

Abstract
The β1-adrenergic receptor (β1AR; ADRB1) polymorphism Arg389Gly is located in an intracellular loop and is associated with distinct human and mouse cardiovascular phenotypes. To test the hypothesis that β1-Arg389 and β1-Gly389 alleles could differentially couple to pathways beyond that of classic Gs-adenylyl cyclase (AC)/cAMP signaling, we performed comparative gene expression profile analyses on hearts from wild-type and transgenic mice that expressed either human β1-Arg389 or β1-Gly389 receptors, or AC5, sampling at an early age prior to the onset of pathological features. All three models upregulated the expression of genes associated with RNA metabolism and translation and downregulated genes associated with mitochondria and energy metabolism, consistent with shared cAMP-driven increase in cardiac contractility, protein synthesis, and compensatory downregulation of mitochondrial energy production. Both β1AR alleles activated additional genes associated with other pathways. Uniquely, β1-Arg389 hearts exhibited upregulated expression of genes associated with inflammation, programmed cell death, and extracellular matrix. These observations expand the scope of 7-transmembrane domain receptor signaling propagation beyond known cognate G protein couplings. Moreover, they implicate alterations of a repertoire of processes evoked by a single amino acid variation in the cardiac β1AR that might be exploited for genotype-specific heart failure diagnostics and therapeutics.
Keywords: polymorphism, adrenergic, heart failure, β-blocker, microarrays
the β1-adrenergic receptor (β1AR) is polymorphic in the human population, with the major variation being at amino acid 389, which can be either Arg or Gly (rs1801253). In recombinantly expressing cell lines (12) and transgenic mice (14), the β1-Arg389 has been found to exhibit increased stimulation of adenylyl cyclase compared with β1-Gly389. In addition, in a model cell-based system β1-Arg389 underwent greater agonist-promoted desensitization [of the adenylyl cyclase (AC) response] compared with β1-Gly389 (17). This polymorphic variation of the β1AR has been associated with a number of human cardiovascular phenotypes including risk for hypertension (1), exercise capacity in heart failure (30), the inotropic response to dobutamine (3, 8), ventricular remodeling in heart failure (29), β-blocker survival in heart failure (11, 14), and the blood pressure response to β-blockers in hypertension (7, 26). The most simplistic mechanism that explains these phenotypes is based on the AC/cAMP coupling of β1-Arg389, enhancing cardiac inotropy and chronotropy or renal renin release, thus culminating in physiologic sequelae, or, providing a higher hemodynamic “baseline” and thus the greatest potential for change upon treatment with β-blockers. And indeed, in young transgenic mice expressing equivalent levels of cardiac myocyte β1-Arg389 or β1-Gly389, the former mice have enhanced basal and agonist-stimulated cardiac contractility and AC activities, develop age-dependent ventricular dysfunction, and exhibit the greatest decrease in heart rate and contractility during β-blocker exposure (14). However, increasing AC activity by transgenically overexpressing AC (types 5 or 6) in the heart does not evoke age-dependent ventricular dysfunction (21, 28). And yet, contractility is persistently increased in such mice, to a level equivalent to that of β1-Arg389 (28). It has also become evident in cell-based studies, that βAR may communicate with pathways unrelated to receptor-Gs coupling or activation of AC (10). We have considered, then, that the nonconservative variation in amino acid 389 may have signaling and pathological consequences that are distinct for the Arg or Gly variant and that these may be independent of the signaling pathways traditionally ascribed to the β1AR. To investigate this, we performed genome-wide expression profiling on hearts from young (90 days of age) β1-Arg389, β1-Gly389, and AC5 transgenic mice, comparing each to their nontransgenic littermates at a stage prior to development of any defined physiological dysfunction or pathological features. Given that the two receptors have different cardiac risk profiles, we hypothesized that they might exhibit correspondingly different expression profiles that would shed light on β1AR signaling implications evoked by this common polymorphic variation.
MATERIALS AND METHODS
Transgenic mice.
The α-myosin heavy chain promoter was utilized to direct expression of the human β1AR (Arg389), human β1AR (Gly389), and human AC5 cDNAs to cardiac myocytes. The generation and characterization of these mice have been described previously (14, 28). The study was approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee. Transgenic lines expressed Arg- and Gly389 β1ARs is at equivalent levels (∼1,000 fmol/mg) as determined by radioligand binding (14); AC5 expression was twofold over endogenous expression as determined by catalytic activity (28). These animals are referred to as β1-Arg389, β1-Gly389, and AC5 mice. All were maintained in identical environments, along with nontransgenic littermates, and euthanized by CO2 narcosis at exactly 90 days of age. Hearts were rapidly removed and immediately frozen in TRIzol (Invitrogen, Carlsbad, CA) for batch RNA extraction.
DNA microarrays and analysis.
Total RNA was prepared from each of six individual hearts with the following genotypes: nontransgenic, β1-Arg389, β1-Gly389, and AC5 using methods previously described (13). The quality of the samples was verified using an Agilent Bioanalyzer 2100 (Agilent Technologies, Palo Alto, CA). Differential gene expression was ascertained using Affymetrix MOE 430 plus 2.0 chips (45,000 probe sets, 39,000 transcripts) according to the manufacturer's protocol. Normalization was performed using robust multiple-array average (RMA) as implemented in GeneSpring 7.3.1 (Agilent). Gene expression levels for each probe set in each individual mouse were referenced relative to the median of its intensity value in corresponding nontransgenic littermate hearts. Quantitative real-time PCR was utilized to confirm appropriate normalization based on relative expression of 10 representative genes. The correlation coefficient for expression determined by the arrays vs. quantitative real-time PCR was found to be R2 = 0.966 (Supplementary Fig. S1).1 Using GeneSpring, we then performed a general ANOVA for the four groups to identify genes that exhibited significant differences between genotypes with the Student's assumption of equal between-group variance and Benjamini-Hochberg false discovery rate cutoff of 5%. The major sets of comparisons were thus between the group of hearts with a given genetic alteration and the group corresponding to nontransgenic littermate hearts. Then, sorting of the data from these comparisons to nontransgenics provided for those transcripts uniquely regulated by only the indicated mouse (i.e., transcripts regulated by β1-Arg389 vs. nontransgenics that were not found to be regulated by β1-Gly389 or AC5). The relative expression patterns of differentially expressed transcripts was determined using the hierarchical tree clustering algorithm as implemented in GeneSpring using Pearson correlation applied to the log ratio of gene expression values. GeneSet Enrichment Analysis (GSEA) for categorical feature overrepresentation in each gene list from our analysis was performed using Toppgene (http://toppgene.cchmc.org/). A significance value (P) from GSEA is calculated based on the number of identified genes in a particular category relative to the total number of genes in that category on the array, to indicate whether there are more genes with changes identified in a particular category than one would expect by chance. A P-value cutoff of 0.05 was utilized, and the minimum number of genes in a category to be included in further analysis was set to 3. To identify the most significant gene networks that distinguished Arg vs. Gly regulation, we prioritized Arg-specific overexpressed genes compared with those activated in Gly using both GSEA and Ingenuity Pathway Analysis approaches (http://analysis.ingenuity.com).
RESULTS
Raw data from the Affymetrix MOE 430 plus 2 GeneChips for the 24 mouse hearts are deposited in Gene Expression Omnibus (GEO) with accession number GSE11887. The genome-wide expression profile analyses of these hearts revealed substantial expression pattern differences between β1-Arg389, β1-Gly389, and AC5 transgenic mice (Fig. 1). Using a statistical cutoff applied to raw signal intensity filtered transcripts (5% false discovery for probe sets with RMA >6.0 and differential expression among at least one of the nontransgenic, AC5, β1-Gly389, or β1-Arg389 group), we identified >3,000 probe sets that exhibited model-dependent expression differences. The number of upregulated and downregulated genes, and their common and unique distributions are shown in Fig. 2. Of these, compared with nontransgenic, 2,926 were up- or downregulated in the β1-Arg389 hearts; 2,067 for β1-Gly389 hearts, and 924 in AC5. The overlaps among genes that were regulated between hearts expressing one of the β1ARs and AC5 or the two β1ARs - that could be due to a common cAMP-dependent mechanism - amounted to 1,986 probe sets. Of these, 1,048 were regulated by both β1-Arg389 and β1-Gly389, but not by AC5 overexpression. This is consistent with the likelihood that β1AR signaling propagates to non-cAMP/PKA events, and, that for these model-affected genes, these events are not dependent on whether the position 389 amino acid is Arg or Gly. However, as shown in Fig. 2, a substantial number of genes are specifically regulated by β1-Arg389 or β1-Gly389 that are not affected by AC5. By far, most of these genes were associated with the β1-Arg389 hearts and amounted to 1,041 vs. the 245 unique genes regulated in β1-Gly389 hearts (P < 1E−9). Downregulation was more prevalent (by ∼50% or more) compared with upregulation. This was primarily due to a dominance of downregulated genes that were in common with either of the β1ARs or a β1AR and AC5. Within the unique genes that were regulated in β1-Arg389 and β1-Gly389 hearts, the distribution was very similar between up- vs. downregulated genes, and thus the dominance of unique β1-Arg389 evoked changes in gene expression was apparent regardless of the direction of the change.
Fig. 1.
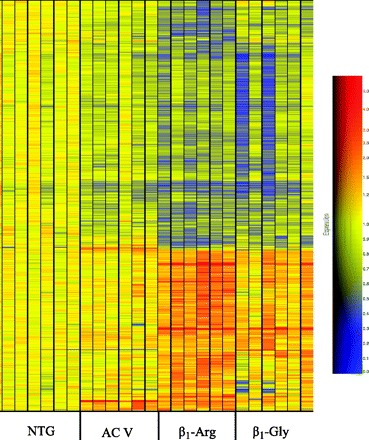
Overall heat map of the expressed genes in the 4 groups of mice. Each group consisted of 6 hearts from the indicated mice, and the colors are scaled to changes compared with the nontransgenic (NTG) hearts.
Fig. 2.
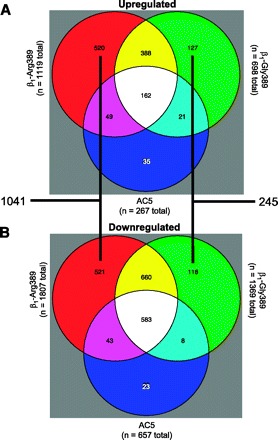
Distribution of altered transcripts from hearts of β1-Arg389, β1-Gly389, and AC5 mice by overlapping and unique gene sets. The numbers in each region represent the number of genes significantly up- (A) or downregulated (B) compared with NTG hearts, sorted by the indicated partitions to indicate genes that were regulated in common with and, distinct from, those from other transgenic mice. Sizes of the partitions are not to scale.
To identify the biological themes that are reflected by the various overlapping and unique sets of genes, we undertook an integrative GSEA in which the different groups of ANOVA-derived genes upregulated or downregulated in each of the transgenic models were subjected to GeneSet enrichment using the ToppGene system previously described by Chen et al. (4). For each enriched gene set, i.e., gene ontology, protein interaction, PubMed article, the enrichment P-value for nonrandom list intersection (using Benjamini-Hochberg false discovery rate P-value correction) was converted to a significance score [S = −log (pval)], and a matrix was constructed composed of the six lists of genes (up- and downregulated for each transgenic model) as illustrated in the heat map of Fig. 3. (The full heat map with annotations is provided in Supplementary Table S1.) Our primary goal was to define common themes among the various overlapping and unique gene sets. Since both β1ARs couple to AC and cAMP signaling, genes regulated in common with overexpression of both receptors and AC5 represented a distinct set of interest. The GO categories of the genes that were regulated in common with both of the allelic forms of the β1AR and AC5 are provided in Supplementary Table S2. As noted above, the shared signatures were usually downregulated, and these were dominated by pathways involved with mitochondrial-based energy generation including fatty acid oxidation and cellular respiration (e.g., ABCB10, ACADVL, ATP5A1, ATP5B, ATP5S, CPT2, CRAT, ETFDH, FECH, HCCS, IMMT, MPV17, NDUFA10, NDUFA9, NDUFAF1, NDUFS1, NDUFS2, NDUFS4, NDUFV1, OPA1, PMPCA, SDHA, SDHD, SLC25A11, SLC25A13, SURF1, TIMM22, TIMM44, UQCRC2), consistent with the phenotypes imposed by all three transgenes that we have observed in cardiac functional studies (14, 28). Given that the physiological, pharmacological, and mouse model phenotypes previously observed associated with the β1-Arg389 variant, hearts from these animals exhibited the most number of transcripts that underwent expression changes. We therefore sought to distinguish informative differences between this group of genes versus the other models. Of the 1,041 β1-Arg389-specific genes that underwent expression changes (Supplementary Table S3) several GO groups were dominant including well-established cardiac pathology-associated genes involved in extracellular matrix, inflammation, and programmed cell death. The most up- and downregulated genes (15 of each) within the β1-Arg389-specific groups are shown in Table 1. As expected, network-based pathway analyses performed using ToppGene and Ingenuity Pathway Analysis revealed several statistically significant networks consistent with the patterns of gene expression evoked in β1-Arg389 hearts, with the top-ranked most significant of these (P = 1E−15) primarily involving regulation of extracellular matrix-associated genes known to be activated directly or indirectly by TGFB1, TGFB2, and TGFB3 activation and signaling (Fig. 4).
Fig. 3.
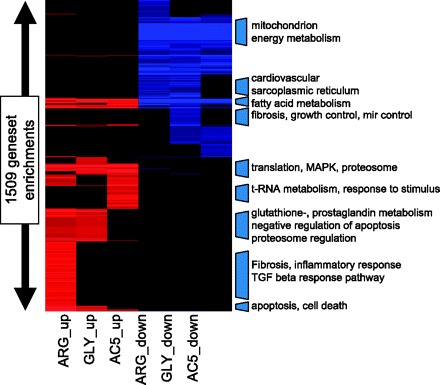
GeneSet Enrichment Analysis. Genes overexpressed or underexpressed in each model were separately subjected to GeneSet Enrichment Analysis for overrepresented gene annotation features. Red blocks are highly significant categories enriched in one or more upregulated gene group. Blue blocks are highly enriched in category among the various downregulated genes. Complete details of each category and the lists of genes that overlap with the category are provided in Supplementary Table S1.
Table 1.
Genes with the greatest degree of up- and downregulation that is unique to β1-Arg389 hearts
| Gene | Fold Change | Affymetrix | Accession No. | Description |
|---|---|---|---|---|
| Spp1 | 6.90 | 1449254_at | NM_009263 | secreted phosphoprotein 1, osteopontin |
| Ccl9 | 3.86 | 1448898_at | NM_011338 | chemokine (C-C motif) ligand 9 |
| Itgb1bp3 | 3.45 | 1453898_at | XM_125745 | integrin beta 1 binding protein 3 |
| C3ar1 | 3.10 | 1442082_at | NM_009779 | complement component 3a receptor 1 |
| Tgfb2 | 2.79 | 1450922_a_at | NM_009367 | transforming growth factor, beta 2 |
| Ccl6 | 2.67 | 1420249_s_at | NM_009139 | chemokine (C-C motif) ligand 6 |
| Fcgr2b | 2.63 | 1451941_a_at | NM_010187 | Fc receptor, IgG, low affinity IIb |
| Ctss | 2.53 | 1448591_at | NM_021281 | cathepsin S |
| Gdf15 | 2.47 | 1418949_at | NM_011819 | growth differentiation factor 15 |
| Thbs1 | 2.42 | 1421811_at | NM_011580 | thrombospondin 1 |
| Arnt2 | 2.28 | 1446894_at | NM_007488 | Aryl hydrocarbon receptor nuclear translocator 2 (Arnt2) |
| Ifi205 | 2.26 | 1452349_x_at | NM_172648 | interferon activated gene 205 |
| Nppa | 2.26 | 1456062_at | NM_008725 | natriuretic peptide precursor type A |
| Cilp | 2.23 | 1457296_at | NM_173385 | cartilage intermediate layer protein, nucleotide pyrophosphohydrolase |
| Hist1 h2ad | 2.23 | 1438009_at | NM_178188 | histone cluster 1, H2ad |
| Impa2 | 0.61 | 1418665_at | NM_053261 | inositol (myo)-1(or 4)-monophosphatase 2 |
| Tef | 0.60 | 1450184_s_at | NM_017376 | thyrotroph embryonic factor |
| Dbp | 0.60 | 1438211_s_at | NM_016974 | D site albumin promoter binding protein |
| Stk23 | 0.60 | 1447806_s_at | NM_019684 | serine/threonine kinase 23 |
| Errfi1 | 0.59 | 1416129_at | NM_133753 | ERBB receptor feedback inhibitor 1 |
| Inmt | 0.59 | 1418697_at | NM_009349 | indolethylamine N-methyltransferase |
| Ccnt2 | 0.58 | 1435445_at | NM_028399 | cyclin T2 |
| Ppp1r10 | 0.57 | 1426727_s_at | NM_175934 | protein phosphatase 1, regulatory subunit 10 |
| Per1 | 0.56 | 1449851_at | NM_011065 | period homolog 1 (Drosophila) |
| Scn4b | 0.51 | 1434008_at | NM_1013390 | sodium channel, type IV, beta |
| Jarid2 | 0.51 | 1422697_s_at | NM_021878 | jumonji, AT rich interactive domain 2 |
| Slc36a2 | 0.45 | 1436521_at | NM_153170 | solute carrier family 36 (proton/amino acid symporter), member 2 |
| Car3 | 0.39 | 1449434_at | NM_007606 | carbonic anhydrase 3 |
| Acot1 | 0.37 | 1449065_at | NM_012006 | acyl-CoA thioesterase 1 |
| Dnajb1 | 0.36 | 1416755_at | NM_018808 | DnaJ (Hsp40) homolog, subfamily B, member 1 |
Fig. 4.
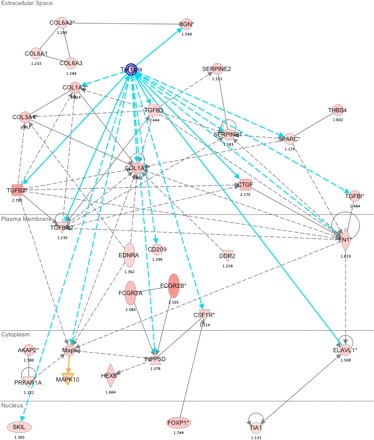
Ingenuity Pathway Analysis (IPA) of genes specifically overexpressed in β1-Arg389 transgenic mouse hearts. Shown is a TGFB1, TGFB2, and TGFB3 centered network, the top-ranked gene network based on the IPA scoring algorithm. The symbols, lines, and colors are the default settings (see http://analysis.ingenuity.com).
DISCUSSION
As introduced earlier, the Arg and Gly moieties at amino acid position 389 of the human β1AR impart differential coupling of the β1AR to its cognate G protein, Gs. This results in the β1-Arg389 receptor having a small increased basal, and agonist-promoted, AC activity and cAMP levels compared with the β1-Gly389 receptor. Multiple clinical gene variant association studies (reviewed in Refs. 9, 25) are consistent with this phenotype, and indeed ex vivo contractile responses to isoproterenol are greater in human trabeculae from normal (and failing) hearts with β1-Arg389 compared with analogous trabeculae with the -Gly389 genotype (14). Thus, one mechanism of the molecular phenotype of the Arg variant has been considered to be a gain-of-function within the AC/cAMP/PKA axis. However, the increase in AC activities (14, 28), as well as contractility, in the transgenic Arg389 hearts compared with the Gly389 hearts is relatively small (∼25%). And AC5 transgenic mice have similar levels of enhanced contractility as the β1-Arg389 mice but do not exhibit the drug response and other features found with the β1-Arg389 mice (28). In addition, it is now recognized that G protein-coupled receptors can signal to the cell interior by multiple mechanisms, such as coupling to unexpected G proteins, direct interactions leading to signaling in the absence of G protein activation, formation of heterodimer “units” with other receptors, and β-arrestin-mediated scaffolding of proteins to facilitate local signaling. Interestingly, in transfected fibroblasts the β1-Arg389 receptor undergoes greater G protein-coupled receptor kinase-mediated desensitization (17), (which is the trigger for β-arrestin), compared with the Gly389 variant. In addition, the two β1ARs differ in their conformational changes induced by agonists and some other complex ligands (12, 19). Such recombinantly expressed model cell-based approaches are suggestive of events that are evoked by activation of the two receptors but do not take into account cell-type or the effects of regulatory events that occur in the intact organ. We have recently reviewed the mechanisms by which signaling diversity can be achieved by this class of receptors (10) and have noted that virtually all studies have been with isolated model cells rather than intact organs. Thus the mechanism by which the β1-Arg389 and β1-Gly389 exert their respective phenotypes in the heart remains unclear, particularly given the aforementioned lack of effects in similarly AC-activated mice, such as the AC5 transgenics. We thus considered whether there were unique signaling events promoted by the two genetic forms of the β1AR in the heart. We studied transgenic mice with cardiomyocyte overexpression of β1-Arg389, β1-Gly389, and AC5, the latter to help to differentiate cAMP/PKA-dependent events. The mice were 3 mo of age, a time that precedes loss of ventricular function (∼6–8 mo) and development of cardiomyopathy (∼9–12 mo) (for β1-Arg389). Intact heart RNA was isolated and utilized in the expression arrays. Although transgene expression was specific for the cardiomyocyte, we recognize that alterations in gene expression within other cells of the heart (such as fibroblasts, vascular endothelial and smooth muscle cells) due to cardiomyocyte expression of β1AR can occur. However, this cell-to-cell communication is part of the milieu of the intact heart functioning in vivo, and thus identification of all signals that were evoked by one or more transgenes was sought so as to provide a global, whole organ, analysis.
We indeed found a large number of transcripts that were regulated in the β1-Arg389 hearts, but not the hearts of β1-Gly389 or AC5 mice. These included genes associated with the extracellular matrix, inflammation, and programmed cell death. Each is discussed below. Because the β1-Arg389 animals progress to cardiomyopathy by 9–12 mo, we have sought to identify early signatures, and thus it is particularly interesting that prior to the development of pathology, the most statistically significant result in both GeneSet Enrichment and Ingenuity Pathway analysis consisted of strong and pervasive induction of extracellular matrix (ECM) pathway genes. This pathway is shown in Fig. 4 in which the activation of a series of genes known to associate with the TGFB signaling family is found. As shown, in the extracellular space, increased TGFB evokes collagen genes COL1A1, COL1A2, COL3A1 and via increased expression of biglycan (BGN), COL6A1-A3. Collagen synthesis is a hallmark of many heart failure models and the human syndrome (2). This effect would be enhanced by increased expression of the plasma membrane TGFB receptor TGFBR2, which is indeed upregulated. Also, TGFBR2 activation by TGFB leads to apoptosis via the SMAD pathway. Other plasma membrane genes that are upregulated are fibronectin (FN1) which can upregulate some collagen genes, and the endothelin receptor (EDNRA), which can cause the same effect. Both FN1 and EDNRA activate MAP kinases in the cytoplasm as do TGFB, the A-kinase anchoring protein 2, which has increased expression exclusively in the Arg389 mice, and activation of EDNRA. FN1 induced upregulation of ELAVL1 in the cytoplasm can lead to enhanced TIA1 expression in the nucleus. This RNA binding protein controls the splicing of FAS, which is proapoptotic. The increased expression of nuclear SKIL (the transcription factor SnON) would be expected to decrease TGFB production, and thus appears to be a compensatory response. On the other hand, enhanced expression of the transcription factor FOXP1 has been shown to be associated with prohypertrophic gene responses and cardiac hypertrophy in humans (6). A number of other ECM genes were regulated that have established relationships to cardiac phenotypes. Among these, the ECM protein osteopontin (Opn), encoded by the gene Spp1 (secreted phosphoprotein 1), was noted to undergo the greatest increase in expression of all genes unique to β1-Arg389 (6.9-fold, Table 1). The diverse functions of Opn include tissue remodeling, cell adhesion, and chemotactic activity (15). Opn binds some integrins, certain variants of CD44, and ECM components (23). In humans, Opn expression has been associated with dilated cardiomyopathy (22, 27), hypertrophic cardiomyopathy, and ventricular tachycardia (15). Of note, Opn is necessary for increased atrial natriuretic factor (ANF) production in mice at an early stage of cardiac hypertrophy from chronic pressure overload (33). ANF transcripts (encoded by the Nppa gene) also were increased in the β1-Arg389 hearts (Table 1). Interestingly, Nupr1 is increased in β1-Arg389 hearts, and this transcription factor is also known to increase ANF (5). Thus along with EDNRA, a link between the ECM pathway and neurohumoral signaling is observed.
We also noted a number of inflammation and immune response genes that were uniquely regulated by the Arg variant. For example, chemokines Ccl9, Ccl6, and Ccl2 were upregulated specifically in β1-Arg389 hearts (Table 1 and Supplementary Tables S1 and S3). Chemokine Ccl6, also known as C10, is expressed by macrophages and is likely involved in chronic inflammatory responses (32). Chemokines Ccl9, Ccl6, Ccl2, and Ccl8 have been shown to recruit macrophages to skeletal muscle in a mouse model of muscular dystrophy (mdx). In the mdx mice, Ccl6 and Ccl2 were found in muscle fibers (16). The chemokine Cc18 was substantially upregulated in β1-Arg389 hearts (10.9-fold over nontransgenic) and was also upregulated in β1-Gly389 hearts, but only by 2.9-fold. Thus while this gene is not uniquely regulated by the Arg genotype, the change is somewhat greater than with Gly, which may contribute to the phenotype based on the differences in the degree of upregulation. No other chemokines were uniquely upregulated in the β1-Arg389 hearts. It has been reported that IL-6 is upregulated in other β1AR-overexpressing mice at 8 wk of age (20). However, we did not observed any change in expression in our 3-mo-old mice relative to the nontransgenic mice for IL-6 expression or for any other interleukin, except IL-15. which had slightly lower expression in our β1-Arg389 hearts (Supplementary Table S3). No changes in expression were seen for interferons or tumor necrosis factor-α. In addition to C3ar1, several other Complement Component genes, C1qa, C1qb, C1qg, C3, and C5r1 (Supplementary Table S1), were uniquely upregulated ∼2-fold in the β1-Arg389 mice. Ctss (cathepsin S) is a proteinase involved in antigen processing and is upregulated 2.53-fold in these hearts. Ctss is expressed in macrophage (24) and in myoblasts (31) under inflammatory conditions. Fcgr2b, the Fc fragment of IgG receptor 2b, is also uniquely upregulated 2.63-fold in the β1-Arg389 hearts (Table 1). This receptor is expressed on macrophage, β-cells, neutrophils, and mast cells (18). Fcgr2b is an inhibitory receptor, antagonizing inflammatory signals in these cells by inhibiting calcium signaling and its downstream events such as cytokine release. These increases in expression of Fcgr2b, the chemokine genes, and the Complement Component genes suggest an inflammatory component to the β1-Arg389 phenotype.
The genes regulated under programmed cell death that are specific for β1-Arg389 include those for apoptotic and nonapoptotic death, cell survival, and cell growth and are summarized in Table 2. An intriguing set of altered genes involved in proapoptotic, antiapoptotic, and p53 signaling was noted. The NF-κB-dependent processes associated with the observed increases in CD14, LY86, TNFRSF1A, TNFRSF1B, and TNFRSF12A and the decrease in TNFAIP8 are all highly proapoptotic. However, the β1-Arg389-specific downregulation of TNFRSF21, CRADD, and MAP3K5, which are also within this same pathway, mitigates against apoptosis. Within the p53 response (the intrinsic apoptosis pathway), upregulation of TIA1, NUPR1 (p8), DYRK2, SCOTIN, DRAM, and INPP5D enhances apoptosis. However, also within the intrinsic pathway, we found an apparent compensatory downregulation of BCL2L11, BAG1, SON, CIDEB, DAP, MTP18, and PEKHHF1, all of which would be expected to be antiapoptotic. These changes suggest a highly adapted response that appears to ultimately maintain a normal cell-death pattern without an overrepresentation of proapoptotic evoked events in β1-Arg389 hearts. And indeed, this is consistent with our previous studies of young β1-Arg389 hearts, which show no evidence of apoptosis (14).
Table 2.
Changes in expression of Programmed Cell Death genes unique to the β1-Arg389 hearts
| Genes | Factor | Function |
|---|---|---|
| RTN4 (Nogo) | 1.88 | inhibits regeneration of the central nervous system (Rho-ROCK pathway) |
| RhoA | 1.29 | activated by various receptors (e.g., angiotensin II, α1 adrenoreceptors); transmits signals from growth inhibitors |
| ANGPTL4 | 1.3 | inhibits lipoprotein-derived fatty acid delivery in the heart |
| PURB | 1.26 | represses expression of alpha-myosin heavy chain |
| SOCS3 | 2.38 | suppressor of cytokine signaling; inhibits proinflammatory signaling |
| TNFRSF1A | 1.23 | tumor necrosis factor receptor superfamily; mediator of apoptosis and regulator of inflammation |
| TNFRSF1B | 1.24 | tumor necrosis factor receptor superfamily; mediator of apoptosis and regulator of inflammation |
| TNFRSF12A | 1.99 | tumor necrosis factor receptor superfamily; promotes apoptosis, regulation of inflammation and the immune response |
| CCL2 (MCP-1) | 1.8 | regulator of angiotensin II-induced target damage in hypertensive heart disease |
| TGF-beta2 | 2.79 | regulation of cell growth, differentiation and repair |
| ZBTB16 | 1.57 | BTB/POZ-zing finger type transcription factor involved in development, proliferation and apoptosis |
| INHB4 | 1.31 | inhibits gonodal glycoprotein hormones |
| LY86 | 1.28 | Toll-like receptor; activated by liposaccharide (LPS) |
| CD14 | 1.95 | innate immune gene; binds to Toll-like receptors; overexpression amplifies cardiomyopathic inflammatory process by stimulating proinflammatory cytokines |
| LITAF | 1.61 | LSP-induced transcription factor; controls the expression of TNF-alpha and other cytokines |
| ANXA1 | 1.49 | mediator of anti-inflammatory actions of glucocorticoids |
| ANXA5 | 1.33 | antiatherothrombic agent (binds PS, inhibits apoptotic cell intake by macrophages) |
| ANXA4 | 1.43 | cholesterol ester transport from caveolae, apoptotic and anti-inflammatory properties |
| DYRK2 | 1.89 | enhances p53 response |
| SCOTIN | 1.36 | novel p53-inducible proapoptotic protein |
| DRAM | 1.32 | damage regulated autophagy regulator; link between autophagy, p53 and programmed cell death |
| INPP5D | 1.37 | SHIP inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5 phosphates (cells attenuate growth factor or cytokine-induced mobilization of calcium, p53 response) |
| TIA1 | 1.13 | cytotoxic effector molecule; mediates RNA triage during stress |
| YWHAH | 1.25 | signaling protein 14-3-3-eta; regulation of cell cycle arrest after DNA damage |
| YWHAZ | 1.31 | signaling protein 14-3-3-zeta; neuroprotection |
| NUPR1 | 1.80 | homologue of p8; involved in events linked to cardiomyocyte hypertrophy and cardiac fibroblasts matrix metalloproteases production |
| ACTN1 | 1.44 | organization of the cytoskeleton and cell binding |
| ITG-beta2 | 1.66 | binding to intracellular adhesion molecule; innate immunity |
| NOL3 | 1.30 | apoptosis repressor protein; protects against oxidative stress and induced apoptosis |
| SFRP1 | 2.00 | secreted Wnt antagonist; overexpression reduces infarct size, improves cardiac recovery, modifies inflammatory response and amplifies angiogenesis |
| SHB | 1.70 | novel SHRC homolog (SH2)-containing protein; interacts with some ligand-activated tyrosine kinase receptors (growth factor, cytokines, hormones) |
| CALR | 1.46 | calcium-binding protein |
| SUL1 | 1.28 | heparan sulfate sulfatation enzyme; cell signaling |
| MGMT | 0.80 | DNA repair protein; prevents mutation and cell death by aberrant alkylation of DNA |
| BAG1 | 0.80 | athanogene 1; leads to the phosphorylation and inhibition of the proapoptotic protein Bad |
| MTP18 | 0.66 | regulates the mitochondrial morphology |
| TNFAIP8 | 0.65 | NF-κB-inducible transcription factor; antiapoptotic and oncogenic |
| PPT1 | 0.87 | palmitoyl-protein thioesterase neuronal ceroid-lipofuscinoses; lysosome dynamics proline |
| PRODH | 0.65 | dehydrogenase (immune, signaling and networking deficits/regulation by hypoxia) |
| BCL2L11 | 0.78 | proapoptotic, BH3-only member of the Bcl-2 family |
| SON | 0.77 | proapoptotic |
| CIDEB | 0.70 | proapoptotic cell death-inducing DFF45 (DNA fragmentating factor 45) |
| DAP | 0.80 | death associated protein |
| PEKHHF1 | 0.67 | lysosome-associated apoptosis-inducing protein |
| CRADD | 0.77 | caspase and RIP adaptor with death domain (via TNF/caspase 2) |
| TNFRSF21 | 0.79 | tumor necrosis factor superfamily receptor 21; inflammation, immune regulation |
| MAP3K5 | 0.73 | apoptosis signal-regulating kinase 1 (ASK1); stress, apoptosis, immune and angiotensin II responses |
| STAT5A | 0.79 | mediate signaling by cytokines (JAK2) |
Taken together, our findings indicate a large number of transcript changes specifically associated with young, healthy β1-Arg389 mice. Most appear to be upregulated, but as indicated above, certainly not all. These transcripts, which we term allele specific and non-cAMP dependent, can be considered within the paradigm shown in Fig. 5. Here, the expression of genes in the nontransgenic mice is set as the “reference,” and the perturbations imposed by the transgenes considered as four possibilities, initially stratified by cAMP-dependent and -independent events (defined by common expression changes with the AC5 transgenic hearts). Within the cAMP-dependent mechanism, a number of genes are altered in the β1-Arg and β1-Gly hearts, and by definition this group includes all transcripts altered in AC5 hearts. Not surprisingly, the genes in this group are dominated by those of respiration and energy metabolism, given that cAMP/PKA activation is a major mechanism by which cardiac inotropy and chronotropy are increased. In the non-cAMP-dependent set of genes, some are common to both β1-Arg389 and β1-Gly389. These represent, then, pathways that are activated by these receptors in a nonallele-specific manner, which does not involve cAMP signaling, and are likely not pathogenic or drug-response genes. The genes whose transcripts were altered in a non-cAMP, allele-specific manner constitute unique signaling that is dependent on the single amino acid difference in the β1AR at position 389. Those imposed by β1-Gly389 are not readily associated with physiological parameters or drug response, since such phenotypes are not observed in these mice or in the limited number of studies with human heart failure. The current experiments were performed in young mice prior to development of any anatomic or physiological abnormalities. Yet the character of some of the changes in expression of certain genes suggests that they may be compensatory in nature, acting to minimize or negate potential pathological events. Of note, we recognize that alterations in gene expression in β1-Arg389 or -Gly389 hearts, but not AC hearts, might nevertheless have processes that involve cAMP that act in conjunction with the unique signaling properties of one or the other β1AR. That is, all changes that occur with β1AR expression (of either genotype) are in the background of elevated cAMP.
Fig. 5.
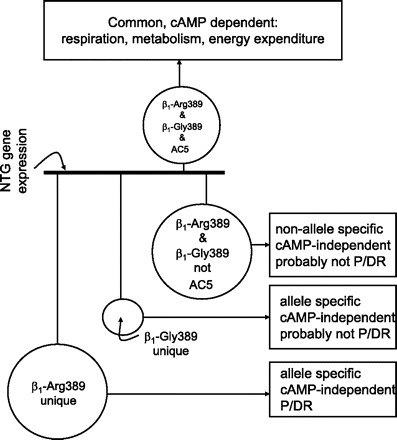
Mechanistic depiction of regulated genes in hearts of β1-Arg389, β1-Gly389, and AC5 mice. The reference (nontransgenic) gene expression is indicated by the dark line in the center, with genes above this line representing those in common with all three transgenics, which are considered cAMP-dependent. Below the baseline are three types of distributions: those common to β1-Arg389 and -Gly389, but not AC5, those unique to β1-Gly389, and those unique to β1-Arg389. Each has a specific set of descriptors as indicated in the boxes. The sizes of the circles are proportional to the number of genes in each pool. P/DR, pathogenic or drug response loci.
In summary, we have identified a substantial group of genes uniquely regulated by a polymorphic variant of the β1AR due to a single amino acid change (Gly to Arg). They consist primarily of genes involved in extracellular matrix, inflammation, and programmed cell death signaling. These results expand the notion that 7-transmembrane domain receptors can alter intracellular events in a manner that may be independent of coupling to G proteins, provide genes and pathways previously unrecognized as potentially interacting with β1ARs, and identify allele-specific pathways where intervention may be therapeutic in heart failure in those with the β1-Arg389 variant.
GRANTS
Supported by National Heart, Lung, and Blood Institute Grant HL-071609 and the Computational Medicine Center, a State of Ohio Third Frontier Wright Center of Innovation.
Supplementary Material
Acknowledgments
We thank Esther Moses for manuscript preparation.
Address for reprint requests and other correspondence: S. B. Liggett, 20 Penn St., HSF-II, Rm. S-114, Baltimore, MD 21201-1075 (e-mail: sligg001@umaryland.edu).
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The online version of this article contains supplemental material.
REFERENCES
- 1.Bengtsson K, Melander O, Orho-Melander M, Lindblad U, Ranstam J, Råstam L, Groop L. Polymorphism in the β1-adrenergic receptor gene and hypertension. Circulation 104: 187–190, 2001. [DOI] [PubMed] [Google Scholar]
- 2.Brower GL, Gardner JD, Forman MF, Murray DB, Voloshenyuk T, Levick SP, Janicki JS. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg 30: 604–610, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Bruck H, Leineweber K, Temme T, Weber M, Heusch G, Philipp T, Brodde OE. The Arg389Gly beta1-adrenoceptor polymorphism and catecholamine effects on plasma-renin activity. J Am Coll Cardiol 46: 2111–2115, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Xu H, Aronow BJ, Jegga AG. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics 8: 392–2007. [DOI] [PMC free article] [PubMed]
- 5.Goruppi S, Patten RD, Force T, Kyriakis JM. Helix-loop-helix protein p8, a transcriptional regulator required for cardiomyocyte hypertrophy and cardiac fibroblast matrix metalloprotease induction. Mol Cell Biol 27: 993–1006, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hannenhalli S, Putt ME, Gilmore JM, Wang J, Parmacek MS, Epstein JA, Morrisey EE, Margulies KB, Cappola TP. Transcriptional genomics associates FOX transcription factors with human heart failure. Circulation 114: 1269–1276, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Johnson JA, Zineh I, Puckett BJ, McGorray SP, Yarandi HN, Pauly DF. Beta 1-adrenergic receptor polymorphisms and antihypertensive response to metoprolol. Clin Pharmacol Ther 74: 44–52, 2003. [DOI] [PubMed] [Google Scholar]
- 8.La Rosee K, Huntgeburth M, Rosenkranz S, Bohm M, Schnabel P. The Arg389Gly beta1-adrenoceptor gene polymorphism determines contractile response to catecholamines. Pharmacogenetics 14: 711–716, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Liggett SB Genetic, molecular, and clinical characterization of adrenergic receptor polymorphisms. In: The Adrenergic Receptors in the 21st Century, edited by Perez DM. Totowa. NJ: Humana Press, 2005, 339–364.
- 10.Liggett SB Cardiac 7-transmembrane-spanning domain receptor portfolios: diversify, diversify, diversify. J Clin Invest 116: 875–877, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liggett SB, Mialet-Perez J, Thaneemit-Chen S, Weber SA, Greene SM, Hodne D, Nelson B, Morrison J, Domanski MJ, Wagoner LE, Abraham WT, Anderson JL, Carlquist JF, Krause-Steinrauf HJ, Lazzeroni LC, Port JD, Lavori PW, Bristow MR. A polymorphism within a conserved β1-adrenergic receptor motif alters cardiac function and β-blocker response in human heart failure. Proc Natl Acad Sci USA 103: 11288–11293, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mason DA, Moore JD, Green SA, Liggett SB. A gain-of-function polymorphism in a G-protein coupling domain of the human β1-adrenergic receptor. J Biol Chem 274: 12670–12674, 1999. [DOI] [PubMed] [Google Scholar]
- 13.McGraw DW, Fogel KM, Kong S, Litonjua AA, Kranias EG, Aronow BJ, Liggett SB. Transcriptional response to persistent β2-adrenergic receptor signaling reveals regulation of phospholamban which alters airway contractility. Physiol Genomics 27: 171–177, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Mialet-Perez J, Rathz DA, Petrashevskaya NN, Hahn HS, Wagoner LE, Schwartz A, Dorn GW II, Liggett SB. β1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med 9: 1300–1305, 2003. [DOI] [PubMed] [Google Scholar]
- 15.Okamoto H Osteopontin and cardiovascular system. Mol Cell Biochem 300: 1–7, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Porter JD, Guo W, Merriam AP, Khanna S, Cheng G, Zhou X, Andrade FH, Richmonds C, Kaminski HJ. Persistent over-expression of specific CC class chemokines correlates with macrophage and T-cell recruitment in mdx skeletal muscle. Neuromuscul Disord 13: 223–235, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Rathz DA, Gregory KN, Fang Y, Brown KM, Liggett SB. Hierarchy of polymorphic variation and desensitization permutations relative to β1- and β2-adrenergic receptor signaling. J Biol Chem 278: 10784–10789, 2003. [DOI] [PubMed] [Google Scholar]
- 18.Ravetch JV, Lanier LL. Immune inhibitory receptors. Science 290: 84–89, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Rochais F, Vilardaga JP, Nikolaev VO, Bunemann M, Lohse MJ, Engelhardt S. Real-time optical recording of beta1-adrenergic receptor activation reveals supersensitivity of the Arg389 variant to carvedilol. J Clin Invest 117: 229–235, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohrbach S, Engelhardt S, Lohse MJ, Werdan K, Holtz J, Muller-Werdan U. Activation of AP-1 contributes to the beta-adrenoceptor-mediated myocardial induction of interleukin-6. Mol Med 13: 605–614, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roth DM, Gao MH, Lai NC, Drumm J, Dalton N, Zhou L, Zhu J, Entrikin D, Hammond HK. Cardiac-directed adenylyl cyclase expression improves heart function in murine cardiomyopathy. Circulation 99: 3099–3102, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Satoh M, Nakamura M, Akatsu T, Shimoda Y, Segawa I, Hiramori K. Myocardial osteopontin expression is associated with collagen fibrillogenesis in human dilated cardiomyopathy. Eur J Heart Fail 7: 755–762, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol 27: 2302–2309, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Shi GP, Munger JS, Meara JP, Rich DH, Chapman HA. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J Biol Chem 267: 7258–7262, 1992. [PubMed] [Google Scholar]
- 25.Small KM, Perez JM, Wagoner LE, Liggett SB. Adrenergic receptor polymorphisms in heart failure; molecular and physiologic phenotypes. In: Molecular Mechanisms of Cardiac Hypertrophy and Failure. Abingdon, UK: Taylor & Francis, 2005, p. 611–624.
- 26.Sofowora GG, Dishy V, Muszkat M, Xie HG, Kim RB, Harris PA, Prasad HC, Byrne DW, Nair UB, Wood AJJ, Stein CM. A common β1-adrenergic receptor polymorphism (Arg389Gly) affects blood pressure response to β-blockade. Clin Pharmacol Ther 73: 366–371, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Stawowy P, Blaschke F, Pfautsch P, Goetze S, Lippek F, Wollert-Wulf B, Fleck E, Graf K. Increased myocardial expression of osteopontin in patients with advanced heart failure. Eur J Heart Fail 4: 139–146, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Tepe NM, Lorenz JN, Yatani A, Dash R, Kranias EG, Dorn GW2, Liggett SB. Altering the receptor-effector ratio by transgenic overexpression of type V adenylyl cyclase: enhanced basal catalytic activity and function without increased cardiomyocyte β-adrenergic signalling. Biochemistry 38: 16706–16713, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Terra SG, Hamilton KK, Pauly DF, Lee CR, Patterson JH, Adams KF, Schofield RS, Belgado BS, Hill JA, Aranda JM, Yarandi HN, Johnson JA. Beta1-adrenergic receptor polymorphisms and left ventricular remodeling changes in response to beta-blocker therapy. Pharmacogenet Genomics 15: 227–234, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Wagoner LE, Craft LL, Zengel P, McGuire N, Rathz DA, Dorn GWI, Liggett SB. Polymorphisms of the β1-adrenergic receptor predict exercise capacity in heart failure. Am Heart J 144: 840–846, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Wiendl H, Lautwein A, Mitsdorffer M, Krause S, Erfurth S, Wienhold W, Morgalla M, Weber E, Overkleeft HS, Lochmuller H, Melms A, Tolosa E, Driessen C. Antigen processing and presentation in human muscle: cathepsin S is critical for MHC class II expression and upregulated in inflammatory myopathies. J Neuroimmunol 138: 132–143, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Wu Y, Prystowsky MB, Orlofsky A. Sustained high-level production of murine chemokine C10 during chronic inflammation. Cytokine 11: 523–530, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Xie Z, Singh M, Singh K. Osteopontin modulates myocardial hypertrophy in response to chronic pressure overload in mice. Hypertension 44: 826–831, 2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.