

Abstract
Acute lung injury in humans is characterized histopathologically by neutrophilic alveolitis, injury of the alveolar epithelium and endothelium, hyaline membrane formation, and microvascular thrombi. Different animal models of experimental lung injury have been used to investigate mechanisms of lung injury. Most are based on reproducing in animals known risk factors for ARDS, such as sepsis, lipid embolism secondary to bone fracture, acid aspiration, ischemia-reperfusion of pulmonary or distal vascular beds, and other clinical risks. However, none of these models fully reproduces the features of human lung injury. The goal of this review is to summarize the strengths and weaknesses of existing models of lung injury. We review the specific features of human ARDS that should be modeled in experimental lung injury and then discuss specific characteristics of animal species that may affect the pulmonary host response to noxious stimuli. We emphasize those models of lung injury that are based on reproducing risk factors for human ARDS in animals and discuss the advantages and disadvantages of each model and the extent to which each model reproduces human ARDS. The present review will help guide investigators in the design and interpretation of animal studies of acute lung injury.
Keywords: acute respiratory distress syndrome, human lung injury
THE CHALLENGE OF MODELING HUMAN LUNG INJURY
Acute lung injury (ALI) and its most severe manifestation, the acute respiratory distress syndrome (ARDS), is a clinical syndrome defined by acute hypoxemic respiratory failure, bilateral pulmonary infiltrates consistent with edema, and normal cardiac filling pressures (5, 235). The fundamental mechanisms that initiate and propagate the lung injury have not been defined completely, despite almost 35 years of intense investigation. Human studies have provided important descriptive information about the onset and evolution of the physiological and inflammatory changes in the lungs. This information has led to hypotheses about mechanisms of injury, but for the most part, these hypotheses have been difficult to test in humans because of the many clinical variables that are difficult to control in critically ill patients.
Animal models provide a bridge between patients and the laboratory bench. Hypotheses generated in human studies can be tested directly in animal models, and the results of studies in more simple in vitro systems can be tested in animal models to assess their relevance in intact living systems. Mechanistic studies can be performed either by treating normal animals with inhibitors that block key steps in specific pathways or by creating animals with specific gene alterations to test the importance of single gene products or pathways. Without animal models there would be no way to test clinical hypotheses generated in patients using intact biological systems, and there would be no way to validate the importance of fundamental laboratory findings without going directly to human experimentation.
Animal model studies are most helpful if the characteristics of the model are directly relevant to humans. Animal model studies are also valuable if they show that a concept proven in cellular or subcellular systems is relevant in a complex animal, even if the human relevance of the specific animal model is uncertain. This is often the case when basic laboratory observations are tested in mice. For example, showing that an inhibitor of a specific cellular pathway protects mice from sepsis or oxidant-induced lung injury suggests that the pathway is important in living animals, but the findings need to be tested further in models closer to humans. Animal studies are hierarchical, as studies with mice or lower animals are relatively simple and rapid to do, but the relevance for humans is limited. On the other hand, studies in primates are relevant for humans, but are complex to organize, require scarce resources, and are expensive.
The purpose of this review is to summarize the strengths and weaknesses of different types of animal models that have been used to study the mechanisms and treatment of ALI. We will review the characteristics of human lung injury that need to be modeled in animal systems and then discuss the characteristics of different animals that need to be considered in designing experimental studies. We will summarize model systems that have been used and the major experimental findings. Last, we will suggest new directions that need to be considered in refining the use of animal models for the study of ALI in humans.
What Should A Model Model?
Ideally, animal models of ALI should reproduce the mechanisms and consequences of ALI in humans, including the physiological and pathological changes that occur (Table 1). Because the responses of the lungs to injury change with time, the evolution of the injury and repair should also be reproduced in the model, even if specific studies focus on a narrow time frame. In humans, the intrapulmonary inflammatory response begins before the onset of clinically defined ALI and is most intense in the first 3 days after the onset of ALI/ARDS (78, 158). The acute inflammatory phase is followed by a chronic fibroproliferative phase in patients who remain intubated and mechanically ventilated. In patients who survive, pulmonary function tests often show restriction, consistent with the parenchymal fibrosis seen in lung biopsies or autopsy specimens (137, 173).
Table 1.
Characteristics of human lung injury
| Clinical features | Acute onset |
| Diffuse bilateral alveolar injury | |
| Acute exudative phase | |
| Repair with fibrosis | |
| Physiological changes | V/Q abnormalities |
| Severe hypoxemia | |
| Decreased compliance | |
| Impaired alveolar fluid clearance | |
| Biological changes | Increased endothelial and epithelial permeability |
| Increase in cytokine concentrations in the lungs | |
| Protease activation | |
| Coagulation abnormalities | |
| Pathological changes | Neutrophilic alveolar infiltrates |
| Intra-alveolar coagulation and fibrin deposition | |
| Injury of the alveolar epithelium with denudation of the basement membrane |
The acute physiological changes at the onset of ALI/ARDS in humans include severe hypoxemia that is usually responsive to positive end-expiratory pressure (PEEP) and a fall in total thoracic compliance caused by reduced lung compliance. A marked increase in extravascular lung water is caused by an increase in endothelial and epithelial permeability, associated with an impairment in the ability of the alveolar epithelium to actively clear alveolar fluid (37, 134, 193, 234). These changes cause severe ventilation/perfusion mismatching and an increase in shunt fraction. Studies that have sampled lung alveolar fluids either by bronchoalveolar lavage (BAL) or by direct aspiration of edema fluid using deep airway suction have shown that the hallmark is an acute neutrophilic inflammatory response with fibrin-rich proteinaceous exudates containing an array of pro- and anti-inflammatory cytokines (reviewed in Ref. 165). ALI/ARDS also occurs in neutropenic patients, when alveolar structures are damaged by ionizing radiation or chemotherapeutic agents (156). Markers of the fibrotic response such as procollagen peptide III are evident as soon as the lung injury begins (29, 130) and are greatest in patients who die (29, 130). Surfactant abnormalities were suspected when ALI/ARDS was first described, and abnormalities of surfactant composition and function have been described in a number of studies (80, 81, 164).
Detailed studies of lung pathology in patients at the onset and during the course of ALI/ARDS are limited, and information is derived from the study of the lungs of patients who died or underwent late open lung biopsy (6, 10). These studies confirm the severe epithelial and endothelial injury and the late fibroproliferative response (Fig. 1). The injury to the epithelium tends to be more pronounced than the endothelial injury (10), but endothelial injury and microvascular thrombi are detectable, supporting angiographic findings in humans with ALI/ARDS (218). The hallmark of the late fibroproliferative response is injury of the alveolar basement membrane, with fibroblast proliferation and increases in types I and III collagen in the alveolar walls and the alveolar spaces (173).
Fig. 1.
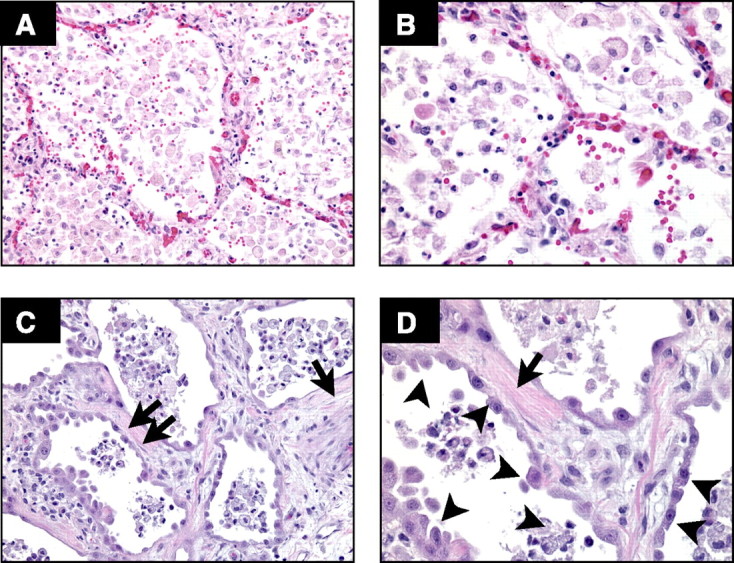
Human ARDS. Photomicrographs from the lungs of 2 different patients with ARDS stained with H&E. The alveolar spaces are filled with a mixed mononuclear/neutrophilic infiltrate, the alveolar walls are thickened, and the septae are edematous. Note the presence of cellular debris and proteinaceous material in the air spaces (A, magnification ×200; B, ×400). In later stages, there is a fibroproliferative response with collagen deposition in the alveolar walls (arrows). Note that the alveolar epithelium has been replaced with cuboidal cells (arrowheads). Magnification in C, ×200; D, ×400.
To simulate human ALI/ARDS, animal models should reproduce the acute injury to the epithelial and endothelial barriers in the lungs and the acute inflammatory response in the air spaces. Ideally, the injury should evolve over time if the animals are supported for prolonged periods. One of the most difficult aspects of modeling human ALI in humans is that the lungs of humans can be affected by the mechanisms involved in the primary illness (e.g., sepsis) and/or they can be affected by therapeutic modalities used for supportive care (e.g., mechanical ventilation) (224). This increases the complexity of the ideal animal model of lung injury, as it requires the incorporation of specific treatment modalities into experimental protocols, adding additional variables to the experimental design.
No single animal model reproduces all of the characteristics of ALI/ARDS in humans, and most of the existing animal models are relevant for only limited aspects of human ALI/ARDS. Nevertheless, if the characteristics of an animal model are well understood, and the results are interpreted within the limits of the specific model, animal studies can provide focused tests of key elements of the lung injury response in humans.
UNIQUE CHARACTERISTICS OF ANIMAL SPECIES IN MODELING HUMAN LUNG INJURY
A number of different animal species have been used to study lung injury, yet there are important and sometimes major differences among animal species in responses to injury, particularly injury in response to microbial products. This section highlights some of the critical species differences that should be considered when modeling human lung injury. In addition, special considerations such as size, availability of species-specific reagents, and the use of genetically modified animals are discussed.
Species Differences in the Innate Immune Response
Species differences in Toll-like receptors.
Early investigators studying the physiological consequences of endotoxin recognized that differences existed between animal species in the development of ALI (123). Because humans and mice diverged as separate species millions of years ago, they have adapted to pathogens that are specific to their unique environmental niches, so it is not surprising that differences exist in the innate immune response of humans and mice to microbes and microbial products. Leukocytes and some mesenchymal cells of humans and lower animals recognize microbial products via a group of specialized pattern recognition receptors called Toll-like receptors. The work of Hajjar and colleagues (83) suggested evolutionary divergence in these receptors by showing that Toll-like receptor 4 (TLR4) from humans and mice recognize different lipopolysaccharide structures. Other examples include species differences in TLR2 (133, 146), TRL3 (89), TLR7, TLR8 (248), and TLR9 (100) (reviewed in Ref. 175). Species differences in innate immune responses need to be taken into account in selecting animal models of ALI for specific experimental protocols.
Species differences in the mononuclear phagocyte system.
The mononuclear phagocyte system (MPS) is composed of macrophages and macrophage precursors such as blood monocytes. Intravascular macrophages directly face the circulation where they can bind to and ingest microorganisms, endotoxin, and particles circulating in blood. The distribution of intravascular macrophages varies across species. In many species, intravascular macrophages are restricted to the spleen and liver, e.g., splenic macrophages and Kupffer cells. But in some species, the lungs contain pulmonary intravascular macrophages (PIM), a resident population of mature macrophages that adhere to endothelial cells in pulmonary capillaries.
The PIM are found in the lungs of many species including sheep, cattle, pigs, cats, goats, horses, and cetaceans. In species that have PIM, particulate matter and LPS in blood are localized to the lungs. In contrast, species that lack PIM such as the dog, rat, mouse, rabbit, and non-human primates, localize particulate matter in blood to the liver and spleen (24). Most evidence suggests that PIM are not part of the MPS in normal humans (236). The role of PIMs in the development of lung injury was investigated in studies through depletion of these macrophages in sheep, which decreased endotoxin-induced lung injury (206). Therefore, the presence of PIM increases the susceptibility of a species to lung injury (Table 2), which should be taken into account when developing animal models of human lung injury (236).
Table 2.
Unique characteristics of animal species relevant to modeling lung injury
| Animal | % Identity with Human TLR4 HVR | Pulmonary Intravascular Macrophages | LPS Sensitivity | Nitric Oxide Production |
|---|---|---|---|---|
| Human | 100% | No | Intermediate | + |
| NHP | 95% | No | Intermediate | + |
| Pig | ND | Yes | High | ++ |
| Dog | ND | No | Low | ++ |
| Sheep | ND | Yes | High | ++ |
| Rabbit | 57% | No | Intermediate | ++ |
| Rat | 48% | No | Low | +++ |
| Mouse | 48% | No | Low | +++ |
HVR, hypervariable region of TLR4; NH, nonhuman primate; ND, not determined.
Species differences in nitric oxide.
Clinical studies and animal models suggest that nitric oxide (NO) is involved in the pathogenesis of lung injury (202, 232), and important species-dependent differences in NO pathways have been described. For example, the production of NO during the TLR-mediated antimicrobial response to Mycobacterium tuberculosis varies among species. In humans, macrophages kill M. tuberculosis through a vitamin D-dependent response (127). In contrast, mice, nocturnal animals with minimal exposure to sunlight, have developed a TLR-mediated pathway in macrophages, which kills M. tuberculosis through NO-dependent mechanisms (216). Additional studies show that human and rodent macrophages differ significantly in their ability to produce NO. Rodent macrophages produce large amounts of NO, whereas normal nonactivated human macrophages make very little NO (187, 188). Macrophages of cattle produce NO, whereas macrophages of hamsters, monkeys, goats, and pigs are more like human macrophages and generate very little NO (48, 104, 106, 188). Variability in the production of NO by macrophages of different species must be taken into consideration when extrapolating results from animal studies to humans (Table 2).
Species differences in chemokines and chemokine receptors.
CXCL8/interleukin-8 is a potent neutrophil chemotactic factor that has been implicated in the pathogenesis of ALI (15, 139). CXCL8 and related CXC-chemokines are produced by all species in response to bacterial products. Mice and rats lack the gene for CXCL8 but produce two related CXC chemokines, KC and macrophage inflammatory protein-2 (MIP-2). KC and MIP-2 share more sequence identity with human CXCL1-3/GROα, β, and γ than they do with CXCL8 (253) but are considered the functional homologs of human CXCL8 because of their critical role in the recruitment of neutrophils into lungs (219). Recent work by Fan and colleagues (58) demonstrated that mice express two CXC-receptors, CXCR1 and CXCR2, similar to humans, and that human CXCL8 and mouse GCP-2/CXCL6 bind murine CXCR1. In contrast, murine MIP-2 and KC activate neutrophils by binding to murine CXCR2 and do not bind to murine CXCR1. Additional work is needed to determine the role of murine GCP-2/CXCL6 in the development of ALI in mice.
Special Considerations
Animal size.
Animal size is an important consideration in selecting animal models of ALI. This is especially true when physiological parameters such as arterial oxygen tension and mean arterial pressure are monitored (135). Until recently, body size has prohibited the measurement of physiological parameters in mice. With the development of small implantable radiotelemetry transmitters, the measurement of systemic blood pressure, heart rate, and temperature is now possible in unrestrained mice, although it remains difficult and expensive (27). Size is also an important consideration for the collection of blood. It is easier to obtain sufficient quantities of blood or to obtain multiple blood samples in larger species such as the rabbit or nonhuman primates. The ability to obtain multiple blood samples is important when the study design requires monitoring of physiological parameters, such as blood gases, plasma cytokines, and leukocyte counts over time. Conversely, cost constraints, the amount of reagent required, and availability can limit the use of larger species.
Species-specific reagents.
Investigators are often faced with a lack of species-specific reagents to measure inflammatory mediators, receptors, or other proteins, which are involved in the pathogenesis of ALI. To circumvent this problem, cross-species reagents have been used. In several studies in which comparisons were made between species-specific and cross-species ELISAs, species-specific ELISAs produced more sensitive and specific results (107, 108). While this does not exclude their use, cross-species ELISA may underestimate the amount of a specific protein in a biological sample.
The Mouse as a Tool to Study the Biology of ALI
Mouse models of human disease are widely used because of the availability of specific reagents and the development of genetically modified mice that can be used to evaluate the physiological function of specific genes. Genetically modified mice have been used extensively in the study of inflammatory processes in the lungs. A number of excellent reviews of the production of genetically modified mice are available (56, 97, 203) (reviewed in Refs. 22, 41, 91, 252). An important consideration when using genetically modified mice is the selection of appropriate controls to eliminate genetic and environmental background effects (200). The best controls are congenic mice, which are essentially isogenic with an inbred strain of mice except for the gene of interest. Another option is to use the offspring of heterozygous breeding pairs, so that age- and sex-matched littermates can be used to minimize environmental and genetic variability. From a practical standpoint it is often difficult to use congenic mice or the offspring of heterozygous breeding pairs because of the number of animals involved. However, it is important to understand how to select appropriate controls for experiments using transgenic mice so that robust conclusions can be drawn about the function of specific genes.
ALI has many etiologies, suggesting that numerous genetic, environmental, and developmental factors are involved in the progression of this clinical syndrome. In recent years, genomic, proteomic, statistical, and computational resources have been applied to developing animal models that can be used to investigate complex genetic traits. Specific examples include association mapping in classic inbred mouse strains (32, 160). By taking full advantage of the genetic power of mice, investigators will be able to better understand complex disease processes such as ALI. However, it is also important to remember that mice and humans have evolved under different evolutionary pressures, so caution is needed when extrapolating findings in mouse models to human disease.
AGENTS TO INDUCE LUNG INJURY/MODEL SYSTEMS
Most animal models of ALI are based on clinical disorders that are associated with ALI/ARDS in humans. These include sepsis, multiple transfusions, multiple trauma, aspiration of gastric contents, and reperfusion of ischemic tissues (95, 163). Exposure to high concentrations of oxygen is another factor associated with ALI, but the data linking exposure to high concentrations of oxygen with the development of human lung injury are limited (12). Investigators have tried to reproduce each of these risk factors in a variety of animals, and the frequency with which each model has been reported in the English literature within the past 5 years is shown in Table 3.
Table 3.
Number of papers indexed in PubMed using animal models of acute lung injury in the English literature from 2003–2007
| n | % | |
|---|---|---|
| Mechanical ventilation | 436 | 30% |
| LPS | 279 | 19% |
| Live bacteria | 224 | 16% |
| Hyperoxia | 175 | 12% |
| Bleomycin | 149 | 10% |
| Oleic acid | 79 | 5% |
| Cecal ligation and puncture | 61 | 4% |
| Acid aspiration | 38 | 3% |
| Total | 1,441 | 100% |
In this section, we will review models of lung injury that target primarily the vascular endothelium, the alveolar epithelium, or more than one site. We will emphasize the similarities and differences between each model and human lung injury, comment on species variability within each model, and identify the major advantages and disadvantages of each model (Table 4). Particular emphasis has been placed on models that attempt to reproduce an acute neutrophilic alveolitis similar to that seen in ARDS.
Table 4.
Animal models of lung injury
| Model (Ref.) | Similarities with ARDS | Differences with ARDS | Technical Issues |
|---|---|---|---|
| Oleic acid (191) | Acute and repair phases with similar histopathological and physiological features to human ARDS | Only a fraction of human ARDS is caused by fat embolism Does not model the physiopathology of septic ARDS | Good reproducibility Requires intravenous injection of oleic acid, which can be difficult in small animals |
| LPS (239) | Neutrophilic inflammatory response with increase in intrapulmonary cytokines | The changes in alveolar-capillary permeability are mild | Very reproducible |
| Acid aspiration (141) | Disruption of the alveolar/capillary barrier with neutrophilic infiltration | Humans aspirate gastric contents, not pure acid | Very reproducible Narrow difference between injurious and noninjurious doses |
| Hyperoxia (67) | Acute phase of epithelial injury and neutrophilic infiltration followed by type II cell proliferation and scarring | In normal human lungs, 100% oxygen has not induced lung injury; it is unclear whether hyperoxia is involved in the pathogenesis of ARDS | Good reproducibility Requires special equipment to administer and monitor the desired gas concentrations |
| Bleomycin (143) | Acute inflammatory injury followed by reversible fibrosis | No formation of hyaline membranes. Physiopathological relevance unclear | Good reproducibility |
| Saline lavage (49, 124) | Depletion of surfactant Decreased lung compliance Impaired gas exchange | Without an additional stimulus, there is minimal impairment of permeability and little PMN recruitment | Animals must be anesthetized, intubated, and ventilated throughout the procedure and afterwards |
| Pulmonary ischemia/reperfusion (186) | Increase in pulmonary vascular permeability PMN infiltration | The injury is usually hemorrhagic | Requires complex animal surgery |
| Nonpulmonary ischemia/reperfusion (120) | Increased microvascular permeability and PMN sequestration in the lungs | The injury is mild, and the inflammatory component mostly limited to the interstitium | Requires complex surgery |
| Intravenous bacteria (39) | Interstitial edema, intravascular congestion, PMN sequestration | Minimal neutrophilic alveolitis; no hyaline membrane formation | Important biological variability |
| Intrapulmonary bacteria (64) | Increased permeability, interstitial edema, neutrophilic alveolitis | Positive cultures rare in early ARDS (14) | Important biological variability |
| Peritonitis (135) | Increased permeability, variable degrees of neutrophilic alveolitis | Minimal hyaline membrane formation | Biological variability Lethal dose close to injury dose |
| Cecal ligation and puncture (223) | Increased permeability, variable neutrophilic alveolitis | Minimal hyaline membrane formation | Biological variability Surgery required |
Models That Target Primarily the Capillary Endothelium
Oleic acid.
Basis for the model: oleic acid (cis-9-octadecenoic acid) is the most common free fatty acid in mammals, representing 60% of the free fatty acid pool (61). Oleic acid also represents ∼50% of the total fatty acids present in pulmonary emboli in patients with long bone trauma (198). The oleic acid model was developed as an attempt to reproduce ALI due to lipid embolism (191).
Mechanism: oleic acid is directly toxic to endothelial cells at concentrations of 5 × 10−4 M in vitro (17). Within 1 min of intravenous oleic acid administration (0.04 ml/kg in dogs), electron microscopy reveals severe vacuolation of endothelial cells, and passage of an intravascular marker, Evans blue dye, into the subendothelial space (144). The damage to the endothelium appears to be due to necrosis, rather than apoptosis (96). Endothelial injury is followed by epithelial injury, with swelling and necrosis of type I cells, but no evidence of apoptosis (96). Within 30 min, oleic acid becomes detectable in the air spaces. Oleic acid is not detected intracellularly and remains in the extracellular space and the air spaces (17). The actual mechanism whereby oleic acid induces cell death is not clear, but direct membrane damage is likely to be an important event.
Technical issues: oleic acid is insoluble in water and must be dissolved in ethanol or emulsified in blood before administration. It can be administered via a peripheral vein, a central vein, or directly into the right atrium or the pulmonary artery. Because of the need for intravenous administration, oleic acid has been used more extensively in rats and larger animals than in mice. Regardless of the route of intravenous administration (peripheral vs. central), the effects of oleic acid are evident almost immediately (247). Single doses of oleic acid lead to ALI that is greatest at 12 h and decreases towards 24 h, whereas repeated doses result in pulmonary fibrosis (44).
Characteristics of the lung injury: oleic-acid lung injury is characterized by an early phase of necrosis and microvascular thrombosis, followed by a repair stage with proliferation of type 2 cells and fibrotic foci in subpleural areas (44). Microscopically, the injury is multifocal and heterogeneous, with some areas showing only minimal changes; other areas show interstitial edema and varying degrees of air space edema, and some areas show extensive hemorrhagic infiltration with fibrin deposition (74, 221) (Fig. 2). There is neutrophilic infiltration, but the injury is not dependent on PMN, suggesting that a direct effect of oleic acid on the endothelium is the critical event (105). Electron microscopic examination reveals necrosis and detachment of alveolar epithelial cells, necrosis of endothelial cells, and exposure of the basement membrane on both the epithelial and the endothelial sides (221). On macroscopic examination, the lungs show patchy areas of hemorrhage and edema, which become confluent in dependent areas. Nondependent areas are usually spared (44, 46).
Fig. 2.

Oleic acid model. Rabbit lungs 6 h after the onset of intravenous infusion of saline (A) or 0.1 ml·kg−1·h−1 oleic acid over 2 h (B). Note the presence of hemorrhage, hyaline membrane formation, and inflammatory infiltrates in the lungs of the rabbit treated with oleic acid. Both rabbits were mechanically ventilated for the duration of the experiment (FiO2 = 0.8, respiratory rate = 30 bpm, PEEP = 2 cmH2O, tidal volume = 10 cc/kg). [From Furue et al. (74).]
The histological changes of oleic acid lung injury are associated with marked functional changes. Pulmonary microvascular permeability is markedly increased, with elevation of extravascular lung water and leakage of protein-rich fluid into the interstitium and the air spaces (46, 247). These changes in alveolar permeability are proportional to the dose of oleic acid, develop within 1 h of administration, and are rapidly reversible, with the alveolar fluid clearance normalizing within 8 h (54). There are also severe alterations in gas exchange, with respiratory acidosis, hypoxemia, and increased alveolar-arterial oxygen difference within 90 min of intravenous administration (40, 46, 170, 221). The gas exchange alterations result from severe ventilation/perfusion (V/Q) mismatching, increased intrapulmonary shunt, and increased dead space ventilation (170). The static lung compliance and functional residual capacity (FRC) fall rapidly, and mean airway pressure rises (40, 125). Systemically, the cellular damage induced by oleic acid becomes evident rapidly, and increases in circulating angiotensin converting enzyme (ACE) can be detected within 2.5 min after intravenous administration of 0.1 ml/kg to dogs (151). The hemodynamic responses are dose dependent and are characterized by myocardial depression, early systemic hypotension, and pulmonary hypertension (40, 46, 125, 190, 231).
Combination with other models: the intravenous administration of small doses of endotoxin markedly worsens gas exchange in the canine oleic acid model, primarily by increasing the distribution of blood flow towards injured areas (192).
Advantages and disadvantages: the oleic acid model produces the basic characteristics of ALI, early and rapidly reversible patchy inflammatory lung injury with permeability changes and impairment in gas exchange and lung mechanics. A major advantage of this model is its reproducibility; the administration of the same dose of oleic acid by the same route to different animals is followed by a reasonably reproducible pulmonary injury. Oleic acid instillation provides an excellent model to study ventilatory strategies, lung mechanics, and V/Q distribution during lung injury in large and small animals. One disadvantage is the requirement for intravenous administration, which requires expertise in small animals like mice. One question is the extent to which this type of injury actually occurs in humans, because few cases of ARDS are associated with long bone trauma or lipid injury (95), and there is no evidence that the pathophysiology of the injury caused by oleic acid (direct toxicity to endothelial cells) is similar to that underlying ARDS associated with sepsis and aspiration. Therefore, the oleic acid model is probably not as appropriate for studying the pathophysiology of ALI due to sepsis, or therapeutic strategies aimed at modifying host inflammatory responses to reduce the severity of lung injury.
Endotoxin.
Basis for the model: LPS is a glycolipid present in the outer membrane of gram-negative bacteria that is composed of a polar lipid head group (lipid A) and a chain of repeating disaccharides (171). Most of the biological effects of LPS are reproduced by lipid A (189), although the presence or absence of the repeating oligosaccharide O antigen influences the magnitude of the response (112). In serum, LPS binds to a specific LPS binding protein (LBP) (131, 217), forming an LPS:LBP complex that activates the CD14/TLR4 receptor structure on monocytes, macrophages, and other cells, triggering the production of inflammatory mediators (211, 244, 249) (Fig. 3, C and D). LPS is an important mediator of sepsis in response to gram-negative bacteria, and systemic administration of LPS was one of the earliest approaches used to model the consequences of bacterial sepsis.
Fig. 3.
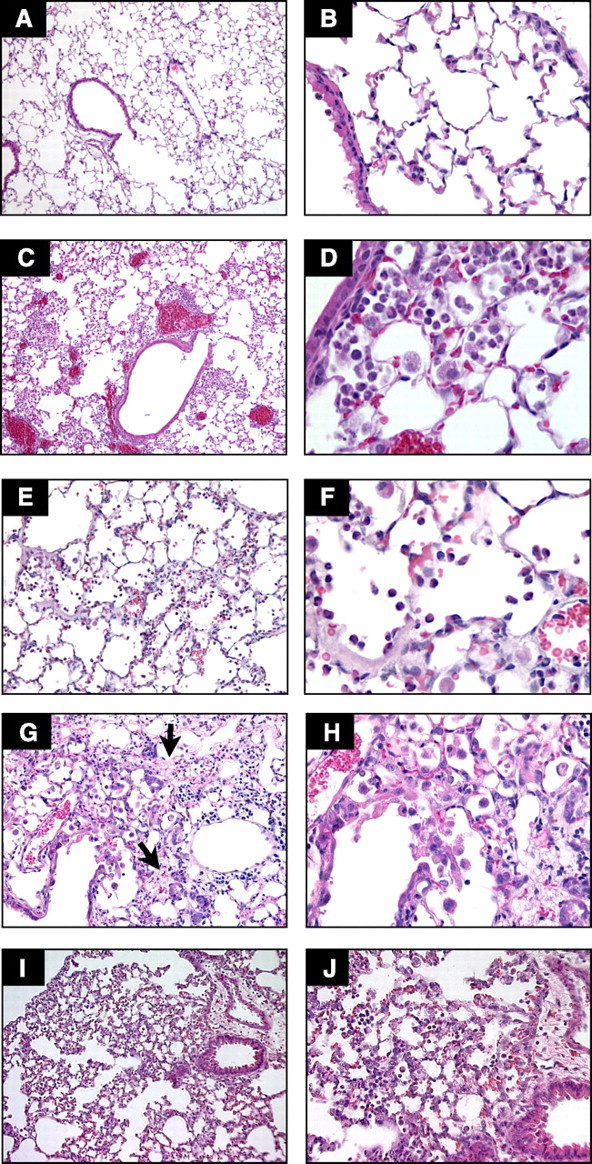
Comparison of selected models of acute lung injury (ALI). A and B: normal mouse lungs. The alveolar walls are very thin, and the majority of the alveoli contain no cells (magnification in A, ×100; B, ×400). C and D: lungs from a mouse euthanized 18 h after intratracheal instillation of 5 ng/g LPS. Note the patchy nature of the injury (C, ×100) and the presence of inflammatory infiltrates and vascular congestion (D, ×400). E and F: lungs from a rabbit euthanized 2 h after exposure to mechanical ventilation with Tv = 25 cc/kg, PEEP = 2.5 cmH2O, FiO2 = 0.5, and RR = 20 bpm. Note the presence of intra-alveolar neutrophilic infiltrates and the deposition of hyaline membranes (E, ×200; F, ×630). G and H: lungs from a mouse euthanized 21 days after the administration of intratracheal bleomycin. Note the presence of fibrotic areas (arrows) (G, ×200; H, ×400). I and J: lungs from a mouse euthanized 12 h after aerosolization of Escherichia coli, 1 × 108 cfu/ml. Note diffuse thickening of the alveolar spaces and intra-alveolar neutrophilic infiltrates (I, ×200; J, ×400). Hematoxylin and eosin.
Mechanism: following intravenous administration of LPS, the capillary endothelium is the initial site of injury. The cellular injury induced by LPS appears to be related to apoptosis, in contrast to the oleic acid model, in which the injury is due to a direct toxic effect resulting in necrosis. Apoptosis of endothelial cells develops rapidly following administration of LPS and precedes other tissue damage (72, 230). Systemic treatment with the broad-spectrum caspase inhibitor Z-VAD.fmk blocks apoptosis and improves survival in mice treated with intravenous LPS (111).
The hemodynamic response to intravenous LPS is characterized by an initial phase of leukopenia, decreased cardiac output, and a fall in arterial pressure. There is an increase in pulmonary arterial pressure, which is due mainly to an increase in the resistance of postcapillary veins (123). This initial phase is followed by slow improvement in leukocyte counts and the hemodynamic profile, over 4–6 h. Changes in the lungs become evident within 2–4 h and include hypoxemia with an increase in the alveolar-arterial oxygen difference (A-a O2 difference). In the lungs, the administration of LPS, either by intravenous or intra-alveolar routes, is followed by changes in PMN deformability and the entrapment of PMN in the pulmonary capillaries (176). However, only small numbers of PMN migrate into the air spaces. PMN entrapment occurs before epithelial permeability changes or disruption of the alveolar/epithelial barrier (9, 239, 241). In contrast, intratracheal LPS is followed by large increases in PMN in the air spaces.
LPS has been administered to humans via the intravenous and the intratracheal routes. Intravenous doses ranging from 2 to 4 ng/kg are associated with evidence of systemic inflammation and priming of alveolar macrophages (92, 169, 205). The ability of intravenous LPS to cause septic shock was evident when a research technician self-administered 1 mg of Salmonella LPS intravenously, and within 3 h developed high-cardiac output shock requiring intravenous pressors, disseminated intravascular coagulation, abnormalities of hepatic and renal function, and noncardiogenic pulmonary edema (213). The intratracheal instillation of LPS at doses ranging from 1 to 4 ng/kg is followed by an early phase characterized by increases in BAL fluid PMN, albumin, and proinflammatory cytokines (TNF-α, IL-1β, IL-6, G-CSF, IL-8, ENA-78, MCP-1, MIP-1α, and MIP-1β) and a later phase 24–48 h after instillation characterized by normalization of the BAL fluid cytokine concentrations and increases in the BAL fluid PMN, monocyte, macrophage, and lymphocyte counts (153) (Fig. 3, C and D).
Technical issues: as discussed in unique characteristics of animal species in modeling human lung injury, species responses to LPS are highly variable and differ in animals with and without PIM. Following treatment with small amounts of LPS, pulmonary inflammation occurs in sheep, calves, pigs, and cats, but not in rodents or dogs (242). Animals with PIM develop manifestations of sepsis and lung injury with very small doses of LPS, e.g., in the μg/kg range. In contrast, animals without PIM require much higher doses, in the range of mg/kg. In addition to species susceptibility, there are also differences in LPS responses between different strains of the same species. For example, BALB/c mice are very sensitive to LPS, whereas C57BL/6 mice are more resistant.
In addition to differences in the susceptibility of different species to LPS, the response of LPS depends on the type of LPS used. LPS from bacteria-forming smooth colonies have the O-chain, consisting of variable numbers of repeating disaccharides, and are less pyrogenic. LPS produced by bacteria growing in rough colonies lacks the O-chain and tends to be more pyrogenic (121). LPS preparations often contain contaminants, such as bacterial lipoproteins, which may influence the biological effects of LPS by interacting with TLRs other than TLR4, which is the primary LPS receptor (211).
Advantages and disadvantages: the use of bacterial LPS has a number of advantages as a method to model the effects of gram-negative bacteria in animals and humans. LPS is easy to administer, and the results tend to be reproducible within experiments. LPS is a potent activator of the innate immune responses via TLR4 pathways and has little direct toxicity to cells in vitro. Thus, the use of LPS provides information about the effects of host inflammatory responses, which occur in bacterial infections. LPS has some significant disadvantages, however. LPS preparations vary in purity and can be contaminated with bacterial lipoproteins and other bacterial materials (211). In general, LPS treatment does not cause the severe endothelial and epithelial injury that occurs in humans with ARDS (239). Bacteria produce exotoxins that are potent cellular toxins (122) and use effector systems such as the type III systems that cause direct cellular lysis (183). Thus, LPS by itself provides an incomplete picture of the effects of live bacteria in the lungs.
Models That Target Primarily the Alveolar Epithelium
Acid aspiration.
Basis for the model: the development of acute respiratory failure following aspiration of gastric contents was first described in 1946 by Mendelson (138), and aspiration of gastric contents is now recognized as an important risk factor for ARDS (50, 95, 163). One of the main characteristics of gastric contents is a low pH, so investigators have used hydrochloric acid (HCl) to simulate lung injury due to gastric acid aspiration in animals. However, it is important to keep in mind that in addition to low pH, gastric contents contain several other products such as food particles, bacterial cell wall products, and cytokines such as IL-1β, and also have a very high osmolarity. All of these factors may contribute to the pathogenesis of aspiration-induced lung injury (118, 172). An important consideration is that a very low pH is required to produce lung injury in animals, e.g., pH 1.5, lower than is typically measured in human gastric contents, but the low pH model remains widely used.
Mechanism: low pH acid aspiration is a neutrophil-dependent form of lung injury that is characterized by injury of the airway and alveolar epithelium, including type I alveolar epithelial cells, followed by a repair process that involves proliferation of alveolar type II cells (60, 117). The evidence supporting the alveolar epithelium as a key target site of acid aspiration includes studies showing that acid aspiration causes an impairment in the alveolar epithelial fluid transport function, resulting in changes in alveolar fluid clearance independently of pulmonary blood flow or vascular filtration (136, 141). However, acid aspiration also results in injury to the capillary endothelium, by a mechanism that appears to require circulating neutrophils (60, 117). Although the exact mechanism of cellular injury is unclear, it is unclear whether the acid itself causes direct injury because it is rapidly neutralized in biological fluids that are buffered by proteins and the bicarbonate system (7).
Technical issues: acid injury is produced by instilling HCl directly into the trachea or bronchi while an animal is mechanically ventilated. Although HCl is rapidly neutralized in the lungs, the concentration of HCl affects the severity of injury. Most studies use 0.1 N HCl, although higher concentrations (up to 0.5 N) have been used. Because this results in a pH that is generally lower than that of gastric juice [most ICU patients have gastric juice pH in the range of 3.0–4.0 (23)], an alternative method is to use 0.3% NaCl titrated with HCl to a pH of 1.2–1.5. This results in a solution with pH and osmolarity approaching the gastric juice of some patients.
Characteristics of the injury: the lung injury is characterized histologically by an acute inflammatory response with patchy areas of neutrophilic inflammation, alveolar hemorrhage, intra-alveolar and interstitial edema, and impairment in the alveolar fluid clearance (Fig. 4) (136, 251). The acute inflammatory response is followed by a fibrotic response that is evident ∼1 wk after instillation (250). The main physiological features are an immediate increase in airway resistance and peak pressures, a fall in lung compliance, and a decrease in the FRC. The vascular response includes elevation of the pulmonary vascular resistance and pulmonary arterial pressure, with an increase in the shunt fraction and dead space. The pulmonary capillary wedge pressure and heart rate remain unchanged, whereas the cardiac output and mean arterial pressure either decrease or remain unchanged (159). The main systemic consequences are leukopenia and thrombocytopenia. Interestingly, the endobronchial administration of 0.1 N HCl is followed by an increase in microvascular permeability in the ileum, which can be blocked with antibodies targeting neutrophil adhesion molecules (anti-CD18) (207), showing that acid aspiration in the lungs is linked to neutrophil-dependent injury to distant organs.
Fig. 4.

Acid aspiration model. Lung tissue sections from a normal mouse (left) and a mouse euthanized 2 h after intratracheal instillation of 1 M HCl, 2 μl/g (pH = 1.5) (right). Note the presence of intra-alveolar proteinaceous deposits (arrow). [From Zarbock et al. (251).]
Advantages/disadvantages: the acid aspiration model is particularly useful for studying hemodynamic and physiological changes of ALI, ventilatory strategies, and mechanisms of neutrophil recruitment. Acid aspiration can also be used in conjunction with other techniques, such as mechanical ventilation, to further reproduce clinically relevant scenarios (60). A limitation of the acid aspiration method is that the difference between injurious and noninjurious acid concentrations is very narrow. Another limitation is that humans do not aspirate HCl, but instead aspirate complex gastric contents, which is a suspension of particulate matter, bacterial products, cytokines, and a pH that is usually higher than 1.5. Further studies are needed to determine whether these differences between gastric fluid and HCl result in a difference between this model and the injury seen in humans following the aspiration of gastric contents.
Hyperoxia.
Basis for the model: in most mammalian species, exposure to 100% O2 eventually results in respiratory distress and death (67). However, the same findings have not been reproduced in humans with normal lungs, who develop only mild increases in alveolar capillary permeability after 24 h of exposure to 100% oxygen, and no clinical or pathological evidence of lung injury when the exposure is extended to several days (12, 42). Many clinicians suspect that oxygen may exacerbate and even cause ALI in critically ill patients, although there still is no conclusive evidence that oxygen either produces or exacerbates ARDS in humans (42). Hyperoxia has been used as a direct cause of injury in animal studies, and as a secondary injury in animals with another underlying injury process, such as cecal ligation and puncture (75).
Mechanism: although the molecular basis of oxygen toxicity is still unclear, it is generally agreed that oxygen damage is mediated by reactive oxygen species (free radicals) derived directly from molecular oxygen, or from interactions with other species, such as NO.
Oxygen radicals have an unpaired orbital electron and act chemically as either reductants (electron donors) or oxidants (electron acceptors) (181). During normal cellular respiration in the mitochondria, a small fraction of oxygen undergoes the addition of a single electron to form the superoxide anion (O2•−). Superoxide anions can be protonated to perhydroxy radicals (HO2·) or can react further to form hydrogen peroxide (H2O2). Hydrogen peroxide and superoxide, in the presence of divalent metals, can react together to form hydroxyl radicals (OH·), which are extremely reactive. Normally, the small amounts of superoxide and hydrogen peroxide produced during cellular respiration are scavenged by antioxidant enzymes, such as superoxide dismutase, catalase, and glutathione peroxidase (69). In the presence of hyperoxia, the mitochondria and other organelles produce increased amounts of superoxide anions (68), which may exceed the capacity of antioxidant enzymes and consume cellular glutathione. Furthermore, superoxide anions react with NO to form peroxynitrite, a potent oxidizing molecule. There is evidence that peroxynitrite is formed in the lungs of hyperoxic humans (202) and animals and that induction of NO synthesis worsens hyperoxic lung injury (90).
The accumulation of free hydroxyl radicals and peroxynitrite results in oxidation of proteins and peroxidation of membrane lipids and nucleic acids (68). The mechanism of cellular injury appears to be a combination of necrosis and apoptosis (11, 93), but the importance of apoptosis in the pathogenesis of oxygen-induced injury remains unclear. The lung epithelial cell line A549 becomes apoptotic in vitro when exposed to oxidants such as H2O2 (65). However, preexposure to oxygen inhibits the apoptotic response to H2O2, and this is mediated by activation of NF-κB (65). In mice exposed to hyperoxia, neither the number of apoptotic cells nor the severity of injury is attenuated by caspase inhibitors, and Fas-null (lpr) mice and p53 knockout mice are not protected from hyperoxic lung injury (11). However, disruption of the proapoptotic CD40-CD40 ligand system and overexpression of IL-6 each reduce the number of apoptotic cells and prevent hyperoxic lung injury (2, 233).
Hyperoxia increases NF-κB translocation in lung mononuclear cells and the production of proinflammatory mediators such as TNF-α, IFN-γ, and IL-1β (152, 196). However, the role of inflammatory cells such as PMN in hyperoxic injury is controversial. Although PMN accumulate in hyperoxic injury (63), neutrophil depletion does not prevent either oxygen-induced lung injury or mortality in a rats, rabbits, mice, or lambs (174, 204). Furthermore, hyperoxia enhances ventilation-induced lung injury by a mechanism independent of cytokine production and lipid peroxidation but involving changes in focal adhesion proteins (45, 201).
Technique: to create hyperoxia as a cause of ALI, the animals are housed in a sealed cage, with an inflow of O2 sufficient to keep the ambient Po2 at or above the desired level (as measured by an oximeter) and CO2 at ambient levels. The ideal cage allows the investigator to introduce food and remove waste through airlocks without altering the composition of gas in the cage.
Characteristics of the injury: the exposure to normobaric oxygen is followed within 3–4 days by an exudative phase in the lungs, characterized by death of alveolar type I cells, swelling, and necrosis of endothelial cells, interstitial edema, and filling of alveoli with an exudative fluid (38, 109). The initial event appears to be damage to the capillary endothelium, followed by platelet adhesion and PMN accumulation in the capillary vessels and interstitium (13, 82). Eventually, alveolar epithelial injury occurs, with an increase in microvascular permeability measured as wet-to-dry (W/D) ratios and migration of neutrophils into the air spaces, as evidenced by increase in total numbers in BAL fluid (18, 20, 102, 148). The pulmonary architecture is preserved during this early phase (82). The initial exudative phase is followed by a proliferative phase, characterized by proliferation of endothelial cells and type 2 pneumocytes, which cover the denuded alveolar basement membrane. Fibroblast proliferation occurs while the endothelial damage persists. Recovery from oxygen injury is characterized by a normal-appearing endothelium and epithelium, with only some areas of interstitial scarring (109). Interestingly, there is synergism between hyperoxia and infection, and the presence of bacterial pneumonia increases the morphological and physiological changes associated with hyperoxia (33).
In rats and mice, exposure to 100% O2 for 40–50 h results in ALI, and longer exposure (60–70 h) results in death (13, 18, 63, 148). Survival after exposure to O2 is age dependent, as neonates are more tolerant to hyperoxia and survive for up to 7 days (67). Older rats (24 mo) are also more tolerant to O2 toxicity (31). Interestingly, exposure to lower levels of O2 (FiO2 = 0.8 or less) is associated with minimal mortality (13). Rats exposed to 85% O2 for 5–7 days can survive subsequent exposures to 100% O2 (13). This tolerance is not due to a decrease in the production of oxygen radicals (68) but is associated with an increase in the pulmonary concentration of antioxidant enzymes, such as manganese superoxide dismutase and glutathione reductase (67).
Studies in rodents have shown important strain-dependent differences in susceptibility to hyperoxia. Exposure to >95% O2 for 48 and 60 h results in larger pleural effusions with higher total protein content in Fischer rats compared with Sprague-Dawley rats (209). Similarly, C57BL/6 mice are more susceptible to hyperoxia than C3H/HeJ mice. This difference in susceptibility has recently been linked to the Nrf2I gene (30).
Advantages/disadvantages: the overall relevance of the hyperoxia injury model to human disease is a matter of controversy because exposure of humans to 100% oxygen for up to 3 days does not result in lung injury (12). Furthermore, some studies suggest that BAL fluid from patients with ARDS has antioxidant activity and inhibits lipid peroxidation in vitro (129), whereas other studies report the opposite effect (36). Despite these limitations, the hyperoxic model, with its well-defined exudative and proliferative phases, provides an excellent model to study lung repair following lung injury. This model is reproducible but requires specialized equipment and expertise to ensure the delivery of the appropriate concentration of oxygen.
Surfactant depletion by saline lavage.
Basis for the model: the saline lavage model was developed by Lachmann et al. (124) in 1979 based on the observation that ARDS is associated with depletion of surfactant from the air spaces and reduced concentrations of surfactant-associated proteins in BAL fluid. In this model, warmed isotonic saline solution is instilled into the lungs and then removed by aspiration. The lavage is repeated until a target degree of hypoxemia is reached. Many groups have combined saline lavage with mechanical ventilation, and in many studies it is difficult to determine the extent to which the lung injury is caused by the saline lavage, by the mechanical ventilation, or both.
Mechanism: repeated lavage with saline reduces the surfactant lipid concentration in alveolar lining fluids, altering alveolar surface tension. Pulmonary surfactant is a complex mixture of phospholipids and proteins that has several important functions (88). Surfactant decreases surface tension and prevents collapse of the alveolar spaces at low lung volumes. Surfactant proteins stabilize surfactant and modulate host defenses in the lungs (243). Depletion of surfactant may be associated with lung injury by two mechanisms: first, by facilitating alveolar collapse and increasing the likelihood of mechanical injury to the alveolar walls during repeated cycles of opening/closure during mechanical ventilation, and second, by impairing alveolar host defenses. The administration of bovine surfactant and/or synthetic surfactant-related peptides attenuates the injury produced by saline lavage (35, 229).
Characteristics of the model: saline lavage (0.9% NaCl) leads to almost immediate hypoxemia and widening of the A-a O2 difference. These findings are rapidly reversible by recruitment maneuvers such as sustained inflation, suggesting that the gas exchange abnormalities reflect collapsed alveoli with otherwise intact alveolar walls (99, 182). The saline lavage by itself has little consequence in terms of permeability changes or inflammation (71, 182), although TNF-α is detectable in lavage fluid (98). When saline lavage is followed by mechanical ventilation with high volumes and no PEEP, a type of lung injury results that is very similar to ARDS in humans. The lung injury is dependent on PMN and is characterized by increased protein permeability, PMN infiltration into the air spaces and interstitium, increased cytokine production, and hyaline membrane formation (99, 110, 124). Mechanical ventilation alone, without previous saline lavage, can induce similar findings (53, 145, 220). Instilling LPS into the air spaces intensifies the injury response to surfactant depletion (34). Rabbits subjected to lung lavage with 0.9% NaCl, five aliquots of 30 ml/kg each, followed immediately by instillation of 0.25 μg/kg LPS, and then mechanical ventilation with 100% FiO2, develop minimal inflammation at 4 h but progress to significant neutrophilic alveolitis and protein-rich alveolar edema at later times (34).
Comparisons with human ALI/ARDS: surfactant depletion is an important feature of ALI/ARDS in humans, but unlike newborns, surfactant depletion in adults is usually a consequence rather than a primary cause of lung injury (164). In ALI/ARDS, surfactant abnormalities occur because of injury of the alveolar epithelium and exudation of protein-rich edema fluid into the alveolar spaces. Saline lavage of the lungs results in surfactant depletion in the absence of major alveolar epithelial damage. Epithelial damage occurs only when the saline lavage is followed by an injurious ventilatory strategy. Therefore, surfactant depletion followed by mechanical ventilation simulates established ALI/ARDS and provides information about the consequences of surfactant depletion, but it is less useful in modeling the initial pathophysiological mechanisms that lead to ALI/ARDS.
Advantages and disadvantages: the main technical disadvantage of the saline lavage model is that the animals require intubation, mechanical ventilation, and general anesthesia. The major advantage is that this model provides an ideal way to test the effects of different ventilatory strategies on the development of tissue injury because the tissue injury results more from the ventilatory strategy than from the saline lavage.
Mechanical ventilation.
During the 1980s and 1990s, several studies showed that mechanical ventilation can produce lung inflammation and injury in animals, termed ventilator-induced lung injury (VILI) (49, 52). These studies provided the basis for a large multicenter study comparing different ventilatory strategies in patients with ALI/ARDS, which changed the clinical management of patients in intensive care units (215). The mechanical ventilation model is the only model of lung injury that has affected clinical practice and improved survival in humans.
Most animal models of lung injury were developed by reproducing known risk factors for human ARDS. In contrast, VILI results from applying a treatment, mechanical ventilation. In humans with clinical risk factors for ARDS, mechanical ventilation is superimposed on an ongoing inflammatory process in the lungs. It is necessary to distinguish between ventilator-induced lung injury, in which mechanical ventilation is the sole method used to generate injury, and ventilator-associated lung injury, in which mechanical ventilation modifies lung injury due to a clinically relevant cause, such as acid aspiration or sepsis (66).
Mechanism: the mechanism of VILI involves direct tissue damage due to mechanical stretch and activation of specific intracellular pathways involved in “mechanotransduction” (reviewed in Ref. 224). Overstretching of alveolar walls results in endothelial and epithelial breaks and interstitial edema (70). Detachment of endothelial cells from the basement membrane and death of epithelial cell with denuding of the epithelial basement membrane become obvious after 20 min of mechanical ventilation with very high tidal volumes (produced by peak pressures of 45 cmH2O in rats) (51). However, the development of hyaline membranes and increased permeability require the presence of PMN, suggesting that in addition to mechanical damage, inflammatory damage is also necessary for mechanical ventilation to induce injury (110). Mechanical cell deformation can be converted to biochemical changes, including production of proinflammatory cytokines (168, 225) and changes in lipid trafficking (226). The inflammatory component of VILI remains controversial, as studies by Tremblay et al. (218a) showed increased production of inflammatory cytokines in lungs ventilated without PEEP, by a mechanism different from that of LPS (19), whereas studies by Ricard et al. (178) have reported no association between VILI and production of proinflammatory cytokines in the same animal model.
One important aspect of mechanical ventilation-associated lung injury is that the role of stretch appears to be different in normal vs. inflamed lungs. For example, it is clear that in patients with ARDS, the use of high tidal volumes is associated with higher mortality (215). However, patients requiring chronic ventilation for neuromuscular disease and normal lungs are routinely ventilated with large tidal volumes, e.g., 10 ml/kg or larger, yet they do not develop ALI. Recent studies have demonstrated that the combination of noninjurious mechanical ventilatory strategies with small doses of LPS or bacteria that cause minimal inflammation results in the development of a form of lung injury characterized by cytokine release, neutrophilic alveolitis, and protein leakage into the air spaces, showing that mechanical ventilation and bacterial products are synergistic in causing lung injury (4, 47, 154). The signal transduction pathways activated by mechanical stretch and innate immunity that leads to lung injury need to be clarified.
Technical issues: mechanical ventilation models are challenging, particularly in small animals like mice. Several variables must be taken into account, including technical issues like the method of intubation (endotracheal vs. tracheostomy), the type of ventilator, the appropriate physiological monitoring, and the amount of fluid support. Endotracheal intubation avoids inflammatory responses associated with tracheostomy and can be achieved with practice. The animals must be anesthetized throughout the ventilation protocol, and this may make it difficult to determine whether the mice are alive or dead unless other variables such as blood pressure, heart rate, or end-dial CO2 are monitored. Airway pressures should be monitored, as lung compliance can change as injury develops. Body temperature must be monitored and maintained within a narrow range because mice are extremely sensitive to temperature changes. PEEP can be generated with a PEEP valve attached to the end of the ventilator. Fluids can be delivered intraperitoneally either by injection or using an infusion pump. With adequate monitoring and support, mice can be ventilated for 8 h or longer (47).
Characteristics of the lung injury: the severity of lung injury depends on the ventilatory strategy, in particular the tidal volume delivered and the presence or absence of PEEP. Large tidal volume ventilation results in alveolar hemorrhage, hyaline membrane formation, neutrophilic infiltration, decreased compliance, and gas-exchange abnormalities (Fig. 3, E and F). Mechanical ventilation at tidal volumes of 10 ml/kg and FiO2 = 0.21 is not injurious, but mechanical ventilation combined with low doses of LPS has a synergistic effect, resulting in neutrophilic alveolitis and increased alveolar permeability (3).
Advantages and disadvantages: the main advantage of the mechanical ventilation model is its clinical relevance, which is highlighted by the fact that this is the only model that has led to changes in clinical practice. The main disadvantage is the complexity of the model. Furthermore, animals such as mice can only be ventilated for relatively short periods of time, whereas patients require mechanical ventilation for days or weeks.
Intratracheal bleomycin.
The bleomycin model is usually considered a model of pulmonary fibrosis, but its administration is also associated with features of ALI, and thus we will briefly review it here. The bleomycin model has been reviewed in more detail elsewhere in this series (143). Bleomycin is an antineoplastic antibiotic drug isolated in 1966 from the actinomycete Streptomyces verticillus (222). Bleomycin forms a complex with oxygen and metals such as Fe2+, leading to the production of oxygen radicals, DNA breaks, and ultimately cell death (26). Bleomycin can be inactivated by bleomycin hydrolase, a cysteine protease that shows variable levels of expression in the lungs (194). The susceptibility of the lungs to bleomycin-induced toxicity is largely dependent on the levels of expression of bleomycin hydrolase in the lungs; species with high levels of expression, such as rabbits, are relatively resistant to bleomycin-induced toxicity, whereas species with low levels of expression, such as C57BL/6 mice, are sensitive (126). In addition to species-related differences in bleomycin susceptibility, there are also differences in strain susceptibility, with C57BL/6 mice being highly sensitive and Balb/c and C3H mice being resistant; however, the differences in strain susceptibility to bleomycin appear to be related more to differential expression of genes involved in apoptosis regulation or oxidative stress than to differences in the expression of bleomycin reductase (87).
Bleomycin can be administered intravenously, intratracheally, intraperitoneally, or subcutaneously (143). The intravenous model mimics the human clinical scenario but requires the administration of the drug twice per week for 4–8 wk (1). In contrast, the intratracheal model requires the administration of one single dose of bleomycin (143). It is important to emphasize that although both models result in lung injury and fibrosis, the site of initial injury is different: the endothelium is the primary target of intravenous bleomycin, and the epithelium is the primary target of intratracheal bleomycin. The dose response and pathological changes of the lung to intravenous bleomycin were first described in 1974, and we refer readers to the original Adamson and Bowden report (1). Here we will focus on the intratracheal model, which is the most widely used due to its lower complexity and high reproducibility.
The administration of intratracheal bleomycin is followed by a well-described sequence of events. Within 24 h of bleomycin administration, there is an increase in BAL neutrophils, which then normalize towards day 11. The lymphocytic response follows, beginning around day 7 and persisting on day 11. The fibrotic response becomes detectable in tissues by day 11 and persists by day 20. As for cytokines, IL-1β, KC, IL-6, TNF, and IL-1R1 peak between 6 and 63 h, TGF-β R1 and R2 peak towards day 7, and G-CF, GM-CSF, IFN-γ, and IL-4 peak towards day 14 (28, 76, 180, 246). At the tissue level, the early phase is characterized by patchy neutrophilic alveolitis and the late phase by patchy areas of fibrosis, but there is no formation of hyaline membranes (Fig. 3, G and H). The extent of fibrosis is proportional to the severity of the initial injury (197). Thus, the bleomycin model is relatively technically easy, very reproducible, well standardized, and reproduces the pattern of ALI, with early inflammation that heals with fibrosis. The main disadvantage is that the physiological relevance of bleomycin is often questioned, with the argument that intratracheal instillation of bleomycin is an “overwhelming stimulus” that bears little relevance to the clinical setting. Despite these caveats, the bleomycin model remains one of the most commonly used models of ALI in mice.
Models Targeting the Epithelium and Endothelium
Ischemia/reperfusion models.
Ischemia followed by reperfusion (I/R), either in the lungs or in distant vascular beds, can lead to lung injury. A classic form of injury secondary to lung I/R is the “reimplantation response,” a form of lung injury following lung transplantation. The reimplantation response is characterized by non-cardiogenic pulmonary edema, inflammatory infiltrates, and hypoxia, and is unrelated to rejection (167, 199). Lung injury is also associated with ischemia/reperfusion at distant, nonpulmonary sites. For example, a 20–40% incidence of ALI has been reported following repair of thoracoabdominal aortic aneurysms, which involves visceral ischemia and reperfusion (237). This type of injury affects the alveolar epithelium and the capillary endothelium. It is unclear whether the epithelium and the endothelium are both injured simultaneously or whether the injury occurs primarily in endothelial or epithelial cells. Studies in humans and rabbits show that both types of cells have features compatible with apoptosis and necrosis (59, 186, 208).
Pulmonary models of ischemia reperfusion: in pulmonary models of I/R, the lung is subjected to ischemia for varying periods of time before circulation is reestablished. The lungs are perfused by two separate vascular systems, the pulmonary circulation and the bronchial circulation. Ischemia can be produced by clamping the pulmonary artery, which preserves the bronchial circulation, or by clamping the hilum, which stops all circulation. In a variant of the lung I/R model, ischemia is achieved by lung collapse, followed by reexpansion (101). Electron microscopic examination of cat lungs treated with 2 h of ischemia (inflated lungs) and 2 h of reperfusion shows structural damage of the alveolar endothelium and epithelium, with separation of type II cells from the basement membrane, thickened vascular endothelium, and disruption of the septal capillary wall (149). An interesting feature of this model is that I/R of one lung results in inflammatory changes not only in the occluded lung but also in the contralateral lung (185). The injury to the contralateral side, when present, is milder and characterized by permeability changes and PMN infiltration (84, 157).
Five main variables must be considered in pulmonary I/R models: the inflation state of the lung (deflated vs. inflated), the extent of the ischemic bed (pulmonary, bronchial circulation, venous return), the duration of ischemia and reperfusion, the experimental preparation (in vivo vs. isolated perfused lungs), and the animal species.
Lung inflation: the extent of lung inflation during the ischemic period affects the severity of the injury. Occlusion of the pulmonary circulation for as long as 7 days results in minimal permeability changes and no macroscopic injury as long as the lungs are kept inflated (16, 94, 162). In contrast, if the lungs are kept deflated during the ischemic period, the resulting injury is severe and characterized by hemorrhage, edema, and inflammation, and there is a marked decrease in bronchial blood flow (21). Persistent lung inflation can also prevent reperfusion injury after interruption of both the pulmonary and bronchial circulation with a hilar clamp (85, 186).
When lung ischemia is followed by reperfusion, inflation of the lung also is an important factor. Sakuma et al. (186) showed that in rabbits, 4 h of unilateral hilar occlusion with lung deflation followed by 1 h of reperfusion resulted in fulminant pulmonary edema and death. The development of injury in this model was prevented by inflation of the lungs, either with oxygen or nitrogen. Similar effects have been shown in dogs and rats (85, 227).
Extent of the ischemic bed: because the lungs are perfused by two separate systems, the pulmonary circulation and the bronchial circulation, occlusion of the pulmonary artery alone results in milder injury than occlusion of both the pulmonary artery and the bronchial circulation (85, 161). Reverse pulmonary venous flow from the left atrium can occur if the hilum is not clamped, leading to less injury than complete occlusion of the hilum (43, 84, 155). The extent of ischemia is also affected by whether oxygenated air continues to ventilate the lungs while the circulation is blocked.
Duration of ischemic and reperfusion periods: although the effect of the duration of the ischemia and reperfusion periods is difficult to evaluate because of the many different protocols used, some generalizations can be made. Ischemia alone, in the absence of reperfusion, may or may not result in injury, depending on the extent of inflation of the ischemic bed (21, 94). When ischemic time is varied (45 min to 4 h) and the reperfusion time remains constant, longer ischemic times result in worse lung injury (186, 227). When the ischemic time is kept constant and the reperfusion time is varied (30 min to 2 h), the severity of injury increases with longer reperfusion times (113, 149). Thus, ischemia and reperfusion times each affect the severity of the subsequent injury.
Experimental preparation: I/R can be performed either in vivo or in isolated lung preparations, and the experimental method does not seem to affect the development of injury. in vivo models allow the study of secondary changes in the contralateral lung.
Species variability: the results of I/R injury vary among species. Rabbits have been most widely used in pulmonary I/R models, followed by rats and dogs; sheep, pigs, cats, and ferrets have also been used. In the dog, the major change after lung reperfusion is a large increase in pulmonary postcapillary resistance (101). In rats, pulmonary edema is due mostly to an increase in the permeability of the postalveolar venules (114). Rabbits seem to be more sensitive than dogs to I/R.
In summary, lung ischemia followed by reperfusion results in lung injury characterized by increased pulmonary vascular permeability and edema, PMN infiltration, and sometimes hemorrhage. The severity of injury is worse when the lungs remain deflated during the ischemic period and when the pulmonary and bronchial circulations are occluded together.
Nonpulmonary I/R: the nonpulmonary I/R model has been used mainly in rats and mice, although there are some studies in rabbits (8, 150) and sheep (116, 195). Models differ in terms of the anatomic area subjected to ischemia, e.g., gut, liver, kidneys, lower torso, the duration of the ischemic period, and the duration of reperfusion. In general, the severity of lung injury is more closely related to the volume of ischemic tissue than to the duration of ischemia. Furthermore, the lung injury is mild and temporary unless a second intrapulmonary stimulus such as intratracheal LPS is added (120).
Technical considerations: I/R in nonpulmonary vascular beds is generally achieved by isolating and clamping a specific artery for a period of time and then removing the clamp and restoring perfusion. The most commonly used vascular bed in large animals is the superior mesenteric artery, whereas in mice hindlimb ischemia has been achieved either by surgical occlusion of the supraceliac aorta or without surgery by applying tourniquets to the extremities (57, 73, 103, 116, 119, 237). These methods require that the animal be anesthetized. In abdominal I/R, the abdominal cavity is left open during the ischemic period and covered by a plastic wrap. At the end of the ischemic period, the clamp is removed, and reperfusion is initiated. Surgery can be avoided by applying tourniquets to the extremities to create variable periods of tissue ischemia.
Specific models: in nonpulmonary I/R models in rats, mice, sheep, and other species, reperfusion following ischemic periods ranging from 30 to 120 min is associated with a systemic response characterized by decreased cardiac output and hypotension, metabolic acidosis, and PMN activation (73, 119) (245). In the lungs, PMN sequestration begins within 90 min of reperfusion (245) and is followed 1 h later by increases in pulmonary microvascular permeability, which peak after 4–6 h (119, 212). After 12–18 h, lung permeability returns to baseline levels (120, 212). Lung histology shows microvascular sequestration of PMN and proteinaceous alveolar exudates (116). The lung injury in this model depends on circulating PMN (132).
The administration of 2.5 mg/kg iv LPS to rats 6 h after 45 min of mesenteric ischemia results in a more severe form of injury, with a sustained increase in alveolar permeability for up to 18 h following reperfusion, and the development of alveolar edema, alveolar hemorrhage, and fibrinous exudates (120, 212). As in models of VILI, nonpulmonary ischemia and circulating LPS appear to have synergistic effects on the development of lung injury.
Advantages and disadvantages: the main advantage of the nonpulmonary ischemia reperfusion models is that they reproduce a known clinical phenomenon, the development of lung injury after intestinal or peripheral ischemia and reperfusion in humans. I/R can be performed in a variety of large and small animal species, including mice. However, when I/R of nonpulmonary beds is the only stimulus, the lung injury is mild, and secondary stimuli (such as LPS) are required to cause significant lung injury. Abdominal surgery is required in the intestinal I/R model, which induces an inflammatory response that must be controlled for in the experimental design.
Models of lung injury due to sepsis.
Sepsis is one of the main risk factors for ARDS (95), and many animal models of sepsis have been developed to study ALI. Three main strategies have been used to induce sepsis: 1) the administration of live bacteria, 2) the creation of an endogenous infection, e.g., cecal ligation and puncture, and 3) the administration of bacterial products, such as endotoxin. The following sections address models that use live bacteria, because LPS models have been discussed in Endotoxin. Important variables in live bacteria models of ALI include: the route of administration, the size of the bacterial inoculum, the bacterial species, and the animal species. In general, more severe responses are seen in species with PIM than in those without PIM.
Intravenous bacteria: intravenous bacteria can be administered as a bolus or as a continuous infusion. The administration of an intravenous bolus of bacteria is followed within an hour by an initial phase of hypotension and leukopenia, which can progress to septic shock, intravascular coagulation, and death if the inoculum is high. If the animals survive, the initial phase is followed between 1 and 3 h later by a brief period of hemodynamic stabilization and then by a phase of microvascular injury with PMN sequestration in the lungs, an increase in pulmonary microvascular permeability, interstitial edema, an increase in shunt fraction and pulmonary arterial pressure, and intravascular thrombosis (238). However, the alveolar epithelium is relatively resistant to intravenous bacteria, and unless the inoculum is very high, there is little neutrophilic alveolitis or intra-alveolar edema formation (166, 239). Thus, experimental bacteremia is not associated with the full histopathological picture of ARDS, including epithelial damage and hyaline membrane formation. In humans, the relationship between bacteremia, sepsis, and ARDS is not clear. Although sepsis is a major risk factor for ARDS, less than 50% of patients with clinically defined sepsis syndrome have detectable bacteremia, and when present, the bacteremia is usually less severe than that seen in most animal models (142). Furthermore, bacteremia alone (in the absence of sepsis syndrome) is associated with an extremely low risk of ARDS (62). Thus, the relevance of bacteremia models for human lung injury is unclear. Investigators have tried to address these concerns by inducing primary infections that lead to sepsis and lung injury.
Models of lung injury secondary to peritonitis: cecal ligation and puncture.
Because many cases of human sepsis follow intraperitoneal infections (25), animal models have been developed in which peritonitis is followed by sepsis and lung injury. Experimental peritonitis can be induced surgically by ligating and perforating the cecum (cecal ligation and puncture; CLP), or, alternatively, the bacteria can be incorporated into a carrier vehicle such as a fibrin clot or a sponge placed in the peritoneum (55, 135). The inoculation of live bacteria directly into the peritoneal cavity is seldom used because the bacteria are either cleared rapidly by local host defense systems, or, if the inoculum is high, cause rapid death by sepsis (115). In either case, there is very little lung injury.
CLP is the most widely used model of peritonitis. In CLP, the cecum is ligated and punctured three to five times with a needle. The severity of the injury depends on the number of holes in the cecum and on the size of the needle used to make the holes (228). In contrast to models using LPS and live bacteria, in which the effects are almost immediate, the effects of CLP develop over days, and the onset is less abrupt. Leukopenia develops within 24–30 h of surgery; pulmonary hypertension develops within 24–30 h; and lung injury develops within 18–72 h (79). The main features of CLP-associated lung injury are hypoxemia, neutrophilic inflammation, and interstitial and alveolar edema (77, 128, 223) (Fig. 5). Blood cultures are usually positive for multiple organisms, the most common being enteric gram-negative rods such as serratia, enterobacter, and bacteroides species (77). Mortality is high, ranging from 25% at 18 h to 70–90% within 30 h of the operation (79, 223).
Fig. 5.

Cecal ligation and puncture (CLP). Lungs from mice following sham surgery (left) or from mice subjected to 90 min of hemorrhagic shock (MAP = 30 mmHg) followed 24 h later by CLP (right). The lungs were stained for neutrophil-specific esterase (red). [From Lomas-Neira et al. (128).]
Thus, CLP results in mild lung injury similar to ALI/ARDS, although with less impressive intra-alveolar inflammation and hyaline membrane formation. It is probably the single best animal model of sepsis and organ injury, but the main disadvantage is the requirement for major surgery. In addition, the actual bacterial inoculum in CLP is unknown, and the model could be affected by differences in colonic flora among animals, resulting in significant variability.
Local administration of live bacteria into the lungs: pulmonary infections are the main risk factor of ALI/ARDS in 46–51% of patients (184). In animals, local administration of bacteria into the lungs can be achieved by using an intratracheal catheter, by transtracheal instillation, by direct intrabronchial instillation using a bronchoscope, or by aerosolization (64, 86, 147). Intratracheal instillation of bacteria results in a localized area of pneumonia, whereas aerosolization results in diffuse infection (Fig. 3, E and F). Aerosolization has two main advantages: there is no need to anesthetize the animal, and a large number of animals can be treated simultaneously in an aerosol chamber (179). A recent article in this series has extensively reviewed animal models of pneumonia (140), and, therefore, here we will only present a very succinct commentary of some key features of this model.
The local administration of bacteria results in pneumonia, and, depending on the size of the bacterial inoculum, systemic manifestations of sepsis. In rabbits, the intrapulmonary instillation of Escherichia coli at doses ranging from 1 × 107 to 1 × 1010 CFU/ml into the right lower lobe via a catheter produced local and systemic inflammatory responses (64). At low bacterial doses, the systemic changes were minimal. At high bacterial doses, the animals developed severe localized pneumonia with epithelial injury and empyema and severe systemic manifestations including shock (64, 240). Interestingly, bacterial pneumonia due to Pseudomonas aeruginosa and also to E. coli is associated with an increase in alveolar fluid clearance, indicating that alveolar epithelial function is partly preserved (177, 210). Histologically, the intratracheal administration of bacteria results in localized pneumonia (rather than ARDS) in the instilled lung. However, unilateral administration of bacteria can result in lung injury in the contralateral lung, depending on the bacterial dose. In rats, the administration of 1 × 106 CFU/ml P. aeruginosa to the right lung results 4 h later in an ipsilateral increase in inflammatory cytokines (TNF-α and CINC) but not in neutrophil sequestration or permeability changes (214). A higher dose, 1 × 107 CFU/ml, results in bilateral increases in permeability and PMN sequestration as well as an increase in plasma cytokines.
SUMMARY AND CONCLUSIONS
In summary, this review discusses several animal models of ALI with the goal of helping investigators choose the model best suited to specific scientific questions. Important information has been derived from each of these animal models. However, none of these models of ALI adequately reproduces the full characteristics of human ALI/ARDS, namely an acute neutrophilic alveolitis with hyaline membrane formation and intravascular microthrombi, and, therefore, the choice of a particular model should be made after considering the specific features of each model.
Several limitations are common to all models of ALI. Most models are based on one, or, at most, two methods to cause injury. However, human ALI/ARDS is seldom caused by any single event. More often, ARDS in humans is associated with complex interactions between primary risk factors such as sepsis and oropharyngeal aspiration; comorbidities, such as diabetes, alcoholism, and underlying lung disease; and additional factors inherent to the host, such as genetic determinants. Another shortcoming of animal models is that interspecies variability in innate immunity and other responses may affect the relevance of animal data for humans. Finally, most models target a primary tissue, the alveolar epithelium, the alveolar endothelium, or both, but the primary target tissue in human ALI/ARDS remains unknown. These shortcomings, alone or in combination, are probably responsible for the fact that animal models have often yielded different or even contradictory results and that therapeutic approaches effective in animals are not always successful in humans. However, despite these limitations, studies using animal models of ALI/ARDS remain essential because at the present time there is no substitute for animal models as tools to generate information about the pathophysiology of lung injury and to test novel therapeutic interventions in complex biological systems.
To overcome these shortcomings, investigators must pay special attention to choosing the model best suited to test their hypothesis and should be aware of the differences between the model used and human ALI/ARDS. Of the models discussed in this review, the models that most consistently approach the features of human ARDS, at least those observed during the acute phase, include oleic acid instillation, acid aspiration, and hyperoxia. Unfortunately, only a fraction of cases of ARDS is caused by long bone trauma (modeled by oleic acid instillation), humans seldom aspirate gastric juice with pH 1.5 or less, and hyperoxia has not been shown to cause ALI/ARDS in humans (42). Surfactant depletion by saline lavage results in a decrease in lung compliance and hypoxemia, but not in alveolar epithelial injury or neutrophilic alveolitis unless a second injury, such as bacterial endotoxin or mechanical ventilation, is added. Sepsis is one of the most common causes of ARDS in humans, but attempts at modeling sepsis by treating animals with bacteria or bacterial products systemically is usually associated with only mild alveolar neutrophilia and minimal changes in alveolar-capillary permeability.
Because of the difficulties in modeling human ALI/ARDS in animals, and because of differences among species, extrapolating the results of animal studies to humans must be done with a great deal of care. Ideally, findings from animal models should be reproduced in more than one mammalian species before human experimentation. This is particularly relevant for studies in mice, which differ in important ways from humans, as described in unique characteristics of animal species in modeling human lung injury. In addition, scientific reports should pay special attention to describing the actual features of the “lung injury” observed in the model, including measurements of alveolar-capillary permeability and the inflammatory response.
In conclusion, studies in animal models are essential to our understanding of the pathophysiology of ALI and the development of novel therapeutic strategies for human ALI/ARDS even though none of the animal models available fully reproduces the findings of human disease. Further attempts to develop standardized criteria for defining lung injury in animals would help in the selection of animal models for specific experimental protocols and improve the usefulness of data generated in different animal models of lung injury.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-083044 (G. Matute-Bello) and HL-081764 (T. R. Martin) and the Research Service of the Veterans Administration Puget Sound Healthcare System.
Acknowledgments
We thank Drs. Michael Matthay and Robb Glenny for useful suggestions and comments in the preparation of this manuscript.
REFERENCES
- 1.Adamson IY, Bowden DH. The pathogenesis of bloemycin-induced pulmonary fibrosis in mice. Am J Pathol : 185–197, 1974. [PMC free article] [PubMed] [Google Scholar]
- 2.Adawi A, Zhang Y, Baggs R, Finkelstein J, Phipps RP. Disruption of the CD40-CD40 ligand system prevents an oxygen-induced respiratory distress syndrome. Am J Pathol : 651–657, 1998. [PMC free article] [PubMed] [Google Scholar]
- 3.Altemeier WA, Matute-Bello G, Frevert CW, Kawata Y, Kajikawa O, Martin TR, Glenny RW. Mechanical ventilation with moderate tidal volumes synergistically increases lung cytokine response to systemic endotoxin. Am J Physiol Lung Cell Mol Physiol : L533–L542, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, Liles WC. Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J Immunol : 4069–4075, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet : 319–323, 1967. [DOI] [PubMed] [Google Scholar]
- 6.Ashbaugh DG, Maier RV. Idiopathic pulmonary fibrosis in adult respiratory distress syndrome. Diagnosis and treatment. Arch Surg : 530–535, 1985. [DOI] [PubMed] [Google Scholar]
- 7.Awe WC, Fletcher WS, Jacob SW. The pathophysiology of aspiration pneumonitis. Surgery : 232–239, 1966. [Google Scholar]
- 8.Axon RN, Baird MS, Lang JD, Brix AE, Nielsen VG. PentaLyte decreases lung injury after aortic occlusion-reperfusion. Am J Respir Crit Care Med : 1982–1990, 1998. [DOI] [PubMed] [Google Scholar]
- 9.Azghani AO, Miller EJ, Peterson BT. Virulence factors from Pseudomonas aeruginosa increase lung epithelial permeability. Lung : 261–269, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med : 35–56, 1982. [PubMed] [Google Scholar]
- 11.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF. Oxygen toxicity in mouse lungs: pathways to cell death. Am J Respir Cell Mol Biol : 573–581, 1998. [DOI] [PubMed] [Google Scholar]
- 12.Barber RE, Hamilton WK. Oxygen toxicity in man. A prospective study in patients with irreversible brain damage. N Engl J Med : 1478–1484, 1970. [DOI] [PubMed] [Google Scholar]
- 13.Barry BE, Crapo JD. Patterns of accumulation of platelets and neutrophils in rat lungs during exposure to 100% and 85% oxygen. Am Rev Respir Dis : 548–555, 1985. [DOI] [PubMed] [Google Scholar]
- 14.Bauer TT, Valencia M, Badia JR, Ewig S, Gonzalez J, Ferrer M, Torres A. Respiratory microbiology patterns within the first 24 h of ARDS diagnosis: influence on outcome. Chest : 273–279, 2005. [DOI] [PubMed] [Google Scholar]
- 15.Baughman RP, Gunther KL, Rashkin MC, Keeton DA, Pattishall EN. Changes in the inflammatory response of the lung during acute respiratory distress syndrome: prognostic indicators. Am J Respir Crit Care Med : 76–81, 1996. [DOI] [PubMed] [Google Scholar]
- 16.Becker PM, Pearse DB, Permutt S, Sylvester JT. Separate effects of ischemia and reperfusion on vascular permeability in ventilated ferret lungs. J Appl Physiol : 2616–2622, 1992. [DOI] [PubMed] [Google Scholar]
- 17.Beilman G. Pathogenesis of oleic acid-induced lung injury in the rat: distribution of oleic acid during injury and early endothelial cell changes. Lipids : 817–823, 1995. [DOI] [PubMed] [Google Scholar]
- 18.Bellmeyer A, Martino JM, Chandel NS, Scott Budinger GR, Dean DA, Mutlu GM. Leptin resistance protects mice from hyperoxia-induced acute lung injury. Am J Respir Crit Care Med : 587–594, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, Phillips RJ, Strieter RM. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest : 1703–1716, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhandari V, Choo-Wing R, Homer RJ, Elias JA. Increased hyperoxia-induced mortality and acute lung injury in IL-13 null mice. J Immunol : 4993–5000, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Bishop MJ, Holman RG, Guidotti SM, Alberts MK, Chi EY. Pulmonary artery occlusion and lung collapse depletes rabbit lung adenosine triphosphate. Anesthesiology : 611–617, 1994. [DOI] [PubMed] [Google Scholar]
- 22.Bockamp E, Maringer M, Spangenberg C, Fees S, Fraser S, Eshkind L, Oesch F, Zabel B. Of mice and models: improved animal models for biomedical research. Physiol Genomics : 115–132, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Bonten MJ, Gaillard CA, van der Geest S, van Tiel FH, Beysens AJ, Smeets HG, Stobberingh EE. The role of intragastric acidity and stress ulcus prophylaxis on colonization and infection in mechanically ventilated ICU patients. A stratified, randomized, double-blind study of sucralfate versus antacids. Am J Respir Crit Care Med : 1825–1834, 1995. [DOI] [PubMed] [Google Scholar]
- 24.Brain JD, Molina RM, DeCamp MM, Warner AE. Pulmonary intravascular macrophages: their contribution to the mononuclear phagocyte system in 13 species. Am J Physiol Lung Cell Mol Physiol : L146–L154, 1999. [DOI] [PubMed] [Google Scholar]
- 25.Brun-Buisson C, Doyon J, Carlet P, Dellamonica F, Gouin A, Lepoutre J, Mercier C, Offenstadt G, Regnier B. Incidence, risk factors and outcome of severe sepsis and septic shock in adults. A multicenter prospective study in intensive care units. JAMA : 968–974, 1995. [PubMed] [Google Scholar]
- 26.Burger RM, Peisach J, Horwitz SB. Activated bleomycin. A transient complex of drug, iron, and oxygen that degrades DNA. J Biol Chem : 11636–11644, 1981. [PubMed] [Google Scholar]
- 27.Butz GM, Davisson RL. Long-term telemetric measurement of cardiovascular parameters in awake mice: a physiological genomics tool. Physiol Genomics : 89–97, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Cavarra E, Carraro F, Fineschi S, Naldini A, Bartalesi B, Pucci A, Lungarella G. Early response to bleomycin is characterized by different cytokine and cytokine receptor profiles in lungs. Am J Physiol Lung Cell Mol Physiol : L1186–L1192, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Chesnutt AN, Matthay MA, Tibayan FA, Clark JG. Early detection of type III procollagen peptide in acute lung injury. Pathogenetic and prognostic significance. Am J Respir Crit Care Med : 840–845, 1997. [DOI] [PubMed] [Google Scholar]
- 30.Cho HY, Jedlicka AE, Reddy SPM, Zhang LY, Kensler TW, Kleeberger SR. Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am J Respir Cell Mol Biol : 42–51, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Choi AM, Sylvester S, Otterbein L, Holbrook NJ. Molecular responses to hyperoxia in vivo: relationship to increased tolerance in aged rats. Am J Respir Cell Mol Biol : 74–82, 1995. [DOI] [PubMed] [Google Scholar]
- 32.Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, Bleich A, Bogue M, Broman KW, Buck KJ, Buckler E, Burmeister M, Chesler EJ, Cheverud JM, Clapcote S, Cook MN, Cox RD, Crabbe JC, Crusio WE, Darvasi A, Deschepper CF, Doerge RW, Farber CR, Forejt J, Gaile D, Garlow SJ, Geiger H, Gershenfeld H, Gordon T, Gu J, Gu W, de Haan G, Hayes NL, Heller C, Himmelbauer H, Hitzemann R, Hunter K, Hsu HC, Iraqi FA, Ivandic B, Jacob HJ, Jansen RC, Jepsen KJ, Johnson DK, Johnson TE, Kempermann G, Kendziorski C, Kotb M, Kooy RF, Llamas B, Lammert F, Lassalle JM, Lowenstein PR, Lu L, Lusis A, Manly KF, Marcucio R, Matthews D, Medrano JF, Miller DR, Mittleman G, Mock BA, Mogil JS, Montagutelli X, Morahan G, Morris DG, Mott R, Nadeau JH, Nagase H, Nowakowski RS, O'Hara BF, Osadchuk AV, Page GP, Paigen B, Paigen K, Palmer AA, Pan HJ, Peltonen-Palotie L, Peirce J, Pomp D, Pravenec M, Prows DR, Qi Z, Reeves RH, Roder J, Rosen GD, Schadt EE, Schalkwyk LC, Seltzer Z, Shimomura K, Shou S, Sillanpaa MJ, Siracusa LD, Snoeck HW, Spearow JL, Svenson K, Tarantino LM, Threadgill D, Toth LA, Valdar W, de Villena FP, Warden C, Whatley S, Williams RW, Wiltshire T, Yi N, Zhang D, Zhang M, Zou F; Complex Trait Consortium. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet : 1133–1137, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Coalson JJ, King RJ, Winter VT, Prihoda TJ, Anzueto AR, Peters JI, Johanson WG Jr. O2- and pneumonia-induced lung injury. I. Pathological and morphometric studies. J Appl Physiol : 346–356, 1989. [DOI] [PubMed] [Google Scholar]
- 34.Cochrane CG, Revak SD. Surfactant lavage treatment in a model of respiratory distress syndrome. Chest : 85S–86S, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Cochrane CG, Revak SD, Merritt TA, Schraufstatter IU, Hoch RC, Henderson C, Andersson S, Takamori H, Oades ZG. Bronchoalveolar lavage with KL4-surfactant in models of meconium aspiration syndrome. Pediatr Res : 705–715, 1998. [DOI] [PubMed] [Google Scholar]
- 36.Cochrane CG, Spragg R, Revak SD. Pathogenesis of the adult respiratory distress syndrome: evidence of oxidant activity in bronchoalveolar lavage fluid. J Clin Invest : 754–761, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Constantin JM, Cayot-Constantin S, Roszyk L, Futier E, Sapin V, Dastugue B, Bazin JE, Rouby JJ. Response to recruitment maneuver influences net alveolar fluid clearance in acute respiratory distress syndrome. Anesthesiology : 944–951, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Crapo JD, Barry BE, Foscue HA, Shelburne J. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis : 123–143, 1980. [DOI] [PubMed] [Google Scholar]
- 39.Cross AS, Opal SM, Sadoff JC, Gemski P. Choice of bacteria in animal models of sepsis. Infect Immun : 2741–2747, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curtis SE, Peek JT. Effects of progressive intratracheal administration of perflubron during conventional gas ventilation in anesthetized dogs with oleic acid injury. Adv Exp Med Biol : 51–58, 1994. [DOI] [PubMed] [Google Scholar]
- 41.Davey RA, MacLean HE. Current and future approaches using genetically modified mice in endocrine research. Am J Physiol Endocrinol Metab : E429–E438, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Davis WB, Rennard SI, Bitterman PB, Crystal RG. Pulmonary oxygen toxicity. Early reversible changes in human alveolar structures induced by hyperoxia. N Engl J Med : 878–883, 1983. [DOI] [PubMed] [Google Scholar]
- 43.Deffebach ME, Lakshminarayan S, Kirk W, Butler J. Bronchial circulation and cyclooxygenase products in acute lung injury. J Appl Physiol : 1083–1088, 1987. [DOI] [PubMed] [Google Scholar]
- 44.Derks CM, Jacobovitz-Derks D. Embolic pneumopathy induced by oleic acid A systematic morphologic study. Am J Pathol : 143–158, 1977. [PMC free article] [PubMed] [Google Scholar]
- 45.Desai LP, Sinclair SE, Chapman KE, Hassid A, Waters CM. High tidal volume mechanical ventilation with hyperoxia alters alveolar type II cell adhesion. Am J Physiol Lung Cell Mol Physiol : L769–L778, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Devitt H, Burka J, Jones R, Amy R, King E. Hemodynamic and pathologic effects of prostacyclin on oleic acid-induced pulmonary injury. Surgery : 213–220, 1988. [PubMed] [Google Scholar]
- 47.Dhanireddy S, Altemeier WA, Matute-Bello G, O'Mahony DS, Glenny RW, Martin TR, Liles WC. Mechanical ventilation induces inflammation, lung injury, and extra-pulmonary organ dysfunction in experimental pneumonia. Lab Invest : 790–799, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Dorger M, Jesch NK, Rieder G, Hirvonen MR, Savolainen K, Krombach F, Messmer K. Species differences in NO formation by rat and hamster alveolar macrophages in vitro. Am J Respir Cell Mol Biol : 413–420, 1997. [DOI] [PubMed] [Google Scholar]
- 49.Dos Santos CC, Slutsky AS. Cellular responses to mechanical stress: invited review. Mechanisms of ventilator-induced lung injury: a perspective. J Appl Physiol : 1645–1655, 2000. [DOI] [PubMed] [Google Scholar]
- 50.Doyle RL, Szaflarski N, Modin GW, Wiener-Kronish JP, Matthay MA. Identification of patients with acute lung injury. Predictors of mortality. Am J Respir Crit Care Med : 1818–1824, 1995. [DOI] [PubMed] [Google Scholar]
- 51.Dreyfuss D, Basset G, Soler P, Saumon G. Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am Rev Respir Dis : 880–884, 1985. [DOI] [PubMed] [Google Scholar]
- 52.Dreyfuss D, Saumon G. Ventilator-induced Lung Injury. Lessons from experimental studies. Am J Respir Crit Care Med : 294–323, 1998. [DOI] [PubMed] [Google Scholar]
- 53.Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis : 1159–1164, 1988. [DOI] [PubMed] [Google Scholar]
- 54.Ehrhart IC, Hofman WF. Oleic acid dose-related edema in isolated canine lung perfused at constant pressure. J Appl Physiol : 1115–1120, 1981. [DOI] [PubMed] [Google Scholar]
- 55.Eichacker PQ, Waisman Y, Natanson C, Farese A, Hoffman WD, Banks SM, MacVittie TJ. Cardiopulmonary effects of granulocyte colony-stimulating factor in a canine model of bacterial sepsis. J Appl Physiol : 2366–2373, 1994. [DOI] [PubMed] [Google Scholar]
- 56.Elias JA, Zheng T, Lee CG, Homer RJ, Chen Q, Ma B, Blackburn M, Zhu Z. Transgenic modeling of interleukin-13 in the lung. Chest : 339S–345S, 2003. [PubMed] [Google Scholar]
- 57.Engles RE, Huber TS, Zander DS, Hess PJ, Welborn MB, Moldawer LL, Seeger JM. Exogenous human recombinant interleukin-10 attenuates hindlimb ischemia-reperfusion injury. J Surg Res : 425–428, 1997. [DOI] [PubMed] [Google Scholar]
- 58.Fan X, Patera AC, Pong-Kennedy A, Deno G, Gonsiorek W, Manfra DJ, Vassileva G, Zeng M, Jackson C, Sullivan L, Sharif-Rodriguez W, Opdenakker G, Van Damme J, Hedrick JA, Lundell D, Lira SA, Hipkin RW. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 AND IL-8/CXCL8. J Biol Chem : 11658–11666, 2007. [DOI] [PubMed] [Google Scholar]
- 59.Fischer S, Cassivi SD, Xavier AM, Cardella JA, Cutz E, Edwards V, Liu M, Keshavjee S. Cell death in human lung transplantation: apoptosis induction in human lungs during ischemia and after transplantation. Ann Surg : 424–431, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Folkesson HG, Matthay MA, Hebert CA, Broaddus VC. Acid aspiration induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest : 107–116, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fonte DA, Hausberger FX. Pulmonary free fatty acids in experimental fat embolism. J Trauma : 668–672, 1971. [DOI] [PubMed] [Google Scholar]
- 62.Fowler AA, Hamman RF, Good JT, Benson KN, Baird M, Eberle DJ, Petty TL, Hyers TM. Adult respiratory distress syndrome: risk with common predispositions. Ann Intern Med : 593–597, 1983. [DOI] [PubMed] [Google Scholar]
- 63.Fox RB, Hoidal JR, Brown DM, Repine JE. Pulmonary inflammation due to oxygen toxicity: involvement of chemotactic factors and polymorphonuclear leukocytes. Am Rev Respir Dis : 521–523, 1981. [DOI] [PubMed] [Google Scholar]
- 64.Fox-Dewhurst R, Alberts M, Kajikawa O, Caldwell E, Johnson MC II, Skerrett SJ, Goodman RB, Ruzinski JT, Wong VA, Chi EY, Martin TR. Pulmonary and systemic inflammatory responses in rabbits with gram-negative pneumonia. Am J Respir Crit Care Med : 2030–2040, 1997. [DOI] [PubMed] [Google Scholar]
- 65.Franek WR, Horowitz S, Stansberry L, Kazzaz JA, Koo HC, Li Y, Arita Y, Davis JM, Mantell AS, Scott W, Mantell LL. Hyperoxia inhibits oxidant-induced apoptosis in lung epithelial cells. J Biol Chem : 569–575, 2001. [DOI] [PubMed] [Google Scholar]
- 66.Frank JA, Gutierrez JA, Jones KD, Allen L, Dobbs L, Matthay MA. Low tidal volume reduces epithelial and endothelial injury in acid-injured rat lungs. Am J Respir Crit Care Med : 242–249, 2002. [DOI] [PubMed] [Google Scholar]
- 67.Frank L, Bucher JR, Roberts RJ. Oxygen toxicity in neonatal and adult animals of various species. J Appl Physiol : 699–704, 1978. [DOI] [PubMed] [Google Scholar]
- 68.Freeman BA, Crapo JD. Biology of disease: free radicals and tissue injury. Lab Invest : 412–426, 1982. [PubMed] [Google Scholar]
- 69.Fridovich I. The biology of oxygen radicals. Science : 875–880, 1978. [DOI] [PubMed] [Google Scholar]
- 70.Fu Z, Costello ML, Tsukimoto K, Prediletto R, Elliott AR, Mathieu-Costello O, West JB. High lung volume increases stress failure in pulmonary capillaries. J Appl Physiol : 123–133, 1992. [DOI] [PubMed] [Google Scholar]
- 71.Fujino Y, Goddon S, Dolhnikoff M, Hess D, Amato MB, Kacmarek RM. Repetitive high-pressure recruitment maneuvers required to maximally recruit lung in a sheep model of acute respiratory distress syndrome. Crit Care Med : 1579–1586, 2001. [DOI] [PubMed] [Google Scholar]
- 72.Fujita M, Kuwano K, Kunitake R, Hagimoto N, Miyazaki H, Kaneko Y, Kawasaki M, Maeyama T, Hara N. Endothelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Int Arch Allergy Immunol : 202–208, 1998. [DOI] [PubMed] [Google Scholar]
- 73.Fullerton DA, Eisenach JH, Friese RS, Agrafojo J, Sheridan BC, McIntyre RC Jr. Impairment of endothelial-dependent pulmonary vasorelaxation after mesenteric ischemia/reperfusion. Surgery : 879–884, 1996. [DOI] [PubMed] [Google Scholar]
- 74.Furue S, Kuwabara K, Mikawa K, Nishina K, Shiga M, Maekawa N, Ueno M, Chikazawa Y, Ono T, Hori Y, Matsukawa A, Yoshinaga M, Obara H. Crucial role of group IIA phospholipase A2 in oleic acid-induced acute lung injury in rabbits. Am J Respir Crit Care Med : 1292–1302, 1999. [DOI] [PubMed] [Google Scholar]
- 75.Garner WL, Downs JB, Reilley TE, Frolicher D, Kargi A, Fabri PJ. The effects of hyperoxia during fulminant sepsis. Surgery : 747–751, 1989. [PubMed] [Google Scholar]
- 76.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, Ryffel B, Couillin I. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest : 3786–3799, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gnidec AG, Sibbald WJ, Cheung H, Craig A. Ibuprofen reduces the progression of permeability edema in an animal model of hyperdynamic sepsis. J Appl Physiol : 1024–1032, 1988. [DOI] [PubMed] [Google Scholar]
- 78.Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med : 602–611, 1996. [DOI] [PubMed] [Google Scholar]
- 79.Goya T, Abe M, Shimura H, Torisu M. Characteristics of alveolar macrophages in experimental septic lung. J Leukoc Biol : 236–243, 1992. [DOI] [PubMed] [Google Scholar]
- 80.Greene KE, Wright JR, Steinberg KP, Ruzinski JT, Caldwell E, Wong WB, Hull W, Whitsett JA, Akino T, Kuroki Y, Hagae H, Hudson LD, Martin TR. Serial changes in surfactant-associated proteins in lung and serum before and after the onset of ARDS. Am J Respir Crit Care Med : 1843–1850, 1999. [DOI] [PubMed] [Google Scholar]
- 81.Gunther A, Siebert C, Schmidt R, Ziegler S, Grimminger F, Yabut M, Temmesfeld B, Walmrath D, Morr H, Seeger W. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med : 176–184, 1996. [DOI] [PubMed] [Google Scholar]
- 82.Haddad IY, Crow JP, Hu P, Ye Y, Beckman J, Matalon S. Concurrent generation of nitric oxide and superoxide damages surfactant protein A. Am J Physiol Lung Cell Mol Physiol : L242–L249, 1994. [DOI] [PubMed] [Google Scholar]
- 83.Hajjar AM, Ernst RK, Tsai JH, Wilson CB, Miller SI. Human Toll-like receptor 4 recognizes host-specific LPS modifications. Nat Immunol : 354–359, 2002. [DOI] [PubMed] [Google Scholar]
- 84.Hamvas A, Park CK, Palazzo R, Liptay M, Cooper J, Schuster DP. Modifying pulmonary ischemia-reperfusion injury by altering ventilatory strategies during ischemia. J Appl Physiol : 2112–2119, 1992. [DOI] [PubMed] [Google Scholar]
- 85.Hamvas A, Schuster DP. Bronchial and reverse pulmonary venous blood flow protect the lung from ischemia-reperfusion injury. J Appl Physiol : 731–736, 1994. [DOI] [PubMed] [Google Scholar]
- 86.Hashimoto S, Pittet JF, Hong K, Folkesson H, Bagby G, Kobzik L, Frevert C, Watanabe K, Tsurufuji S, Wiener-Kronish J. Depletion of alveolar macrophages decreases neutrophil chemotaxis to Pseudomonas air space infections. Am J Physiol Lung Cell Mol Physiol : L819–L828, 1996. [DOI] [PubMed] [Google Scholar]
- 87.Haston CK, Tomko TG, Godin N, Kerckhoff L, Hallett MT. Murine candidate bleomycin induced pulmonary fibrosis susceptibility genes identified by gene expression and sequence analysis of linkage regions. J Med Genet : 464–473, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hawgood S, Clements JA. Pulmonary surfactant and its apoproteins. J Clin Invest : 1–6, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heinz S, Haehnel V, Karaghiosoff M, Schwarzfischer L, Muller M, Krause SW, Rehli M. Species-specific regulation of Toll-like receptor 3 genes in men and mice. J Biol Chem : 21502–21509, 2003. [DOI] [PubMed] [Google Scholar]
- 90.Heremans H, Dillen C, Groenen M, Matthys P, Billiau A. Role of interferon-gamma and nitric oxide in pulmonary edema and death induced by lipopolysaccharide. Am J Respir Crit Care Med : 110–117, 2000. [DOI] [PubMed] [Google Scholar]
- 91.Hoebe K, Beutler B. Unraveling innate immunity using large scale N-ethyl-N-nitrosourea mutagenesis. Tissue Antigens : 395–401, 2005. [DOI] [PubMed] [Google Scholar]
- 92.Hollenstein U, Homoncik M, Stohlawetz PJ, Marsik C, Sieder A, Eichler HG, Jilma B. Endotoxin down-modulates granulocyte colony-stimulating factor receptor (CD114) on human neutrophils. J Infect Dis : 343–346, 2000. [DOI] [PubMed] [Google Scholar]
- 93.Howlett CE, Hutchison JS, Veinot JP, Chiu A, Merchant P, Fliss H. Inhaled nitric oxide protects against hyperoxia-induced apoptosis in rat lungs. Am J Physiol Lung Cell Mol Physiol : L596–L605, 1999. [DOI] [PubMed] [Google Scholar]
- 94.Huber GL, Edmunds LH Jr. Pulmonary artery occlusion. II. Morphologic studies. J Appl Physiol : 1002–1011, 1967. [DOI] [PubMed] [Google Scholar]
- 95.Hudson LD, Milberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am J Respir Crit Care Med : 293–301, 1995. [DOI] [PubMed] [Google Scholar]
- 96.Hussain N, Wu F, Zhu L, Thrall RS, Kresch MJ. Neutrophil apoptosis during the development and resolution of oleic acid-induced acute lung injury in the rat. Am J Respir Cell Mol Biol : 867–874, 1998. [DOI] [PubMed] [Google Scholar]
- 97.Ikegami M, Scoville EA, Grant S, Korfhagen T, Brondyk W, Scheule RK, Whitsett JA. Surfactant protein-D and surfactant inhibit endotoxin-induced pulmonary inflammation. Chest : 1447–1454, 2007. [DOI] [PubMed] [Google Scholar]
- 98.Imai T, Fujita T. Unilateral lung injury caused by ischemia without hypoxia in isolated rat lungs perfused with buffer solution. J Lab Clin Med : 830–836, 1994. [PubMed] [Google Scholar]
- 99.Imai Y, Nakagawa S, Ito Y, Kawano T, Slutsky AS, Miyasaka K. Comparison of lung protection strategies using conventional and high-frequency oscillatory ventilation. J Appl Physiol : 1836–1844, 2001. [DOI] [PubMed] [Google Scholar]
- 100.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol : 987–995, 2004. [DOI] [PubMed] [Google Scholar]
- 101.Jackson RM, Russell WJ, Veal CF. Endogenous and exogenous catalase in reoxygenation lung injury. J Appl Physiol : 858–864, 1992. [DOI] [PubMed] [Google Scholar]
- 102.Jain D, Atochina-Vasserman E, Kadire H, Tomer Y, Inch A, Scott P, Savani RC, Gow AJ, Beers MF. SP-D-deficient mice are resistant to hyperoxia. Am J Physiol Lung Cell Mol Physiol : L861–L871, 2007. [DOI] [PubMed] [Google Scholar]
- 103.Javadpour M, Kelly CJ, Chen G, Stokes K, Leahy A, Bouchier-Hayes DJ. Thermotolerance induces heat shock protein 72 expression and protects against ischaemia-reperfusion-induced lung injury. Br J Surg : 943–946, 1998. [DOI] [PubMed] [Google Scholar]
- 104.Jesch NK, Dorger M, Enders G, Rieder G, Vogelmeier C, Messmer K, Krombach F. Expression of inducible nitric oxide synthase and formation of nitric oxide by alveolar macrophages: an interspecies comparison. Environ Health Perspect , Suppl 5: 1297–1300, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Julien M, Hoeffel JM, Flick MR. Oleic acid lung injury in sheep. J Appl Physiol : 433–440, 1986. [DOI] [PubMed] [Google Scholar]
- 106.Jungi TW, Adler H, Adler B, Thony M, Krampe M, Peterhans E. Inducible nitric oxide synthase of macrophages. Present knowledge and evidence for species-specific regulation. Vet Immunol Immunopathol : 323–330, 1996. [DOI] [PubMed] [Google Scholar]
- 107.Kajikawa O, Goodman RB, Johnson MC II, Martin TR. Sensitive and specific immunoassays for rabbit IL-8 and MCP-1: two important cytokines that regulate leukocyte migration in the lungs. J Immunol Methods : 19–29, 1996. [DOI] [PubMed] [Google Scholar]
- 108.Kajikawa O, Johnson MC II, Goodman RB, Frevert CW, Martin TR. A sensitive immunoassay to detect the alpha-chemokine GRO in rabbit blood and lung fluids. J Immunol Methods : 135–143, 1997. [DOI] [PubMed] [Google Scholar]
- 109.Kapanci Y, Weibel ER, Kaplan HP, Robinson FR. Pathogenesis and reversibility of the pulmonary lesions of oxygen toxicity in monkeys. II. Ultrastructural and morphometric studies. Lab Invest : 101–118, 1969. [PubMed] [Google Scholar]
- 110.Kawano T, Mori S, Cybulsky M, Burger R, Ballin A, Cutz E, Bryan AC. Effect of granulocyte depletion in a ventilated surfactant-depleted lung. J Appl Physiol : 27–33, 1987. [DOI] [PubMed] [Google Scholar]
- 111.Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, Maeyama T, Hara N. Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am J Pathol : 597–603, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kelly NM, Young L, Cross AS. Differential induction of tumor necrosis factor by bacteria expressing rough and smooth lipopolysaccharide phenotypes. Infect Immun : 4491–4496, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khimenko PL, Moore TM, Taylor AE. Blocked ETA receptors prevent ischemia and reperfusion injury in rat lungs. J Appl Physiol : 203–207, 1996. [DOI] [PubMed] [Google Scholar]
- 114.Khimenko PL, Taylor AE. Segmental microvascular permeability in ischemia-reperfusion injury in rat lung. Am J Physiol Lung Cell Mol Physiol : L958–L960, 1999. [DOI] [PubMed] [Google Scholar]
- 115.Kim YM, Hong SJ, Billiar TR, Simmons RL. Counterprotective effect of erythrocytes in experimental bacterial peritonitis is due to scavenging of nitric oxide and reactive oxygen intermediates. Infect Immun : 3074–3080, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Klausner JM, Anner H, Paterson IS, Kobzik L, Valeri CR, Shepro D, Hechtman HB. Lower torso ischemia-induced lung injury is leukocyte dependent. Ann Surg : 761–767, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Knight PR, Druskovich G, Tait AR, Johnson KJ. The role of neutrophils, oxidants, and proteases in the pathogenesis of acid pulmonary injury. Anesthesiology : 772–778, 1992. [DOI] [PubMed] [Google Scholar]
- 118.Knight PR, Rutter T, Tait AR, Coleman E, Johnson K. Pathogenesis of gastric particulate lung injury: a comparison and interaction with acidic pneumonitis. Anesth Analg : 754–760, 1993. [DOI] [PubMed] [Google Scholar]
- 119.Koike K, Moore EE, Moore FA, Kim FJW, Carl VS, Banerjee A. Gut phospholipase A2 mediates neutrophil priming and lung injury after mesenteric ischemia-reperfusion. Am J Physiol Gastrointest Liver Physiol : G397–G403, 1995. [DOI] [PubMed] [Google Scholar]
- 120.Koike K, Moore FA, Moore EE, Poggetti RS, Tuder RM, Benarjee A. Endotoxin after gut ischemia/reperfusion causes irreversible lung injury. J Surg Res : 656–662, 1992. [DOI] [PubMed] [Google Scholar]
- 121.Komuro T, Yomota C, Kimura T, Galanos C. Comparison of R- and S-form lipopolysaccharides fractionated from Escherichia coli UKT-B lipopolysaccharide in pyrogen and Limulus tests. FEMS Microbiol Lett : 79–83, 1989. [DOI] [PubMed] [Google Scholar]
- 122.Kudoh I, Wiener-Kronish JP, Hashimoto S, Pittet JF, Frank D. Exoproduct secretions of Pseudomonas aeruginosa strains influence severity of alveolar epithelial injury. Am J Physiol Lung Cell Mol Physiol : L551–L556, 1994. [DOI] [PubMed] [Google Scholar]
- 123.Kuida H, Hinshaw LB, Gilbert RP, Visscher MB. Effect of gram-negative endotoxin on pulmonary circulation. Am J Physiol : 335–344, 1958. [DOI] [PubMed] [Google Scholar]
- 124.Lachmann B, Robertson B, Vogel J. in vivo lung lavage as an experimental model of the respiratory distress syndrome. Acta Anaesthesiol Scand : 231–236, 1980. [DOI] [PubMed] [Google Scholar]
- 125.Lamm WJ, Graham MM, Albert RK. Mechanism by which the prone position improves oxygenation in acute lung injury. Am J Respir Crit Care Med : 184–193, 1994. [DOI] [PubMed] [Google Scholar]
- 126.Lazo JS, Humphreys CJ. Lack of metabolism as the biochemical basis of bleomycin-induced pulmonary toxicity. Proc Natl Acad Sci USA : 3064–3068, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science : 1770–1773, 2006. [DOI] [PubMed] [Google Scholar]
- 128.Lomas-Neira JL, Chung CS, Wesche DE, Perl M, Ayala A. in vivo gene silencing (with siRNA) of pulmonary expression of MIP-2 versus KC results in divergent effects on hemorrhage-induced, neutrophil-mediated septic acute lung injury. J Leukoc Biol : 846–853, 2005. [DOI] [PubMed] [Google Scholar]
- 129.Lykens MG, Davis WB, Pacht ER. Antioxidant activity of bronchoalveolar lavage fluid in the adult respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol : L169–L175, 1992. [DOI] [PubMed] [Google Scholar]
- 130.Marshall RP, Bellingan G, Webb S, Puddicombe A, Goldsack N, McAnulty RJ, Laurent GJ. Fibroproliferation occurs early in the acute respiratory distress syndrome and impacts on outcome. Am J Respir Crit Care Med : 1783–1788, 2000. [DOI] [PubMed] [Google Scholar]
- 131.Martin TR, Mathison JC, Tobias PS, Leturcq DJ, Moriarty AM, Maunder RJ, Ulevitch RJ. Lipopolysaccharide binding protein enhances the responsiveness of alveolar macrophages to bacterial lipopolysaccharide. Implications for cytokine production in normal and injured lungs. J Clin Invest : 2209–2219, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Martinez-Mier G, Toledo-Pereyra LH, McDuffie JE, Warner RL, Ward PA. Neutrophil depletion and chemokine response after liver ischemia and reperfusion. J Invest Surg : 99–107, 2001. [DOI] [PubMed] [Google Scholar]
- 133.Matsuguchi T, Takagi K, Musikacharoen T, Yoshikai Y. Gene expressions of lipopolysaccharide receptors, toll-like receptors 2 and 4, are differently regulated in mouse T lymphocytes. Blood : 1378–1385, 2000. [PubMed] [Google Scholar]
- 134.Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis : 1250–1257, 1990. [DOI] [PubMed] [Google Scholar]
- 135.Matute-Bello G, Frevert CW, Kajikawa O, Skerrett SJ, Goodman RB, Park DR, Martin TR. Septic shock and acute lung injury in rabbits with peritonitis: failure of the neutrophil response to localized infection. Am J Respir Crit Care Med : 234–243, 2001. [DOI] [PubMed] [Google Scholar]
- 136.McAuley DF, Frank JA, Fang X, Matthay MA. Clinically relevant concentrations of beta2-adrenergic agonists stimulate maximal cyclic adenosine monophosphate-dependent airspace fluid clearance and decrease pulmonary edema in experimental acid-induced lung injury. Crit Care Med : 1470–1476, 2004. [DOI] [PubMed] [Google Scholar]
- 137.McHugh LG, Milberg JA, Whitcomb ME, Schoene RB, Maunder RJ, Hudson LD. Recovery of function in survivors of the acute respiratory distress syndrome. Am J Respir Crit Care Med : 90–94, 1994. [DOI] [PubMed] [Google Scholar]
- 138.Mendelson CL. The aspiration of stomach contents into the lungs during obstetric anesthesia. Am J Obstet Gynecol : 191, 1946. [DOI] [PubMed] [Google Scholar]
- 139.Miller EJ, Cohen AB, Matthay MA. Increased interleukin-8 concentrations in the pulmonary edema fluid of patients with acute respiratory distress syndrome from sepsis. Crit Care Med : 1448–1454, 1996. [DOI] [PubMed] [Google Scholar]
- 140.Mizgerd JP, Skerrett SJ. Animal models of human pneumonia. Am J Physiol Lung Cell Mol Physiol : L387–L398, 2008. [DOI] [PubMed] [Google Scholar]
- 141.Modelska K, Pittet JF, Folkesson HG, Courtney Broaddus V, Matthay MA. Acid-induced lung injury. Protective effect of anti-interleukin-8 pretreatment on alveolar epithelial barrier function in rabbits. Am J Respir Crit Care Med : 1450–1456, 1999. [DOI] [PubMed] [Google Scholar]
- 142.Montgomery AB, Stager MA, Carrico CJ, Hudson LD. Causes of mortality in patients with the adult respiratory distress syndrome. Am Rev Respir Dis : 485–489, 1985. [DOI] [PubMed] [Google Scholar]
- 143.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol : L152–L160, 2008. [DOI] [PubMed] [Google Scholar]
- 144.Motohiro A, Furukawa T, Yasumoto K, Inokuchi K. Mechanisms involved in acute lung edema induced in dogs by oleic acid. Eur Surg Res : 50–57, 1986. [DOI] [PubMed] [Google Scholar]
- 145.Muscedere JG, Mullen JB, Gan K, Slutsky AS. Tidal ventilation at low airway pressures can augment lung injury. Am J Respir Crit Care Med : 1327–1334, 1994. [DOI] [PubMed] [Google Scholar]
- 146.Muzio M, Bosisio D, Polentarutti N, D'Amico G, Stoppacciaro A, Mancinelli R, van't Veer C, Penton-Rol G, Ruco LP, Allavena P, Mantovani A. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J Immunol : 5998–6004, 2000. [DOI] [PubMed] [Google Scholar]
- 147.Narita M, Kawashima K, Morozumi T, Takashima H. Effect of physical defenses of the respiratory tract on the development of pneumonia in pigs inoculated endobronchially with Actinobacillus pleuropneumoniae J Vet Med Sci : 839–844, 1995. [DOI] [PubMed] [Google Scholar]
- 148.Naureckas ET, Factor P, Benjaminov O, Hoffer E, Sriram V, Sznajder JI. Pentoxifylline does not protect against hyperoxic lung injury in rats. Eur Respir J : 1397–1402, 1994. [DOI] [PubMed] [Google Scholar]
- 149.Neely CF, Keith IM. A1 adenosine receptor antagonists block ischemia-reperfusion injury of the lung. Am J Physiol Lung Cell Mol Physiol : L1036–L1046, 1995. [DOI] [PubMed] [Google Scholar]
- 150.Nielsen VG, Baird MS, McAdams ML, Freeman BA. Desflurane increases pulmonary alveolar-capillary membrane permeability after aortic occlusion-reperfusion in rabbits: evidence of oxidant-mediated lung injury. Anesthesiology : 1524–1534, 1998. [DOI] [PubMed] [Google Scholar]
- 151.Nukiwa T, Matsuoka R, Takagi H, Ishii Y, Arai T, Kira S. Responses of serum and lung angiotensin-converting enzyme activities in the early phase of pulmonary damage induced by oleic acid in dogs. Am Rev Respir Dis : 1080–1086, 1982. [DOI] [PubMed] [Google Scholar]
- 152.O'Brien-Ladner AR, Nelson ME, Cowley BD Jr, Bailey K, Wesselius LJ. Hyperoxia amplifies TNF-alpha production in LPS-stimulated human alveolar macrophages. Am J Respir Cell Mol Biol : 275–279, 1995. [DOI] [PubMed] [Google Scholar]
- 153.O'Grady NP, Preas HL, Pugin J, Fiuza C, Tropea M, Reda D, Banks SM, Suffredini AF. Local inflammatory responses following bronchial endotoxin instillation in humans. Am J Respir Crit Care Med : 1591–1598, 2001. [DOI] [PubMed] [Google Scholar]
- 154.O'Mahony DS, Liles WC, Altemeier W, Dhanireddy S, Frevert C, Liggitt D, Martin T, Matute-Bello G. Mechanical ventilation interacts with endotoxemia to induce extrapulmonary organ dysfunction. Critical Care : R136, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Obermiller T, Lakshminarayan S, Willoughby S, Mendenhall J, Butler J. Influence of lung volume and left atrial pressure on reverse pulmonary venous blood flow. J Appl Physiol : 447–453, 1991. [DOI] [PubMed] [Google Scholar]
- 156.Ognibene FP, Martin SE, Parker MM, Schlesinger T, Roach P, Burch C, Shelhamer JH, Parrillo JE. Adult respiratory distress syndrome in patients with severe neutropenia. N Engl J Med : 547–551, 1986. [DOI] [PubMed] [Google Scholar]
- 157.Okada M, Yamashita C, Okada K. Contribution of endothelin-1 to warm ischemia/reperfusion injury of the rat lung. Am J Respir Crit Care Med : 2105–2110, 1995. [DOI] [PubMed] [Google Scholar]
- 158.Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F 2nd, Park DR, Pugin J, Skerrett SJ, Hudson LD, Martin TR. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med : 1896–1903, 2001. [DOI] [PubMed] [Google Scholar]
- 159.Pawlik MT, Schreyer AG, Ittner KP, Selig C, Gruber M, Feuerbach S, Taeger K. Early treatment with pentoxifylline reduces lung injury induced by acid aspiration in rats. Chest : 613–621, 2005. [DOI] [PubMed] [Google Scholar]
- 160.Payseur BA, Place M. Prospects for association mapping in classical inbred mouse strains. Genetics : 1999–2008, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pearse DB, Wagner EM. Role of the bronchial circulation in ischemia-reperfusion lung injury. J Appl Physiol : 259–265, 1994. [DOI] [PubMed] [Google Scholar]
- 162.Pearse DB, Wagner EM, Permutt S. Effect of ventilation on vascular permeability and cyclic nucleotide concentrations in ischemic sheep lungs. J Appl Physiol : 123–132, 1999. [DOI] [PubMed] [Google Scholar]
- 163.Pepe PE, Potkin RT, Reus DH, Hudson LD, Carrico CJ. Clinical predictors of the adult respiratory distress syndrome. Am J Surg : 124–130, 1982. [DOI] [PubMed] [Google Scholar]
- 164.Petty TL, Reiss OK, Paul GW, Silvers GW, Elkins ND. Characteristics of pulmonary surfactant in adult respiratory distress syndrome associated with trauma and shock. Am Rev Respir Dis : 531–536, 1977. [DOI] [PubMed] [Google Scholar]
- 165.Pittet JF, Mackersie RC, Martin TR, Matthay MA. Biological markers of acute lung injury: prognostic and pathogenic significance. Am J Respir Crit Care Med : 1187–1205, 1997. [DOI] [PubMed] [Google Scholar]
- 166.Pittet JF, Wiener-Kronish JP, Serikov V, Matthay MA. Resistance of the alveolar epithelium to injury from septic shock in sheep. Am J Respir Crit Care Med : 1093–1100, 1995. [DOI] [PubMed] [Google Scholar]
- 167.Prop J, Ehrie MG, Crapo JD, Nieuwenhuis P, Wildevuur CR. Reimplantation response in isografted rat lungs. J Thorac Cardiovasc Surg : 702–711, 1984. [PubMed] [Google Scholar]
- 168.Pugin J, Dunn I, Jolliet P, Tassaux D, Magnenat JL, Nicod LP, Chevrolet JC. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol Lung Cell Mol Physiol : L1040–L1050, 1998. [DOI] [PubMed] [Google Scholar]
- 169.Pugin J, Widmer MC, Kossodo S, Liang CM, Preas HL II, Suffredini AF. Human neutrophils secrete gelatinase B in vitro and in vivo in response to endotoxin and proinflammatory mediators. Am J Respir Cell Mol Biol : 458–464, 1999. [DOI] [PubMed] [Google Scholar]
- 170.Putensen C, Rasanen J, Dows JB. Effect of endogenous and inhaled nitric oxide on the ventilation-perfusion relationships in oleic-acid lung injury. Am J Respir Crit Care Med : 330–336, 1994. [DOI] [PubMed] [Google Scholar]
- 171.Raetz CR, Ulevitch RJ, Wright SD, Sibley CH, Ding A, Nathan CF. Gram-negative endotoxin: an extraordinary lipid with profound effects on eukaryotic signal transduction. FASEB J : 2652–2660, 1991. [DOI] [PubMed] [Google Scholar]
- 172.Raghavendran K, Davidson BA, Mullan BA, Hutson AD, Russo TA, Manderscheid PA, Woytash JA, Holm BA, Notter RH, Knight PR. Acid and particulate-induced aspiration lung injury in mice: importance of MCP-1. Am J Physiol Lung Cell Mol Physiol : L134–L143, 2005. [DOI] [PubMed] [Google Scholar]
- 173.Raghu G, Striker LJ, Hudson LD, Striker GE. Extracellular matrix in normal and fibrotic human lungs. Am Rev Respir Dis : 281–289, 1985. [DOI] [PubMed] [Google Scholar]
- 174.Raj JU, Hazinski TA, Bland RD. Oxygen-induced lung microvascular injury in neutropenic rabbits and lambs. J Appl Physiol : 921–927, 1985. [DOI] [PubMed] [Google Scholar]
- 175.Rehli M. Of mice and men: species variations of Toll-like receptor expression. Trends Immunol : 375–378, 2002. [DOI] [PubMed] [Google Scholar]
- 176.Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol : 1254–1263, 2007. [DOI] [PubMed] [Google Scholar]
- 177.Rezaiguia S, Garat C, Delclaux C, Meignan M, Fleury J, Legrand P, Matthay MA, Jayr C. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-alpha-dependent mechanism. J Clin Invest : 325–335, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ricard JD, Dreyfuss D, Saumon G. Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med : 1176–1180, 2001. [DOI] [PubMed] [Google Scholar]
- 179.Richardson JD, Woods D, Johanson WG Jr, Trinkle JK. Lung bacterial clearance following pulmonary contusion. Surgery : 730–735, 1979. [PubMed] [Google Scholar]
- 180.Rojas M, Xu J, Woods CR, Mora AL, Spears W, Roman J, Brigham KL. Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am J Respir Cell Mol Biol : 145–152, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rosen H, Klebanoff SJ. Bactericidal activity of a superoxide anion-generating system. A model for the polymorphonuclear leukocyte. J Exp Med : 27–39, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rotta AT, Gunnarsson B, Fuhrman BP, Hernan LJ, Steinhorn DM. Comparison of lung protective ventilation strategies in a rabbit model of acute lung injury. Crit Care Med : 2176–2184, 2001. [DOI] [PubMed] [Google Scholar]
- 183.Roy-Burman A, Savel RH, Racine S, Swanson BL, Revadigar NS, Fujimoto J, Sawa T, Frank DW, Wiener-Kronish JP. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J Infect Dis : 1767–1774, 2001. [DOI] [PubMed] [Google Scholar]
- 184.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med : 1685–1693, 2005. [DOI] [PubMed] [Google Scholar]
- 185.Sakao Y, Kajikawa O, Martin TR, Nakahara Y, Hadden I, Will A, Harmon CL, Miller EJ. Association of IL-8 and MCP-1 with the development of reexpansion pulmonary edema in rabbits. Ann Thoracic Surg : 1825–1832, 2001. [DOI] [PubMed] [Google Scholar]
- 186.Sakuma T, Takahashi K, Ohya N, Kajikawa O, Martin TR, Albertine KH, Matthay MA. Ischemia-reperfusion lung injury in rabbits: mechanisms of injury and protection. Am J Physiol Lung Cell Mol Physiol : L137–L145, 1999. [DOI] [PubMed] [Google Scholar]
- 187.Schneemann M, Schoedon G. Species differences in macrophage NO production are important. Nat Immunol : 102, 2002. [DOI] [PubMed] [Google Scholar]
- 188.Schneemann M, Schoedon G, Hofer S, Blau N, Guerrero L, Schaffner A. Nitric oxide synthase is not a constituent of the antimicrobial armature of human mononuclear phagocytes. J Infect Dis : 1358–1363, 1993. [DOI] [PubMed] [Google Scholar]
- 189.Schromm AB, Brandenburg K, Loppnow H, Moran AP, Koch MH, Rietschel ET, Seydel U. Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. Eur J Biochem : 2008–2013, 2000. [DOI] [PubMed] [Google Scholar]
- 190.Schuster DP, Perez JE, Trulock EP, Williamson JR, Biello DR, Kenzora JL, Amundsen T, Lange LG. Cardiac dysfunction during acute lung injury induced by oleic acid in dogs. Am Rev Respir Dis : 519–525, 1986. [DOI] [PubMed] [Google Scholar]
- 191.Schuster DP. ARDS: clinical lessons from the oleic acid model of acute lung injury. Am J Respir Crit Care Med : 245–260, 1994. [DOI] [PubMed] [Google Scholar]
- 192.Schuster DP, Kozlowski JK, McCarthy T, Morrow J, Stephenson A. Effect of endotoxin on oleic acid lung injury does not depend on priming. J Appl Physiol : 2047–2054, 2001. [DOI] [PubMed] [Google Scholar]
- 193.Schuster DP, Marklin GF. The effect of regional lung injury or alveolar hypoxia on pulmonary blood flow and lung water measured by positron emission tomography. Am Rev Respir Dis : 1037–1042, 1986. [DOI] [PubMed] [Google Scholar]
- 194.Sebti SM, Mignano JE, Jani JP, Srimatkandada S, Lazo JS. Bleomycin hydrolase: molecular cloning, sequencing, and biochemical studies reveal membership in the cysteine proteinase family. Biochemistry : 6544–6548, 1989. [DOI] [PubMed] [Google Scholar]
- 195.Seekamp A, Regel G, Rother K, Jutila M. The effect of anti-L-selectin (EL-246) on remote lung injury after infrarenal ischemia/reperfusion. Shock : 447–454, 1997. [DOI] [PubMed] [Google Scholar]
- 196.Shea LM, Beehler C, Schwartz M, Shenkar R, Tuder R, Abraham E. Hyperoxia activates NF-kappaB and increases TNF-alpha and IFN-gamma gene expression in mouse pulmonary lymphocytes. J Immunol : 3902–3908, 1996. [PubMed] [Google Scholar]
- 197.Shen AS, Haslett C, Feldsien DC, Henson PM, Cherniack RM. The intensity of chronic lung inflammation and fibrosis after bleomycin is directly related to the severity of acute injury. Am Rev Respir Dis : 564–571, 1988. [DOI] [PubMed] [Google Scholar]
- 198.Sherr S, Montemurno R, Raffer P. Lipids of recovered pulmonary fat emboli following trauma. J Trauma : 242–246, 1974. [DOI] [PubMed] [Google Scholar]
- 199.Siegelman S, Sinha SBP, Veith FJ. Pulmonary reimplantation response. Ann Surg : 30–36, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sigmund CD. Viewpoint: are studies in genetically altered mice out of control? Arterioscler Thromb Vasc Biol : 1425–1429, 2000. [DOI] [PubMed] [Google Scholar]
- 201.Sinclair SE, Altemeier WA, Matute-Bello G, Chi EY. Augmented lung injury due to interaction between hyperoxia and mechanical ventilation. Crit Care Med : 2496–2501, 2004. [DOI] [PubMed] [Google Scholar]
- 202.Sittipunt C, Steinberg KP, Ruzinski JT, Myles C, Zhu S, Goodman RB, Hudson LD, Matalon S, Martin TR. Nitric oxide and nitrotyrosine in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med : 503–510, 2001. [DOI] [PubMed] [Google Scholar]
- 203.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol : L143–L152, 2004. [DOI] [PubMed] [Google Scholar]
- 204.Smith LJ, Friedman H, Anderson J. Hyperoxic lung injury in mice: effect of neutrophil depletion and food deprivation. J Lab Clin Med : 449–458, 1988. [PubMed] [Google Scholar]
- 205.Smith PD, Suffredini AF, Allen JB, Wahl LM, Parrillo JE, Wahl SM. Endotoxin administration to humans primes alveolar macrophages for increased production of inflammatory mediators. J Clin Immunol : 141–148, 1994. [DOI] [PubMed] [Google Scholar]
- 206.Sone Y, Serikov VB, Staub NC, Sr. Intravascular macrophage depletion attenuates endotoxin lung injury in anesthetized sheep. J Appl Physiol : 1354–1359, 1999. [DOI] [PubMed] [Google Scholar]
- 207.St. John RC, Mizer LA, Kindt GC, Weisbrode SE, Moore SA, Dorinsky PM. Acid aspiration-induced acute lung injury causes leukocyte-dependent systemic organ injury. J Appl Physiol : 1994–2003, 1993. [DOI] [PubMed] [Google Scholar]
- 208.Stammberger U, Gaspert A, Hillinger S, Vogt P, Odermatt B, Weder W, Schmid RA. Apoptosis induced by ischemia and reperfusion in experimental lung transplantation. Ann Thorac Surg : 1532–1536, 2000. [DOI] [PubMed] [Google Scholar]
- 209.Stenzel JD, Welty SE, Benzick AE, Smith EO, Smith CV, Hansen TN. Hyperoxic lung injury in Fischer-344 and Sprague-Dawley rats in vivo. Free Radic Biol Med : 531–539, 1993. [DOI] [PubMed] [Google Scholar]
- 210.Su X, Robriquet L, Folkesson HG, Matthay MA. Protective effect of endogenous beta-adrenergic tone on lung fluid balance in acute bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol : L769–L776, 2006. [DOI] [PubMed] [Google Scholar]
- 211.Tapping RI, Akashi S, Miyake K, Godowski PJ, Tobias PS. Toll-like receptor 4, but not Toll-like receptor 2, is a signaling receptor for Escherichia and Salmonella lipopolysaccharides. J Immunol : 5780–5787, 2000. [DOI] [PubMed] [Google Scholar]
- 212.Tavaf-Motamen H, Miner TJ, Starnes BW, Shea-Donohue T. Nitric oxide mediates acute lung injury by modulation of inflammation. J Surg Res : 137–142, 1998. [DOI] [PubMed] [Google Scholar]
- 213.Taveira da Silva AM, Kaulbach HC, Chiudian FS, Lambert DR, Suffredini AF, Danner RL. Brief report: shock and multiple-organ dysfunction after self-administration of Salmonella endotoxin. N Engl J Med : 1457–1460, 1993. [DOI] [PubMed] [Google Scholar]
- 214.Terashima T, Matsubara H, Nakamura M, Sakamaki F, Waki Y, Soejima K, Tasaka S, Nakamura H, Sayama K, Ishizaka A, Kanazawa M. Local Pseudomonas instillation induces contralateral lung injury and plasma cytokines. Am J Respir Crit Care Med : 1600–1605, 1996. [DOI] [PubMed] [Google Scholar]
- 215.The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med : 1301–1308, 2000. [DOI] [PubMed] [Google Scholar]
- 216.Thoma-Uszynski S, Stenger S, Takeuchi O, Ochoa MT, Engele M, Sieling PA, Barnes PF, Rollinghoff M, Bolcskei PL, Wagner M, Akira S, Norgard MV, Belisle JT, Godowski PJ, Bloom BR, Modlin RL. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science : 1544–1547, 2001. [DOI] [PubMed] [Google Scholar]
- 217.Tobias PS, Soldau K, Ulevitch RJ. Isolation of a lipopolysaccharide-binding acute phase reactant from rabbit serum. J Exp Med : 777–793, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tomashefski JF Jr, Davies P, Boggis C, Greene R, Zapol WM, Reid LM. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am J Pathol : 112–126, 1983. [PMC free article] [PubMed] [Google Scholar]
- 218a.Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest : 944–952, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tsai WC, Strieter RM, Mehrad B, Newstead MW, Zeng X, Standiford TJ. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect Immun : 4289–4296, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tsuno K, Miura K, Takeya M, Kolobow T, Morioka T. Histopathologic pulmonary changes from mechanical ventilation at high peak airway pressures. Am Rev Respir Dis : 1115–1120, 1991. [DOI] [PubMed] [Google Scholar]
- 221.Ulrich K, Stern M, Goddard ME, Williams J, Zhu J, Dewar A, Painter HA, Jeffery PK, Gill DR, Hyde SC, Geddes DM, Takata M, Alton EWFW. Keratinocyte growth factor therapy in murine oleic acid-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol : L1179–L1192, 2005. [DOI] [PubMed] [Google Scholar]
- 222.Umezawa H. Bleomycin and other antitumor antibiotics of high molecular weight. Antimicrob Agents Chemother : 1079–1085, 1965. [PubMed] [Google Scholar]
- 223.Villar J, Ribeiro SP, Mullen JB, Kuliszewski M, Post M, Slutsky AS. Induction of the heat shock response reduces mortality rate and organ damage in a sepsis-induced acute lung injury model. Crit Care Med : 914–921, 1994. [PubMed] [Google Scholar]
- 224.Vlahakis NE, Hubmayr RD. Cellular stress failure in ventilator-injured lungs. Am J Respir Crit Care Med : 1328–1342, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Vlahakis NE, Schroeder MA, Limper AH, Hubmayr RD. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol : L167–L173, 1999. [DOI] [PubMed] [Google Scholar]
- 226.Vlahakis NE, Schroeder MA, Pagano RE, Hubmayr RD. Deformation-induced lipid trafficking in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol : L938–L946, 2001. [DOI] [PubMed] [Google Scholar]
- 227.Waddell TK, Gorczynski RM, DeCampos KN, Patterson GA, Slutsky AS. Major histocompatibility complex expression and lung ischemia-reperfusion in rats. Ann Thorac Surg : 866–872, 1996. [DOI] [PubMed] [Google Scholar]
- 228.Walley KR, Lukacs NW, Standiford TJ, Strieter RM, Kunkel SL. Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect Immun : 4733–4738, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Walther FJ, Hernandez-Juviel J, Bruni R, Waring AJ. Spiking Survanta with synthetic surfactant peptides improves oxygenation in surfactant-deficient rats. Am J Respir Crit Care Med : 855–861, 1997. [DOI] [PubMed] [Google Scholar]
- 230.Wang HL, Akinci IO, Baker CM, Urich D, Bellmeyer A, Jain M, Chandel NS, Mutlu GM, Budinger GR. The intrinsic apoptotic pathway is required for lipopolysaccharide-induced lung endothelial cell death. J Immunol : 1834–1841, 2007. [DOI] [PubMed] [Google Scholar]
- 231.Wang L, Zhu DM, Su X, Bai CX, Ware LB, Matthay MA. Acute cardiopulmonary effects of a dual-endothelin receptor antagonist on oleic acid-induced pulmonary arterial hypertension in dogs. Exp Lung Res : 31–42, 2004. [DOI] [PubMed] [Google Scholar]
- 232.Wang LF, Patel M, Razavi HM, Weicker S, Joseph MG, McCormack DG, Mehta S. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Respir Crit Care Med : 1634–1639, 2002. [DOI] [PubMed] [Google Scholar]
- 233.Ward NS, Waxman AB, Homer RJ, Mantell LL, Einarsson O, Du Y, Elias JA. Interleukin-6-induced protection in hyperoxic acute lung injury. Am J Respir Cell Mol Biol : 535–542, 2000. [DOI] [PubMed] [Google Scholar]
- 234.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med : 1376–1383, 2001. [DOI] [PubMed] [Google Scholar]
- 235.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med : 1334–1349, 2000. [DOI] [PubMed] [Google Scholar]
- 236.Warner AE. Pulmonary intravascular macrophages. Role in acute lung injury. Clin Chest Med : 125–135, 1996. [DOI] [PubMed] [Google Scholar]
- 237.Welborn MB 3rd, Douglas WG, Abouhamze Z, Auffenburg T, Abouhamze AS, Baumhofer J, Seeger JM, Pruitt JH, Edwards PD, Chizzonite R, Martin D, Moldawer LL, Harward TR. Visceral ischemia-reperfusion injury promotes tumor necrosis factor (TNF) and interleukin-1 (IL-1) dependent organ injury in the mouse. Shock : 171–176, 1996. [PubMed] [Google Scholar]
- 238.Welty-Wolf KE, Carraway MS, Ortel TL, Ghio AJ, Idell S, Egan J, Zhu X, Jiao Ja Wong HC, Piantadosi CA. Blockade of tissue factor-factor X binding attenuates sepsis-induced respiratory and renal failure. Am J Physiol Lung Cell Mol Physiol : L21–L31, 2006. [DOI] [PubMed] [Google Scholar]
- 239.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J Clin Invest : 864–875, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wiener-Kronish JP, Sakuma T, Kudoh I, Pittet JF, Frank D, Dobbs L, Vasil ML, Matthay MA. Alveolar epithelial injury and pleural empyema in acute P. aeruginosa pneumonia in anesthetized rabbits. J Appl Physiol : 1661–1669, 1993. [DOI] [PubMed] [Google Scholar]
- 241.Wiggs BR, English D, Quinlan WM, Doyle NA, Hogg JC, Doerschuk CM. Contributions of capillary pathway size and neutrophil deformability to neutrophil transit through rabbit lungs. J Appl Physiol : 463–470, 1994. [DOI] [PubMed] [Google Scholar]
- 242.Winkler GC. Review of the significance of pulmonary intravascular macrophages with respect to animal species and age. Exp Cell Biol : 281–286, 1989. [DOI] [PubMed] [Google Scholar]
- 243.Wright JR. Immunomodulatory functions of surfactant. Physiol Rev : 931–962, 1997. [DOI] [PubMed] [Google Scholar]
- 244.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science : 1431–1433, 1990. [DOI] [PubMed] [Google Scholar]
- 245.Xiao F, Eppihimer MJ, Young JA, Nguyen K, Carden DL. Lung neutrophil retention and injury after intestinal ischemia/reperfusion. Microcirculation : 359–367, 1997. [DOI] [PubMed] [Google Scholar]
- 246.Xu J, Mora A, Shim H, Stecenko A, Brigham KL, Rojas M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol : 291–299, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Yamaguchi K, Mori M, Kawai A, Asano K, Takasugi T, Umeda A, Yokoyama T. Attenuation of hypoxic pulmonary vasoconstriction in acute oleic acid lung injury—significance of vasodilator prostanoids. Adv Exp Med Biol : 299–309, 1992. [DOI] [PubMed] [Google Scholar]
- 248.Yamaguchi T, Kawabata K, Koizumi N, Sakurai F, Nakashima K, Sakurai H, Sasaki T, Okada N, Yamanishi K, Mizuguchi H. Role of MyD88 and TLR9 in the innate immune response elicited by serotype 5 adenoviral vectors. Hum Gene Ther : 753–762, 2007. [DOI] [PubMed] [Google Scholar]
- 249.Yang RB, Mark MR, Gray A, Huang A, Xie MH, Zhang M, Goddard A, Wood WI, Gurney AL, Godowski PJ. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature : 284–288, 1998. [DOI] [PubMed] [Google Scholar]
- 250.Yano T, Deterding RR, Simonet WS, Shannon JM, Mason RJ. Keratinocyte growth factor reduces lung damage due to acid instillation in rats. Am J Respir Cell Mol Biol : 433–442, 1996. [DOI] [PubMed] [Google Scholar]
- 251.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest : 3211–3219, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Zhu Z, Zheng T, Lee CG, Homer RJ, Elias JA. Tetracycline-controlled transcriptional regulation systems: advances and application in transgenic animal modeling. Semin Cell Dev Biol : 121–128, 2002. [DOI] [PubMed] [Google Scholar]
- 253.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity : 121–127, 2000. [DOI] [PubMed] [Google Scholar]