

Abstract
Receptor tyrosine kinases (RTKs) are essential components of signal transduction pathways that mediate cell-to-cell communication. These single-pass transmembrane receptors, which bind polypeptide ligands — mainly growth factors — play key roles in processes such as cellular growth, differentiation, metabolism and motility. Recent progress has been achieved towards an understanding of the precise (and varied) mechanisms by which RTKs are activated by ligand binding and by which signals are propagated from the activated receptors to downstream targets in the cell.
Introduction
Tyrosine phosphorylation is a highly regulated post-translational modification that is essential for inter- and intracellular communication in metazoans. The enzymes that catalyze phosphoryl transfer to tyrosine residues in protein substrates, using ATP as a phosphate donor, are the protein tyrosine kinases, of which there are 58 receptor types (RTKs) and 32 non-receptor types in the human genome [1]. The RTK family includes, among others, epidermal growth factor receptor (EGFR), platelet-derived growth factor receptors, fibroblast growth factor receptors (FGFRs), vascular endothelial growth factor receptors, Met (hepatocyte growth factor/scatter factor [HGF/SF] receptor), Ephs (ephrin receptors), and the insulin receptor. RTKs are essential components of cellular signaling pathways that are active during embryonic development and adult homeostasis. Because of their roles as growth factor receptors, many RTKs have been implicated in the onset or progression of various cancers, either through receptor gain-of-function mutations or through receptor/ligand overexpression [2].
RTKs are single-pass, type I receptors resident in the plasma membrane. Generally, RTKs are activated through ligand-induced oligomerization, typically dimerization, which juxtaposes the cytoplasmic tyrosine kinase domains [3]. For most RTKs, this juxtaposition facilitates autophosphorylation in trans of tyrosine residues in the kinase activation loop or juxtamembrane region, inducing conformational changes that serve to stabilize the active state of the kinase [4]. These and other phosphotyrosine residues serve as recruitment sites for a host of down-stream signaling proteins — enzymes and adapter/scaffolding proteins — typically through Src homology-2 (SH2) or phosphotyrosine-binding (PTB) domains, which recognize phosphotyrosine residues in specific sequence contexts [5].
Understanding the precise structural mechanisms by which ligands and co-factors induce RTK oligomerization and activation continues to be an active area of research. In addition, advances in proteomics techniques and live-cell imaging have yielded global spatial and temporal views of cellular signal propagation. This review high-lights the recent advances in these areas.
Mechanisms of activation of receptor tyrosine kinases
Several years ago, the structural mechanisms by which members of the EGFR subfamily of RTKs (EGFR/ErbB1, ErbB2, ErbB3 and ErbB4) undergo ligand-induced dimerization (2:2 ligand–receptor complex) were elucidated through crystallographic studies [6]. Despite these major advances, questions regarding the kinetics of ligand-receptor and receptor-receptor association at the cell surface remained. For example, there is evidence that EGFRs can exist in a ligand-free, pre-dimerized (but inactive) state on the cell surface [7]. Single-molecule fluorescence experiments in living cells [8•] have provided evidence for a kinetic intermediate in EGF-mediated receptor activation, in which a single EGF molecule is bound to a pre-dimerized receptor, which, through positive cooperativity, greatly facilitates binding of a second EGF molecule to the complex to trigger receptor activation. In another EGF–EGFR binding study, mathematical modeling indicates that the difference in EGF binding affinity for the tethered (autoinhibited) versus extended (dimerization-competent) states of EGFR is only two- to three-fold, implying that other autoinhibitory mechanisms are likely to be operative as well [9].
Until recently, the mechanism by which the EGFR kinase domain was activated in a ligand-stabilized receptor dimer was unresolved. All four ErbB family members contain a conserved tyrosine in the kinase activation loop, but this tyrosine (Tyr845 in EGFR) is not an autophosphorylation (in trans) site, and its phosphorylation is not required for kinase activation. Through crystallographic and biochemical studies, Zhang et al. [10••] have demonstrated that the EGFR kinase is activated by an allosteric mechanism, in which the C-terminal lobe (C-lobe) of one kinase domain within the receptor dimer makes specific contacts with the N-lobe of the other kinase domain (forming an asymmetric dimer), activating the latter (Figure 1a). This mechanism is reminiscent of the activation of cyclin-dependent kinase-2 (CDK2), a serine/threonine kinase, by cyclin A [11], and provides a satisfying explanation for how ErbB3, with an intrinsically inactive kinase domain, is still capable of activating other ErbB family members in heterodimers.
Figure 1.

Modes of RTK dimerization and downstream protein recruitment. (a) Structural model of the activated EGF–EGFR complex. On the extracellular side of the plasma membrane (colored gray and shown approximately to scale), the 2:2 EGF:EGFR complex is two-fold symmetric (dyad axis is vertical). The two receptors in the complex are colored cyan and purple (alternating light and dark coloring for the subdomains), and the two EGF molecules are colored orange (ribbon diagram with semi-transparent surface). The four subdomains of the EGFR ectodomain are, sequentially, L1–CR1–L2–CR2. The transmembrane helices are shown as cylinders, and linker segments (juxtamembrane regions [extra- and intracellular] and C-terminal tail) are drawn schematically as thick lines. On the cytoplasmic side, the two tyrosine kinase domains (N- and C-lobes colored dark and light, respectively) form an asymmetric dimer, with the C-lobe of one kinase domain (purple) interacting with the N-lobe of the other kinase domain (cyan). This interaction activates the second kinase domain (cyan) [10••]. The yellow spheres represent phosphotyrosine recruitment sites in the C-terminal tail of the cytoplasmic domain. The structures are derived from PDB codes 1IVO [48] and 1NQL [49] (ectodomain dimer) and 1M14 [50] (kinase dimer). (b) Structural model of the α2β2 insulin receptor with Grb14 bound. The insulin receptor ectodomain is two-fold symmetric (dyad axis is vertical) and consists, sequentially, of subdomains L1–CR1–L2–Fn1–Fn2–Fn3, of which L1–CR1–L2–Fn1–Fn2(N) are on the α chain and Fn2(C)–Fn3 are on the β chain (the chains are not distinguished in the figure). The tyrosine kinase domains (β chain) are colored as in (a). The BPS–SH2 portion of Grb14 (ribbon diagram with semi-transparent surface) binds to the kinase domain (2:2 complex) and inhibits catalytic activity. The SH2 domain (orange) mediates Grb14 dimerization and the BPS region (black) binds as a pseudosubstrate inhibitor in the kinase active site. The structures are derived from PDB codes 2DTG [16••] (ectodomain) and 2AUG and 2AUH [21•] (Grb14-IRK complex). Insulin is not bound to the ectodomain in this structure, but its presumed binding site (one of two equivalent sites) between L1 (α-chain 1) and Fn1 (α-chain 2) is indicated by the arrow. In both (a) and (b), the distance between the transmembrane helices is somewhat arbitrary, owing to the (presumed) flexible linkers connecting CR2 (a) and Fn3 (b).
The RTK Met is activated through binding of HGF/SF. Using electron microscopy and small-angle x-ray scattering to provide low-resolution structural constraints, Gherardi et al. [12•] have proposed a model for the active HGF-Met complex, in which a 2:2 ligand-receptor complex is stabilized through dimerization of the ‘n’ and ‘k1’ domains of HGF, with no direct receptor–receptor interactions. As is the case for most RTKs, the kinase domain of Met is stabilized in a catalytically repressed state prior to activation-loop phosphorylation. A crystal structure of the unphosphorylated Met kinase domain shows that the activation loop makes numerous interactions with residues of the N-lobe to maintain a low basal level of activity [13].
Activation of FGFRs requires both a polypeptide ligand and heparan sulfate proteoglycans [14]. In mammals, there are four receptors and over twenty FGF ligands, with alternative splicing of both ligands and receptors. Recently, the molecular basis for the differential signaling properties of alternatively spliced FGF8, a key mitogenic factor during mid-hindbrain patterning in the developing vertebrate embryo, was revealed. Structural and in vitro binding studies of FGF8a and FGF8b with FGFRs demonstrated that a single residue (Phe32) in the N-terminal region of FGF8b, which is not present in FGF8a, enhances the signaling strength of FGF8b by increasing receptor affinity and, perhaps, by enabling the kinase domains in the FGFR dimer to associate more closely than in a typical 2:2 FGF–FGFR complex [15].
Unlike the majority of RTKs, which are single-chain receptors, the insulin receptor is an α2β2 heterotetramer that undergoes a not-well-characterized structural re-arrangement upon insulin binding. Recently, the entire disulfide-linked ectodomain of the insulin receptor has been crystallized and its structure determined [16••]. The α chain of the ectodomain comprises two leucine-rich (L) domains with an interposed cysteine-rich domain, followed by an intact fibronectin type III (FnIII) domain and a partial FnIII domain. The β chain contains the second half of the second FnIII domain, which is followed by a third FnIII domain before the transmembrane helix. The tyrosine kinase domain resides in the cytoplasmic portion of the β chain. The crystal structure revealed a symmetric, ‘folded-over’ conformation in which the L1 domain of one α chain is juxtaposed with the first FnIII domain from the other α chain to form the high-affinity binding site for insulin (Figure 1b). Although many issues related to the signal transduction mechanism remain unresolved — most fundamentally, the effect of insulin binding on the relative positioning of the cytoplasmic kinase domains — this structure provides a working framework for the spatial organization in the insulin receptor ectodomain.
A subset of RTKs, including Ret and MuSK (muscle-specific kinase), do not bind their ligands directly, but require co-receptors or other accessory proteins for ligand-induced activation. Ret mediates signaling in response to the glial cell-derived neurotrophic factor (GDNF) family of neuronal growth factors, and requires a GPI-linked co-receptor for activation. Schlee et al. [17•] have provided a quantitative description of the assembly of an active GDNF–Ret–co-receptor signaling complex, consisting of one molecule of ligand (a covalent homodimer), two molecules of co-receptor, and two molecules of Ret. Moreover, the experiments revealed the sequence of steps involved in the assembly of the signaling complex and the affinities of the interactions. Crystal structures of the tyrosine kinase domain of Ret in its unphosphorylated and phosphorylated states, along with biochemical data, indicate that phosphorylation is not required for Ret kinase activation [18].
MuSK is critical for neuromuscular junction formation and is activated by neuronally derived agrin, a large heparan sulfate proteoglycan. The accessory molecule/co-receptor in muscle cells that allows MuSK to be activated by agrin has yet to be identified. A recent crystal structure of the first two immunoglobulin-like domains of MuSK, which are the agrin-responsive elements of the MuSK ectodomain, revealed a hydrophobic patch on the surface of Ig-like domain-1, which functional studies showed is important for MuSK activation by agrin [19]. Whether this patch mediates a homotypic (MuSK dimer) or heterotypic (MuSK–agrin/co-receptor) interaction was not distinguished.
Signaling mechanisms downstream of activated receptor tyrosine kinases
In most cases, the phosphotyrosine recruitment sites in RTKs are located in the C-terminal tail of the receptor, the juxtamembrane region, or the kinase insert region. These regions in RTKs are, for the most part, unstructured and are readily accessible to SH2 and PTB domains. In contrast, the phosphorylated activation loop within the insulin receptor kinase domain (IRK) has been shown to be the target for the SH2 domain-containing adapter proteins APS and Grb10/14. The IRK activation loop contains three autophosphorylated (in trans) tyrosine residues and adopts a particular conformation (i.e. is structured) upon phosphorylation. A crystal structure of the complex between phosphorylated IRK and the SH2 domain of APS reveals that the APS SH2 domain adopts a non-canonical dimeric configuration in which each protomer is capable of interacting with two phosphotyrosines in the IRK activation loop [20]. In the case of Grb10/14, the SH2 domain is also dimeric and recognizes two phosphotyrosine residues in the IRK activation loop [21•]. In addition, a ~40 amino acid region (BPS; between PH and SH2) upstream of the SH2 domain in Grb10/14 binds as a pseudosubstrate inhibitor in the peptide binding groove of the kinase [21•] (Figure 1b). As shown by gene deletion studies, Grb14 is a tissue-specific negative regulator of the insulin receptor [22].
Departing from the standard RTK signaling paradigm, studies on ErbB4 (EGFR family) have demonstrated that the extracellular juxtamembrane region of an ErbB4 splice variant (JM-a) is cleaved by a dual-protease system (TACE–presenilin) in a ligand (neuregulin)-dependent manner [23]. Recently, the liberated ErbB4 cytoplasmic domain was shown to form a complex with the adapter TAB2 and co-repressor N-CoR. This complex translocates to the nucleus and acts to repress gene transcription in neuronal precursor cells, preventing differentiation into astrocytes [24].
Proteolysis is also involved in signaling by Ephs, which bind ephrins and mediate cell motility, among other processes [25]. Unlike most RTK ligands, ephrins are attached to the plasma membrane, either by a GPI linkage (type A) or a transmembrane segment (type B). The mechanism by which ephrin–Eph interactions in trans, across opposed cells, lead to repulsion (rather than attraction) of these cells has not been fully understood. Earlier work showed that a cleavage event mediated by the metalloprotease ADAM10 is involved in the process [26]. Biochemical and structural studies [27•] now describe how ADAM10 constitutively associates with the receptor, EphA3, in the same cell and cleaves the ligand, ephrinA5, in the opposing cell upon ternary complex formation.
Global studies of receptor tyrosine kinase signaling
Studies of RTK signaling pathways have begun to move from detailed studies of individual components to system-wide analyses of entire cascades. These advances have been made possible by the development of new technologies in proteomics and functional genomics. Several recent studies have provided ‘panoramic’ views of tyrosine phosphorylation events that would have been unthinkable a few years ago.
Quantitative proteomic strategies have enabled the unbiased analysis of the temporal aspects of phosphotyrosine signaling. The mass-spectrometry-based approach SILAC (stable isotope labeling by amino acids in cell culture) [28] has been applied to the EGFR signaling network. The tyrosine phosphorylation status of 202 proteins was determined in unstimulated cells or in cells stimulated with EGF for 1, 5, 10, or 20 min [29••]. Significant changes were observed in the phosphorylation state of 81 proteins, including some that had not previously been implicated in EGFR signaling. The phosphorylation profiles illuminated the dynamics of signaling in this system. Strikingly, the data showed a sequence of activation events leading from the receptor itself to MAP kinases, with adaptor proteins and guanine nucleotide exchange factors as intermediates. In another approach, tryptic peptides from EGF-stimulated cells were chemically derivatized prior to IMAC phosphopeptide enrichment and mass spectrometry [30•]. This study permitted the identification of changes in phosphorylation at individual sites. The data from these two studies of EGF signaling were largely consistent. The SILAC method was recently extended to measure the phosphorylation of 2244 proteins at 6600 tyrosine, serine and threonine sites after EGF stimulation [31•]. About half of the proteins contained multiple sites of phosphorylation; in many cases, the multiple sites were phosphorylated with different kinetics, consistent with the role of such proteins as integrating devices.
Similar mass spectrometry-based strategies have been applied to other RTKs. Stable isotope labeling was used to identify proteins with increased tyrosine phosphorylation upon ephrinB1 stimulation of cells [32], and to study insulin signaling in 3T3-L1 adipocytes that were treated with insulin for various lengths of time [33]. Both of these studies established the dynamics of phosphorylation for previously identified substrates and for additional proteins that potentially represent novel effectors. For example, four proteins implicated in GLUT4 translocation were tyrosine phosphorylated in response to insulin [33]. Two recent reports have focused on signaling by ErbB2, a protein that is overexpressed in 20–30% of human breast cancers and is associated with a poor prognosis. A large number of phosphorylation sites were identified in ErbB2-expressing cells [34,35]. In both studies, parallel experiments were carried out in the presence of the ErbB2-selective inhibitors Herceptin or PD168393. The inhibitors reversed many of the phosphorylation events observed in ErbB2-expressing cells, confirming their relevance to ErbB2 signaling. These results demonstrate that phosphotyrosine proteomic studies can be used to understand the effects of kinase inhibitors on cellular signaling networks.
Protein microarrays containing immobilized SH2 and PTB domains have also been used in global studies of EGFR signaling. Binding strengths were measured for phosphotyrosine-containing peptides representing the autophosphorylation sites on ErbB1–4. The resulting apparent equilibrium dissociation constants were used to develop a quantitative interaction network for the receptors [36•]. The data reveal the degree of selectivity for different autophosphorylation sites at different thresholds of affinity. ErbB1 (EGFR) and ErbB2 become more promiscuous as the affinity threshold is lowered, suggesting that overexpression of these proteins could trigger signaling through alternative pathways. Another proteomic approach to studying SH2-domain-dependent signaling is to couple the domains to oligonucleotide tags, and to use the binding properties of the tagged domains to profile tyrosine phosphorylation in a sample [37]. The interactions revealed by this approach (termed oligonu-cleotide-tagged multiplex assay or OTM) can then be quantified by PCR. The approach is extremely sensitive, as demonstrated by phosphotyrosine fingerprinting of samples from tumor cell lines and human leukemia patients [37].
The proteomic studies highlighted above focused on tyrosine-phosphorylated proteins or on proteins that interact physically with RTKs or SH2 domains. In a complementary approach aimed at identifying regulatory components that need not interact directly with RTKs, a genome-wide RNA interference-based screen was carried out in D. melanogaster cells [38•]. Following dsRNA knockdown and insulin stimulation, cells were screened using a reporter for the active form of ERK. A number of genes were found to influence, positively or negatively, ERK signaling, including genes involved in trafficking, cytoskeletal maintenance, transcription and cell proliferation. To determine which genes were required in response to specific RTK activation, the initial hits were subsequently re-screened after stimulation of several RTKs in turn. This functional genomic approach provides a global view of the critical components and connections in the RTK regulatory network; data for individual RTKs can now be integrated with the relevant proteomics studies.
Conclusions
Because of their key roles in mediating cellular proliferation, RTKs, as well as non-receptor tyrosine kinases such as Src and Abl, are attractive candidates for therapeutic intervention. Over the past 15 years, much has been learned regarding the regulatory mechanisms that govern RTK expression, ligand activation, downstream signaling and downregulation. This fundamental biochemical information has been crucial in the quest to develop targeted therapeutics, both to the extracellular regions of RTKs using monoclonal antibodies and to the cytoplasmic (kinase) domains using small-molecule inhibitors (Figure 2). With regard to the latter, imatinib (Gleevec) inhibition of Bcr–Abl in chronic myeloid leukemia (CML) continues to serve as a paradigm [39,40].
Figure 2.
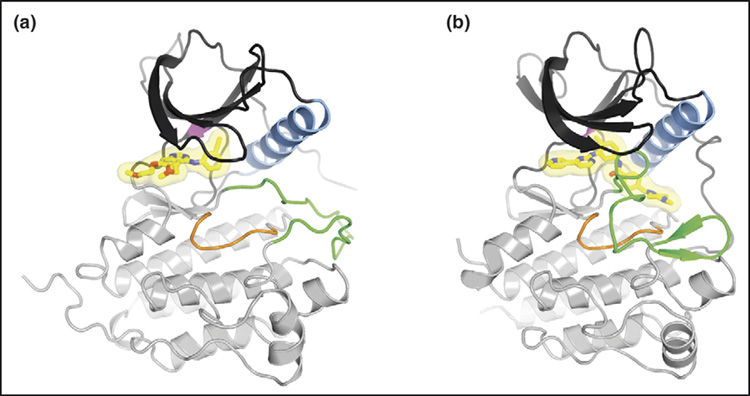
Binding of small-molecule inhibitors to RTKs. The ATP-competitive inhibitors are shown in stick representation with a semi-transparent surface. Carbon atoms are colored yellow, oxygen atoms red, and nitrogen atoms blue. (a) Crystal structure of the tyrosine kinase domain of the EGFR kinase domain in complex with erlotinib (Tarceva) [50]. (b) Crystal structure of the tyrosine kinase domain of Kit in complex with imatinib (Gleevec) [51]. For both (a) and (b), the N-lobe of the kinase is colored dark gray, the C-lobe light gray, α-helix C (N-lobe) light blue, the catalytic loop (C-lobe) orange, and the activation loop (C-lobe) green. The activation loop in (a) is in an active state, whereas the activation loop in (b) is in an autoinhibited, inactive state. The ‘gatekeeper’ residue (Thr790 in EGFR, Thr670 in Kit), which when mutated causes drug resistance to both erlotinib and imatinib, is colored magenta (in the back of the N-lobe).
The ATP-competitive inhibitors gefitinib (Iressa) and erlotinib (Tarceva), which target the EGFR kinase, show promise in the treatment of non-small-cell lung cancer (NSCLC). In clinical trials, dramatic responses to gefitinib were realized by a subset of NSCLC patients, and were correlated with specific point mutations and deletions in EGFR [41–43]. Yet, just as with imatinib treatment of CML patients, NSCLC patients treated with gefitinib are subject to relapse, due to secondary mutation of the ‘gatekeeper’ residue in the ATP binding pocket, which confers resistance to both drugs [44,45].Several compounds have now been identified that are effective against such secondary mutations in Bcr-Abl, EGFR, and Kit [46], and novel strategies for the development of conformation-selective tyrosine kinase inhibitors have been reported [47•], which exploit the generally greater conformational differences between inactive states of kinases than between active states. The next five years or so should provide a good indication as to how well we are able to translate the accrued mechanistic knowledge of RTK signaling into clinical efficacy.
Acknowledgments
Due to space limitations, we regret that we were not able to highlight all of the recent relevant studies on RTK signaling mechanisms. Research support is acknowledged from the National Institutes of Health (DK052916 and NS053414 to SRH; CA058530 to WTM).
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 2.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 3.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 4.Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:464–471. doi: 10.1038/nrm1399. [DOI] [PubMed] [Google Scholar]
- 5.Pawson T, Gish GD, Nash P. SH2 domains, interaction modules and cellular wiring. Trends Cell Biol. 2001;11:504–511. doi: 10.1016/s0962-8924(01)02154-7. [DOI] [PubMed] [Google Scholar]
- 6.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 7.Moriki T, Maruyama H, Maruyama IN. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J Mol Biol. 2001;311:1011–1026. doi: 10.1006/jmbi.2001.4923. [DOI] [PubMed] [Google Scholar]
- 8•.Teramura Y, Ichinose J, Takagi H, Nishida K, Yanagida T, Sako Y. Single-molecule analysis of epidermal growth factor binding on the surface of living cells. EMBO J. 2006;25:4215–4222. doi: 10.1038/sj.emboj.7601308.This single-molecule fluorescence study provides quantitative kinetic parameters for the activation of EGFR by EGF.
- 9.Özcan F, Klein P, Lemmon MA, Lax I, Schlessinger J. On the nature of low- and high-affinity EGF receptors on living cells. Proc Natl Acad Sci USA. 2006;103:5735–5740. doi: 10.1073/pnas.0601469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10• •.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013.This structural and biochemical study establishes that the EGFR kinase is activated through an allosteric mechanism resembling activation of CDK2 by cyclin A. This study also establishes that the inactive state of the EGFR kinase is Src-like.
- 11.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 12•.Gherardi E, Sandin S, Petoukhov MV, Finch J, Youles ME, Ofverstedt LG, Miguel RN, Blundell TL, Vande Woude GF, Skoglund U, et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc Natl Acad Sci USA. 2006;103:4046–4051. doi: 10.1073/pnas.0509040103.Electron microscopy and small-angle x-ray scattering data are used to develop a structural model for the active 2:2 HGF–Met complex.
- 13.Wang W, Marimuthu A, Tsai J, Kumar A, Krupka HI, Zhang C, Powell B, Suzuki Y, Nguyen H, Tabrizizad M, et al. Structural characterization of autoinhibited c-Met kinase produced by coexpression in bacteria with phosphatase. Proc Natl Acad Sci USA. 2006;103:3563–3568. doi: 10.1073/pnas.0600048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 15.Olsen SK, Li JY, Bromleigh C, Eliseenkova AV, Ibrahimi OA, Lao Z, Zhang F, Linhardt RJ, Joyner AL, Mohammadi M. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 2006;20:185–198. doi: 10.1101/gad.1365406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16••.McKern NM, Lawrence MC, Streltsov VA, Lou MZ, Adams TE, Lovrecz GO, Elleman TC, Richards KM, Bentley JD, Pilling PA, et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature. 2006;443:218–221. doi: 10.1038/nature05106.Using two different Fab antibody fragments, the entire ectodomain of the insulin receptor is crystallized and its three-dimensional structure solved. Although the structure lacks insulin, it spatially establishes the domain organization within the ectodomain.
- 17•.Schlee S, Carmillo P, Whitty A. Quantitative analysis of the activation mechanism of the multicomponent growth-factor receptor Ret. Nat Chem Biol. 2006;2:636–644. doi: 10.1038/nchembio823.Using binding and phosphorylation measurements from live cells, along with mathematical modeling, the authors provide a quantitative description of the formation of the ART–Ret–GFRα3 signaling complex.
- 18.Knowles PP, Murray-Rust J, Kjaer S, Scott RP, Hanrahan S, Santoro M, Ibanez CF, McDonald NQ. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem. 2006;281:33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- 19.Stiegler AL, Burden SJ, Hubbard SR. Crystal structure of the agrin-responsive immunoglobulin-like domains 1 and 2 of the receptor tyrosine kinase MuSK. J Mol Biol. 2006;364:424–433. doi: 10.1016/j.jmb.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu J, Liu J, Ghirlando R, Saltiel AR, Hubbard SR. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Mol Cell. 2003;12:1379–1389. doi: 10.1016/s1097-2765(03)00487-8. [DOI] [PubMed] [Google Scholar]
- 21•.Depetris RS, Hu J, Gimpelevich I, Holt LJ, Hubbard SR. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol Cell. 2005;20:325–333. doi: 10.1016/j.molcel.2005.09.001.The crystal structure of the complex between the insulin recepotor kinase and the BPS region of Grb 14 illuminates the mechanism by which Grb 14 functions as a negative regulator of insulin signaling; the BPS region acts as a pseudosubstrate inhibitor.
- 22.Cooney GJ, Lyons RJ, Crew AJ, Jensen TE, Molero JC, Mitchell CJ, Biden TJ, Ormandy CJ, James DE, Daly RJ. Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J. 2004;23:582–593. doi: 10.1038/sj.emboj.7600082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carpenter G. ErbB-4: mechanism of action and biology. Exp Cell Res. 2003;284:66–77. doi: 10.1016/s0014-4827(02)00100-3. [DOI] [PubMed] [Google Scholar]
- 24.Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G. Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell. 2006;127:185–197. doi: 10.1016/j.cell.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 25.Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- 26.Hattori M, Osterfield M, Flanagan JG. Regulated cleavage of a contact-mediated axon repellent. Science. 2000;289:1360–1365. doi: 10.1126/science.289.5483.1360. [DOI] [PubMed] [Google Scholar]
- 27•.Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123:291–304. doi: 10.1016/j.cell.2005.08.014.This study provides mechanistic details governing ADAM10 cleavage of ephrinA5–EphA4, which underlies cell–cell repulsion as mediated by ephrin–Eph interactions.
- 28.Mann M. Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 29••.Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotechnol. 2004;22:1139–1145. doi: 10.1038/nbt1005.This paper describes the first in-depth study of the temporal dimension of the EGFR signaling network, using the SILAC proteomic approach. The authors study the tyrosine phosphorylation status of a number of proteins after stimulation with EGF for various periods of time.
- 30•.Zhang Y, Wolf-Yadlin A, Ross PL, Pappin DJ, Rush J, Lauffenburger DA, White FM. Time-resolved mass spectrometry of tyrosine phosphorylation sites in the epidermal growth factor receptor signaling network reveals dynamic modules. Mol Cell Proteomics. 2005;4:1240–1250. doi: 10.1074/mcp.M500089-MCP200.A chemical derivatization strategy is used to study tyrosine phosphorylated proteins in EGF-stimulated cells.
- 31•.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026.An expanded proteomics study of the EGF signaling network in which the phosphorylation of 2,244 proteins at 6,600 tyrosine, serine, and threonine sites is reported. The data from this study have been compiled into a publicly accessible database called Phosida (http://www.phosida.com).
- 32.Zhang G, Spellman DS, Skolnik EY, Neubert TA. Quantitative phosphotyrosine proteomics of EphB2 signaling by stable isotope labeling with amino acids in cell culture (SILAC) J Proteome Res. 2006;5:581–588. doi: 10.1021/pr050362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmelzle K, Kane S, Gridley S, Lienhard GE, White FM. Temporal dynamics of tyrosine phosphorylation in insulin signaling. Diabetes. 2006;55:2171–2179. doi: 10.2337/db06-0148. [DOI] [PubMed] [Google Scholar]
- 34.Bose R, Molina H, Patterson AS, Bitok JK, Periaswamy B, Bader JS, Pandey A, Cole PA. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc Natl Acad Sci U S A. 2006;103:9773–9778. doi: 10.1073/pnas.0603948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherji M, Brill LM, Ficarro SB, Hampton GM, Schultz PG. A phosphoproteomic analysis of the ErbB2 receptor tyrosine kinase signaling pathways. Biochemistry. 2006;45:15529–15540. doi: 10.1021/bi060971c. [DOI] [PubMed] [Google Scholar]
- 36•.Jones RB, Gordus A, Krall JA, MacBeath G. A quantitative protein interaction network for the ErbB receptors using protein microarrays. Nature. 2006;439:168–174. doi: 10.1038/nature04177.Protein microarrays are used to measure the strength of interaction between purified SH2 or PTB domains and phosphotyrosine-containing peptides representing the autophosphorylation sites on ErbB1-4. The data reveal the degree of selectivity for different autophosphorylation sites at different thresholds of affinity.
- 37.Dierck K, Machida K, Voigt A, Thimm J, Horstmann M, Fiedler W, Mayer BJ, Nollau P. Quantitative multiplexed profiling of cellular signaling networks using phosphotyrosine-specific DNA-tagged SH2 domains. Nat Methods. 2006;3:737–744. doi: 10.1038/nmeth917. [DOI] [PubMed] [Google Scholar]
- 38•.Friedman A, Perrimon N. A functional RNAi screen for regulators of receptor tyrosine kinase and ERK signalling. Nature. 2006;444:230–234. doi: 10.1038/nature05280.A functional genomics study in D. melanogaster cells that identifies genes involved in positive or negative regulation of ERK following RTK stimulation.
- 39.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 40.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science. 2000;289:1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 41.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 42.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 43.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 46.Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK, Milanov ZV, Atteridge CE, Biggs WH, 3rd, Edeen PT, et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA. 2005;102:11011–11016. doi: 10.1073/pnas.0504952102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Okram B, Nagle A, Adrian FJ, Lee C, Ren P, Wang X, Sim T, Xie Y, Xia G, Spraggon G, et al. A general strategy for creating ‘inactive-conformation’ abl inhibitors. Chem Biol. 2006;13:779–786. doi: 10.1016/j.chembiol.2006.05.015.A structure-based drug design study is reported that describes how novel ATP-competitive inhibitors can be designed to target the inactive conformation of protein kinases, in this case Abl; in general, inactive-state inhibitors exhibit higher specificity and affinity than active-state inhibitors.
- 48.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M, et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell. 2002;110:775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 49.Ferguson KM, Berger MB, Mendrola JM, Cho HS, Leahy DJ, Lemmon MA. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol Cell. 2003;11:507–517. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 50.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the EGF receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;23:23. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 51.Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang BC, Wilson KP. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 2004;279:31655–31663. doi: 10.1074/jbc.M403319200. [DOI] [PubMed] [Google Scholar]