Table 1). As described earlier, the λem was 378 nm. The quantum yield, ϕ, was determined to be 0.37 relative to tryptophan (ϕ = 0.14) (45) and 0.33 relative to 2-aminopurine (ϕ = 0.66) (27). These determinations were made on absorbance matched samples in water at pH 7.0 and 25°C.The extended conjugation of 8-vinyladenine makes it a better leaving group than adenine. However, activation of adenine as a leaving group by RTA is proposed to occur via protonation of N7 and/or possibly N3. We were interested in determining the effect of the vinyl group on the pKa values of these ring nitrogens and therefore on leaving group activation. 1H NMR was used to follow the change in chemical shift of the vinyl CH=CH2 proton and the proton at C8 of the purine ring with pH (Figure 10). The former is sensitive to changes in the environment of N9 and the latter to N1 or N3, which are not distinguished in this experiment. The NMR titration data was fitted to a biphasic pKa equation with two pKa’s. The pKa for N9 of 8-vA was found to be 9.5 and that for the N1 -N3 system to be 4.6. These numbers are similar to those of adenine with a pKa of 9.8 for N9. Thus, introduction of the vinyl group only reduces the pKa at N9 by ~ 0.2-0.3 units.
Since the pKa of N7 cannot be determined from the NMR experiment, equilibrium geometries of 2’-deoxyadenosine (dAde) and 8-vinyl-2’-deoxyadenosine were optimized using the B3LYP/6-31G(d,p) level of theory with Gaussian 03 (46). The atomic electrostatic potential (ESP) charges using Merz-Singh-Kollman (MK) scheme were computed at the same level of calculation. Charges at N7 in the two molecules were similar (-0.563 and -0.559 respectively) suggesting that the pKa at N7 might also be similar (Figure S4 in supporting information). The pKa data and charge calculations along with the dynamics simulations suggest that steric factors enforced by the vinyl group orient the ribosyl group such that electronic factors from the enzyme then stabilize the ground state. Thus, electronic properties (of adenine and 8-vA) alone do not bring about the observed difference in catalysis of A-10 versus 8vdA-10.
The similarity between the Kd of 8vdA-10 and the Km of A-10 (also a dissociation constant) suggests that binding is driven by extended structural contacts over the stem-loop (electronic and hydrophobic), whereas catalysis is driven by optimization of electronic interactions (such as H-bonding) to both adenine and ribose at the transition state. Non-enzymatic hydrolysis of 8-vA from 8vdA-10, albeit slow, confirms that it is an activated leaving group. Among the host of transition state analog inhibitors designed for RTA, only those with small hydrophobic leaving groups (such as phenyl) bind more tightly than those with adenine analogs designed to provide a better pKa match (11). This suggests that the high specificity of the RTA active site is due to efficient recognition of subtle electronic and steric properties of the RNA molecules. The extreme specificity of RTA is reflected in its hydrolysis of a single adenosine in the milieu of adenosines in ribosomal RNA.
Rate of Non-Enzymatic Release of 8-Vinyladenine from 8vdA-10
The change in fluorescence intensity of 8vdA-10 was recorded as a function of time to determine the rate of 8-vA release. As mentioned earlier, the fluorescence intensity of the 8-vinyladenine in stem-loop RNA is dependent on the conformation of the tetraloop.
A 270 fold reduction in fluorescence intensity is observed when 8vdA is incorporated in the tetraloop of 8vdA-10. 8-vA release from 8vdA-10 was followed at pH 4.0 (10 mM citrate buffer) over 10 hrs. The rate constant (k4 in Scheme 1) was 6×10-5 nmoles min-1. This rate is ~ 300 fold slower than the release of adenine from the second site (kcat of ~0.02 min-1: k3 in Scheme 1) by RTA. The rate constant was determined from the change in fluorescence intensity of a 5 μM solution of 8vdA-10 at 25 °C (Figure 11).
Figure 11.
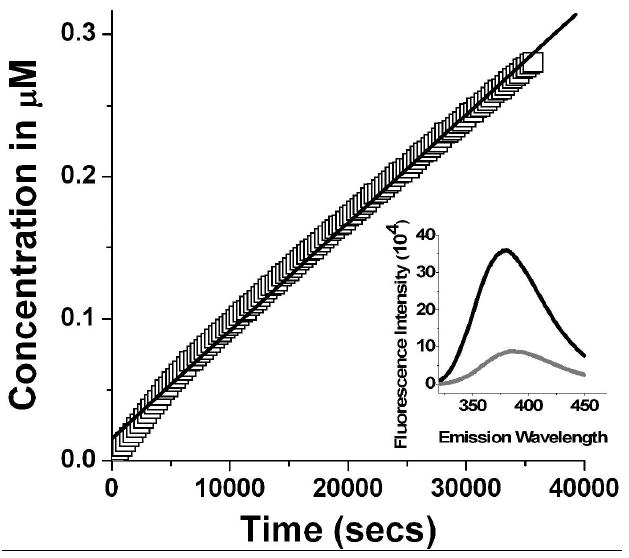
Continuous release of 8-vinyladenine from the stem-loop oligomer, 8vdA-10 over 10 hrs in 10 mM citrate buffer, pH 4.0. The rate constant was derived from a fit to the equation: k = (1/Δα) (ΔF/Δt) where ΔF/Δt is the rate in fluorescence units s-1, Δα = Ftot/[S]tot, where Ftot is the total fluorescence increase for 100% conversion of substrate ([S]tot) to product. Excitation was set at 305 nm and the emission spectrum was recorded in the 320-450 nm range.
Control experiments in a pH 7.6 buffer showed <1% change in fluorescence intensity over 12 hrs consistent with a stable tetraloop conformation and an insignificant hydrolytic rate. A comparison of the non-enzymatic 8-vinyladenine release rate versus enzymatic adenine release established that deadenylation of the tetraloop occurred with 8-vinyl-2’-deoxyadenosine still intact in the loop. A negligible change in fluorescence intensity was observed during the initial time course kinetics of adenine release from the second site in the tetraloop. It must be noted that since the quantum yield of 8-vA is 130 fold greater than that of 8-vinyl-2’-deoxyadenosine within the RNA, it is possible to determine very small release rates.
Kinetics of A-14 turnover by Pokeweed Antiviral Protein (PAP)
Of the various RIP’s, Pokeweed Antiviral Protein (PAP) isolated from the leaves of Phytolacca americana, has been shown to have a broad RNA substrate specificity relative to the GAGA tetraloop specificity of RTA (47). Given this promiscuity of PAP, we decided to test its catalytic activity on 8vdA-10. Rajamohan et al. showed that both wild type and recombinant PAP were able to deadenylate E. coli 23S and 16S rRNA in 25 mM Tris/HCl at pH 7.8 (48). The same pH conditions were used to test 8vdA-10 as a PAP substrate. Before testing the hypothesis of 8vdA-10 turnover by PAP, it was necessary to investigate the depurinating ability of PAP on a small stem-loop SRL RNA mimic, A-14.
PAP was heat treated to remove the non-specific glycosidase/nuclease activity while retaining the RNA N-glycosidase activity (49). Adenine release was detected on incubating A-14 with a commercial source wild type PAP at pH 7.8 (25 mM Tris/HCl buffer). The Km for A-14 was 220 ± 20 μM and the kcat was 2.9 ± 0.2 min-1 (Figure S3B in supporting information). For comparison, the Km and kcat of A-14 hydrolysis by RTA are 8.1 μM and 219 min-1 respectively (12). It is remarkable that RTA has optimal activity at pH 4.0 but is inactive at neutral pH for hydrolysis of small stem-loop RNA whereas PAP can operate at neutral pH albeit not efficiently. Having established that A-14 is a substrate of PAP, we tested its ability to hydrolyze 8vdA-10 under neutral pH conditions.
Hydrolysis of 8vdA-10 by PAP
Incubation of 8vdA-10 with PAP at pH 7.8 (25 mM HEPES buffer) resulted in release of both adenine and of 8-vinyladenine. Adenine release occured at an initial rate of 0.018 min-1. Kinetic characterization of 8vdA-10 binding to PAP indicated a Km > 300 μM for this substrate. The release of 8-vinyladenine, measured by the continuous change in fluorescence intensity of a 5 μM solution of 8vdA-10 on incubation with 1 μM PAP, was determined to be ~1.6×10-3 min-1 which is 10-fold lower than the rate of adenine release from the second site. These results establish that 8-vA is released by PAP, but not as efficiently as adenine from the second site. The RTA active site is highly stringent in comparison and does not catalyze 8-vA release.
Conclusions
We have designed a new RNA/DNA hybrid substrate analog, 8vdA-10, that binds to RTA with a Ki value similar to the Km of the small stem-loop RNA 10-mer, A-10. Despite having an activated leaving group, the N-glycosidic bond of the 8-vinyl-2’-deoxyadenosine in the RNA is not cleaved by RTA. Dynamics calculations suggest that steric factors cause the nucleoside to bind in an orientation where the enzyme destabilizes the formation of the oxacarbenium ion and thus precludes catalysis. However, adenine is released from the second site of 8vdA-10. This deadenylation rate is 2 orders of magnitude slower than that measured for depurination of the first site adenosine in A-10. The binding affinity of 8vdA-10 to the catalytic site that results in the second site depurination is ~ 100 fold lower than to the first, indicating an alternate binding geometry for the modified vinyl RNA. The new stem-loop analog described here thus allows a comparison of the enzymatic specificities for the two adenosines in a tetraloop RNA structure since the first site is still intact when the second site depurination occurs. The results with PAP suggest that second site depurination can also occur at neutral pH. Glycosidic bond scission at a specific site on the 28S rRNA by ribosome binding proteins is stringent under physiological conditions. However, multiple adenine release from truncated stem-loop RNA analogs suggests that non-hydrolyzable adenine isosteres at both sites in the GAGA tetraloop might be of interest for inhibitor design. Our study complements the work of Mély et al. and shows that the 8-vinyl moiety is a useful fluorophore system that can be exploited further to study protein-RNA complexes.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant CA7244. We thank Mr. Matthew Sturm for valuable discussions and Dr. Steven D. Schwartz for providing the computing resource for the molecular dynamics simulations.
Abbreviations
- 8vdA
8-vinyl-2’-deoxyadenosine
- 8-vA
8-vinyladenine
- 8vdA-10
8vdA incorporated into a 10-mer stem-loop RNA with the sequence, 5’-CGCG8vdAGAGCG-3’
- 8vAde
8-vinyladenosine
- RTA
Ricin Toxin A-Chain
- PAP
Pokeweed Antiviral Protein
- KIE’s
Kinetic Isotope Effects
- A-10
stem-loop RNA with the sequence, 5’-CGCGAGAGCG-3’
- A-14
stem-loop RNA with the sequence, 5’-CGCGCGAGAGCGCG-3’
- ApG
adenyl-3,’5’-guanosine
- vApG
8-vinyladenyl-3’,5’-guanosine
- dApG
2’-deoxyadenyl-3,’5’-guanosine
- vdApG
2’-deoxy-8- vinyladenyl-3’,5’-guanosine
References
- 1.Endo Y, Chan YL, Lin A, Tsurugi K, Wool IG. The cytotoxins alpha-sarcin and ricin retain their specificity when tested on a synthetic oligoribonucleotide (35-mer) that mimics a region of 28 S ribosomal ribonucleic acid. J Biol Chem. 1988;263:7917–7920. [PubMed] [Google Scholar]
- 2.Kudlicki W, Kitaoka Y, Odom OW, Kramer G, Hardesty B. Elongation and folding of nascent ricin chains as peptidyl-tRNA on ribosomes: the effect of amino acid deletions on these processes. J Mol Biol. 1995;252:203–212. doi: 10.1006/jmbi.1995.0488. [DOI] [PubMed] [Google Scholar]
- 3.Moazed D, Robertson J, Noller H. Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature. 1988;334:362–364. doi: 10.1038/334362a0. [DOI] [PubMed] [Google Scholar]
- 4.Endo Y, Tsurugi K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J Biol Chem. 1988;263:8735–8739. [PubMed] [Google Scholar]
- 5.Sandvig K, van Deurs B. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002;529:49–53. doi: 10.1016/s0014-5793(02)03182-4. [DOI] [PubMed] [Google Scholar]
- 6.Lord JM, Deeks E, Marsden CJ, Moore K, Pateman C, Smith DC, Spooner RA, Watson P, Roberts LM. Retrograde transport of toxins across the endoplasmic reticulum membrane. Biochem Soc Trans. 2003;31:1260–1262. doi: 10.1042/bst0311260. [DOI] [PubMed] [Google Scholar]
- 7.Eiklid K, Olsnes S, Pihl A. Entry of lethal doses of abrin, ricin and modeccin into the cytosol of HeLa cells. Exp Cell Res. 1980;126:321–326. doi: 10.1016/0014-4827(80)90270-0. [DOI] [PubMed] [Google Scholar]
- 8.Rich V. Murderous experiments of Stalin’s police chief. New Sci. 1992;135:8. [Google Scholar]
- 9.Remnick D. Washington Post. 1992 April 21;:D1. [Google Scholar]
- 10.Ler SG, Lee FK, Gopalakrishnakone P. Trends in detection of warfare agents Detection methods for ricin, staphylococcal enterotoxin B and T-2 toxin. J Chromatogr A. 2006;1133:1–12. doi: 10.1016/j.chroma.2006.08.078. [DOI] [PubMed] [Google Scholar]
- 11.Roday S, Amukele T, Evans GB, Tyler PC, Furneaux RH, Schramm VL. Inhibition of ricin A-chain with pyrrolidine mimics of the oxacarbenium ion transition state. Biochemistry. 2004;43:4923–4933. doi: 10.1021/bi0498499. [DOI] [PubMed] [Google Scholar]
- 12.Chen XY, Link TM, Schramm VL. Ricin A-chain: kinetics, mechanism, and RNA stem-loop inhibitors. Biochemistry. 1998;37:11605–11613. doi: 10.1021/bi980990p. [DOI] [PubMed] [Google Scholar]
- 13.Orita M, Nishikawa F, Kohno T, Senda T, Mitsui Y, Yaeta E, Kazunari T, Nishikawa S. High-resolution NMR study of a GdAGA tetranucleotide loop that is an improved substrate for ricin, a cytotoxic plant protein. Nucleic Acids Res. 1996;24:611–618. doi: 10.1093/nar/24.4.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen XY, Berti PJ, Schramm VL. Ricin A-Chain: Kinetic Isotope Effects and Transition State Structure with Stem-Loop RNA. J Am Chem Soc. 2000;122:1609–1617. [Google Scholar]
- 15.Ben Gaied N, Glasser N, Ramalanjaona N, Beltz H, Wolff P, Marquet R, Burger A, Mély Y. 8-vinyl-deoxyadenosine, an alternative fluorescent nucleoside analog to 2’-deoxyribosyl-2-aminopurine with improved properties. Nucleic Acids Res. 2005;33:1031–1039. doi: 10.1093/nar/gki253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manfredini S, Baraldi PG, Bazzanini R, Marangoni M, Simoni D, Balzarini J, De Clercq E. Synthesis and cytotoxic activity of 6-vinyl- and 6-ethynyluridine and 8-vinyl-and 8-ethynyladenosine. J Med Chem. 1995;38:199–203. doi: 10.1021/jm00001a025. [DOI] [PubMed] [Google Scholar]
- 17.Segel I. Enzyme Kinetics. Wiley Classics Library Edition. John Wiley & Sons; New York: 1993. [Google Scholar]
- 18.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum Publishers; New York: 1999. [Google Scholar]
- 19.Monzingo AF, Robertus JD. X-ray analysis of substrate analogs in the ricin A-chain active site. J Mol Biol. 1992;227:1136–1145. doi: 10.1016/0022-2836(92)90526-p. [DOI] [PubMed] [Google Scholar]
- 20.Rutenber E, Katzin BJ, Ernst S, Collins EJ, Mlsna D, Ready MP, Robertus JD. Crystallographic refinement of ricin to 2.5 Å. Proteins. 1991;10:240–250. doi: 10.1002/prot.340100308. [DOI] [PubMed] [Google Scholar]
- 21.Brook CL, III, Karplus MJ. Deformable stochastic boundaries in molecular dynamics. J Chem Phys. 1983;79:6312–6325. [Google Scholar]
- 22.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 23.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 24.Brook CL, III, Karplus M. Solvent effects on protein motion and protein effects on solvent motion. J Mol Biol. 1989;208:159–181. doi: 10.1016/0022-2836(89)90093-4. [DOI] [PubMed] [Google Scholar]
- 25.Ryckaert J-P, Ciccoti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 26.Alul RH, Singman CN, Zhang GR, Letsinger RL. Oxalyl-CPG: a labile support for synthesis of sensitive oligonucleotide derivatives. Nucleic Acids Res. 1991;19:1527–1532. doi: 10.1093/nar/19.7.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sproat B, Colonna F, Mullah B, Tsou D, Andrus A, Hampel A, Vinayak R. An Efficient Method for the Isolation and Purification of Oligoribonucleotides. Nucleosides Nucleotides. 1995;14:255–273. [Google Scholar]
- 28.Ward DC, Reich E, Stryer L. Fluorescence studies of nucleotides and polynucleotides. I. Formycin, 2-aminopurine riboside, 2,6-diaminopurine riboside, and their derivatives. J Biol Chem. 1969;244:1228. [PubMed] [Google Scholar]
- 29.Jucker FM, Heus HA, Yip PF, Moors EH, Pardi A. A network of heterogeneous hydrogen bonds in GNRA tetraloops. J Mol Biol. 1996;264:968–80. doi: 10.1006/jmbi.1996.0690. [DOI] [PubMed] [Google Scholar]
- 30.Correll CC, Munishkin A, Chan YL, Ren Z, Wool IG, Steitz TA. Crystal structure of the ribosomal RNA domain essential for binding elongation factors. Proc Natl Acad Sci U S A. 1998;95:13436–13441. doi: 10.1073/pnas.95.23.13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kenfack CA, Burger A, Mèly Y. Excited-state properties and transitions of fluorescent 8-vinyl adenosine in DNA. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:26327–26336. doi: 10.1021/jp064388h. [DOI] [PubMed] [Google Scholar]
- 32.Chen XY, Berti PJ, Schramm VL. Transition-State Analysis for Depurination of DNA by Ricin A-Chain. J Am Chem Soc. 2000;122:6527–6534. [Google Scholar]
- 33.Tang S, Xie L, Hou F, Liu WY, Ruan K. Non-specific deadenylation and deguanylation of naked RNA catalyzed by ricin under acidic condition. Biochim Biophys Acta. 2001;1519:192–198. doi: 10.1016/s0167-4781(01)00236-6. [DOI] [PubMed] [Google Scholar]
- 34.Yang X, Gerczei T, Glover LT, Correll CC. Crystal structures of restrictocin-inhibitor complexes with implications for RNA recognition and base flipping. Nat Struct Biol. 2001;8:968–973. doi: 10.1038/nsb1101-968. [DOI] [PubMed] [Google Scholar]
- 35.Olson MA. Ricin A-chain structural determinant for binding substrate analogues: a molecular dynamics simulation analysis. Proteins: Structure, Function, and Genetics. 1997;27:80–95. doi: 10.1002/(sici)1097-0134(199701)27:1<80::aid-prot9>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 36.Kim Y, Mlsna D, Monzingo AF, Ready MP, Frankel A, Robertus JD. Structure of a ricin mutant showing rescue of activity by a noncatalytic residue. Biochemistry. 1992;31:3294–3296. doi: 10.1021/bi00127a035. [DOI] [PubMed] [Google Scholar]
- 37.Singh V, Schramm VL. Transition-state structure of human 5’-methylthioadenosine phosphorylase. J Am Chem Soc. 2006;128:14691–14696. doi: 10.1021/ja065419p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nunez S, Antoniou D, Schramm VL, Schwartz SD. Promoting vibrations in human purine nucleoside phosphorylase. A molecular dynamics and hybrid quantum mechanical/molecular mechanical study. J Am Chem Soc. 2004;126:15720–15729. doi: 10.1021/ja0457563. [DOI] [PubMed] [Google Scholar]
- 39.Sorin EJ, Engelhardt MA, Herschlag D, Pande VS. RNA simulations: probing hairpin unfolding and the dynamics of a GNRA tetraloop. J Mol Biol. 2002;317:493–506. doi: 10.1006/jmbi.2002.5447. [DOI] [PubMed] [Google Scholar]
- 40.Schaerer OD, Verdine GL. A Designed Inhibitor of Base-Excision DNA Repair. J Am Chem Soc. 1995;117:10781–10782. [Google Scholar]
- 41.Walter AE, Turner DH, Kim J, Lyttle MH, Müller P, Mathews DH, Zuker M. Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc Natl Acad Sci USA. 1994;91:9218–9222. doi: 10.1073/pnas.91.20.9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steffen P, Voss B, Rehmsmeier M, Reeder J, Giegerich R. RNAshapes: an integrated RNA analysis package based on abstract shapes. Bioinformatics. 2006;22:500–503. doi: 10.1093/bioinformatics/btk010. [DOI] [PubMed] [Google Scholar]
- 43.Sturm M, Roday S, Schramm VL. 2006 Unpublished results. [Google Scholar]
- 44.Lang P, Magnin G, Mathis G, Burger A, Biellmann J-F. Synthesis of 8-(omega-Hydroxyalkyl)-, 8-(omega-hydroxyalk-1-enyl)-, and 8-(omega-hydroxy-3-alk-1-ynyl)adenines using the tert-butyldimethylsilyloxymethyl group, a new and versatile protecting group of adenine. J Org Chem. 2000;65:7825–7832. doi: 10.1021/jo000841o. [DOI] [PubMed] [Google Scholar]
- 45.Kirby EP, Steiner RF. Influence of solvent and temperature upon the fluorescence of indole derivatives. J Phys Chem. 1970;74:4480–4490. [Google Scholar]
- 46.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr, Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas OJ, Tomasi BV, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Gonzalez C, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian 03, revision B.04. Gaussian; Pittsburgh, PA: 2003. [Google Scholar]
- 47.Parikh BA, Tumer NE. Antiviral activity of ribosome inactivating proteins in medicine. Mini Rev Med Chem. 2004;4:523–543. doi: 10.2174/1389557043403800. [DOI] [PubMed] [Google Scholar]
- 48.Rajamohan F, Mao C, Uckun FM. Binding interactions between the active center cleft of recombinant pokeweed antiviral protein and the alpha-sarcin/ricin stem loop of ribosomal RNA. J Biol Chem. 2001;276:24075–24081. doi: 10.1074/jbc.M011406200. [DOI] [PubMed] [Google Scholar]
- 49.Day PJ, Lord JM, Roberts LM. The deoxyribonuclease activity attributed to ribosome-inactivating proteins is due to contamination. Eur J Biochem. 1998;258:540–545. doi: 10.1046/j.1432-1327.1998.2580540.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.