

Abstract
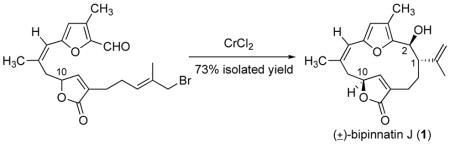
The total synthesis of (±)-bipinnatin J was achieved through a concise route that features the use of a silver ion promoted SN1-type γ-alkylation of a siloxyfuran and a diastereoselective Cr(II)-mediated macrocyclization to provide bipinnatin J (1), wherein the remote furanone stereocenter at C10 induced the relative stereochemistry of the two new stereocenters.
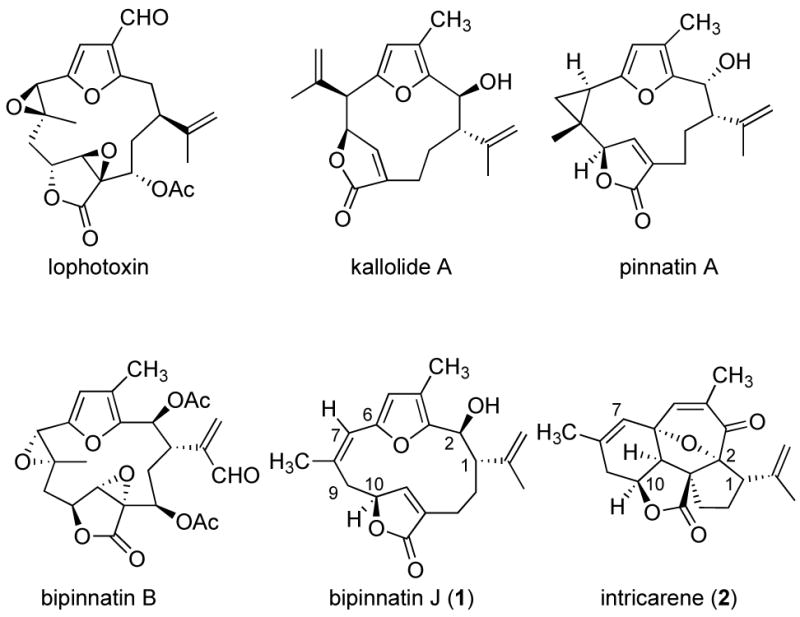
Marine invertebrates have yielded a plethora of structurally diverse natural products possessing a range of interesting biological activities.1 In particular, gorgonian octocorals of the genus Pseudopterogorgia have proven to be particularly fertile producers of diterpenes having the cembrane or pseudopterane skeleta. Members of the first group are characterized by the presence of a 14-membered ring carbon framework and include compounds such as lophotoxin and bipinnatin B, whereas the latter group has a contracted, 12-membered ring framework, as seen in kallolide A and pinnatin A (Figure 1).2 Several of these secondary metabolites exhibit promising pharmacological properties.1–3 For example, bipinnatins A, B, and D display in vitro activity against the P388 murine tumor cell line, with IC50’s of 0.9, 3.2, and 1.5 μg/mL, respectively.2f Additionally, lophotoxin and bipinnatin B are potent neurotoxins that irreversibly block nicotinic acetylcholine receptors.3f Bipinatin I possesses strong cytotoxic action, eliciting significant differential responses at GI50 level for all colon and melanoma cancer cell lines at concentrations of 10−6 M.2h
Figure 1.
Structures of Pseudopterogorgia Metabolites
In connection with our interest in the secondary metabolites of gorgonian corals, and their biosynthetic interrelationships,4 we became interested in the furanocembrane bipinnatin J (1).5–9 Possessing the less common Z-olefin in its macrocyclic ring, this compound may well be a precursor to several more oxidized congeners of the Pseudopterogorgia-derived cembranes. Of special interest is the structural relationship between bipinnatin J and the recently reported pentacyclic diterpene intricarene (2).10 Isolated from Pseudopterogorgia kallos, this aptly named compound may very well arise from bipinnatin J, through oxidation of the furan moiety followed by an oxydopyrylium ion based transannular [5+2] cycloadddion reaction.11 In this letter, we report a simple, convergent route to furanocembranes culminating in the total synthesis of bipinnatin J (1).
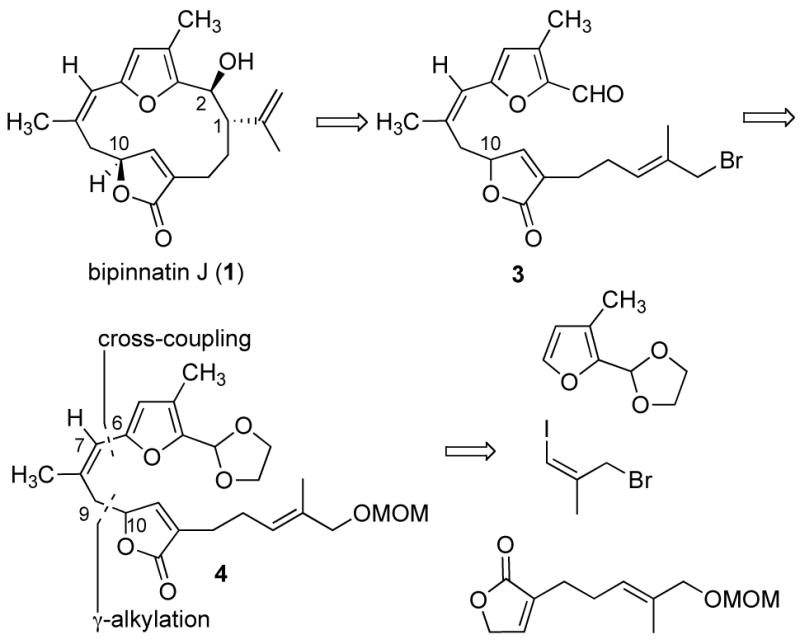
Our strategy to bipinnatin J (1) can be seen through the retrosynthetic analysis presented in Scheme 1. The plan was to construct the 14-membered carbon core of 1 through a metal-promoted macrocyclization of intermediate 3. An analysis of molecular models indicated that the relative stereochemistry at C1 and C2 would be controlled by the sole stereocenter in the furanone unit, located six carbons away. A highly convergent route was devised for the synthesis of the macrocyclization precursor.
Scheme 1.
Retrosynthetic Analysis of Bipinnatin J (1)
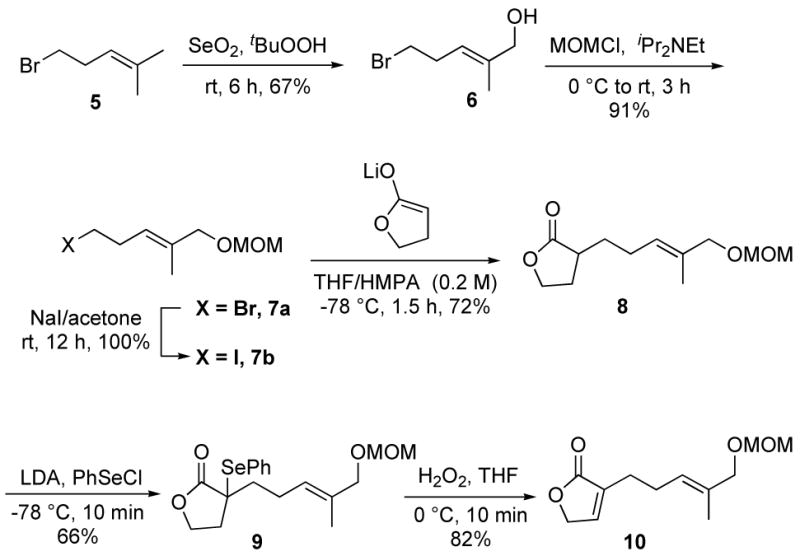
The synthesis of bipinnatin J commenced with the preparation of the substituted butyrolactone unit (Scheme 2). Allylic oxidation of commercially available 5-bromo-2-methylpent-2-ene (5) with SeO2 and t-BuOOH proceeded regioselectively to afford trans-allylic alcohol 6 in 67% isolated yield.12 The hydroxyl group was protected as the MOM ether to afford compound 7a in 91% yield. The cross-coupling of 7a with 3-bromofuran-2(5H)-one to yield the desired alkylated furanone (10) proved low yielding, so a less direct route was utilized. The reaction of γ-butyrolactone enolate with bromide 7a gave the desired alkylation product (8), but in a low yield. The major product was a conjugated diene, presumably arising from E2 elimination of 7a. In order to favor alkylation over elimination, the bromide was exchanged for an iodide. The modified alkylating agent performed as desired and provided the alkylated γ-butyrolactone (8) in 72% yield, along with ~10% of the diene side-product. The required olefin was introduced through a two-step, phenylselenation/selenoxide elimination sequence. The selenoxide elimination proceeded with good regioselectively and yielded primarily the endocyclic olefin-containing product, 10 (17:1 ratio).
Scheme 2.
Synthesis of Fragment 10
The elaboration of compound 10 to the desired cyclization precursor, 3, necessitated alkylation at the y-position of the unsaturated lactone. This transform was achieved through the intermediacy of the corresponding siloxyfuran 11, prepared in quantitative yield by silation of the enolate of 10 (Scheme 3). The desired γ-alkylation was accomplished under SN1 conditions. Treatment of siloxyfuran 11 with the requisite allylic bromide in the presence of Ag(OCOCF3)2 afforded the desired alkylation product (12) in 60% yield.13,14 The required 3-methylfurfural piece was then appended through the Negishi cross-coupling protocol. The reaction of iodide 12 and organozinc compound 13, which was generated in situ by treatment of dioxolane protected 3-methylfurfural with 1.0 equiv of t-BuLi and 1.2 equiv of ZnCl2 in THF at −78 °C, was catalyzed with PdCl2dppf and yielded the coupled product (4) in quantitative yield. Treatment of compound 4 with PPTS in refluxing t-BuOH removed the dioxolane and MOM ether protecting groups to provide aldehyde 14 in 81% isolated yield. It is noteworthy that the same reaction when carried out in MeOH or iPrOH led to decomposition of the starting material, with only trace amounts of the desired product present by 1H NMR. The direct, palladium-mediated reductive cyclization of allylic alcohol 14 was examined, but without success.15
Scheme 3.
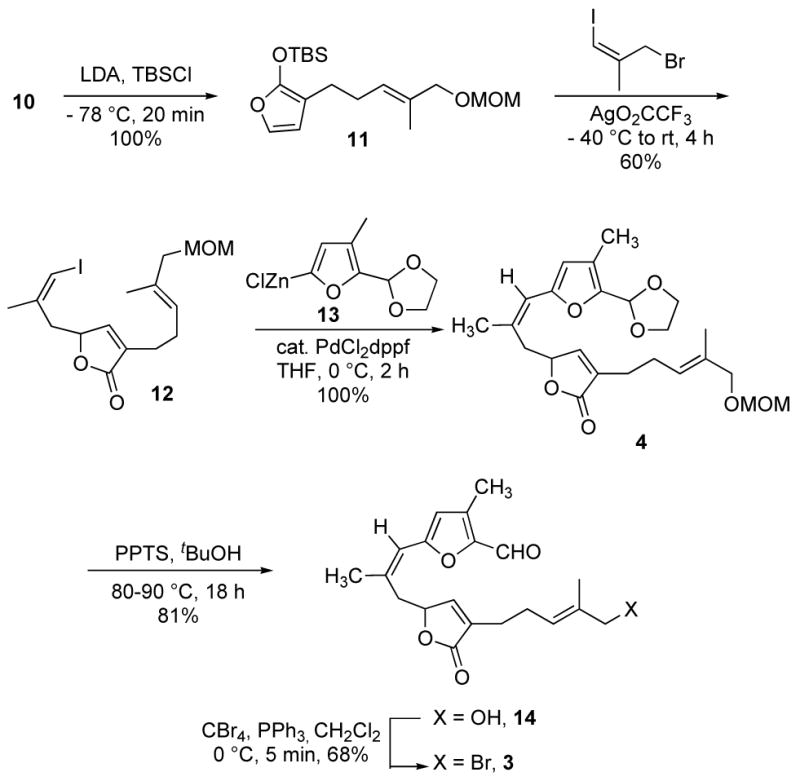
Synthesis of the Macrocyclization Precursor, 3
The Nozaki-Hiyama reaction presented a good alternative for the desired reductive cyclization.16–18 In preparation for this reaction, the hydroxyl group was converted to the bromide using PPh3 and CBr4. The Nozaki-Hiyama macrocyclization of bromide 3 was carried out under dilute reaction conditions (0.429 mmol 3, 20.6 eq CrCl2, 2.0 g activated, powdered 4Å molecular sieves, 300 mL THF) to minimize intermolecular reactions. The reaction went to completion after 16 h and, to our delight, gave bipinnatin J (1), a white solid (mp = 176–178 °C, lit2g = dec. at 141–142 °C), as the major product in 73% isolated yield. The 1H and 13C NMR spectra of the synthetic sample are identical to that reported for the natural product.2g Also isolated from the macrocyclization reaction were two disasteroisomeric cyclization products, 15 and 16, in 12.7% and 5.6% yields, respectively. The structures assigned to these minor products are based on the NMR data and are considered tentative. We are examining the effect of reaction parameters on the diastereoselectivity of the cyclization reaction. The high diastereoselectivity observed in the macrocyclization can be understood by considering the likely transition state for the Nozaki-Hiyama reaction,7,16–18 wherein the remote furanone stereocenter at CIO induces relative stereochemistry of the two new sterocenters.
In summary, we have completed an efficient, convergent total synthesis of bipinnatin J. The longest linear sequence requires 12 steps from the commercially available 5-bromo-2-methylpeten-2-ene. The synthesis features the use of a silver ion promoted SN1-type γ-alkylation of a siloxyfuran and a diastereoselective Cr(II)-mediated macrocyclization, which provides bipinnatin J as the major product. Current efforts are directed toward the asymmetric synthesis of bipinnatin J as well as the biomimetic tranformation of this compound to other Pseudopterogorgia-derived diterpenes, including intricarene (2).
Supplementary Material
General experimental procedures and characterization data for all key intermediates and bipinnatin J. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 4.
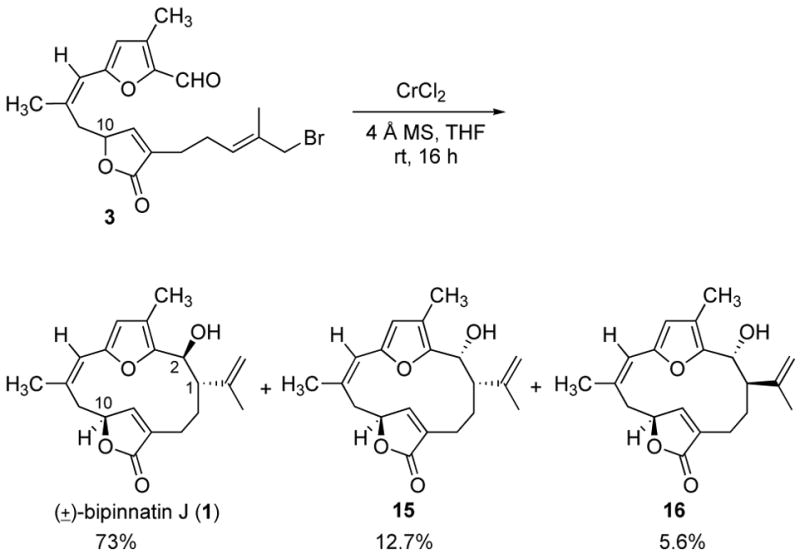
Macrocyclization to Yield Bipinnatin J (1).
Acknowledgments
We are grateful to the National Institutes of Health for financial support of this work.
References
- 1.Faulkner DJ. Nat Prod Rep. 2002;19:1–48. doi: 10.1039/b009029h. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 2.For isolation and bioactivities, see: lophotoxin: Fenical W, Okuda RK, Bandurraga MM, Culver P, Jacobs RS. Science. 1981;212:1512–1514. doi: 10.1126/science.6112796.kallolides: Look SA, Burch MT, Fenical W, Zheng Q, Clardy J. J Org Chem. 1985;50:5741–5746.Rodríguez AD, Shi JG, Shi YP. J Org Chem. 2000;65:3192–3199. doi: 10.1021/jo0001582.pinnatins: Rodríguez AD, Shi JG, Huang SD. J Org Chem. 1998;63:4425–4432.Rodríguez AD, Shi JG. Org Lett. 1999;1:337–340. doi: 10.1021/ol990660c.bipinnatins: Wright AE, Burres NS, Schulte GK. Tetrahedron Lett. 1989;30:3491–3494.Rodríguez AD, Shi JGJ. Org Chem. 1998;63:420–421. doi: 10.1021/jo971884g.Rodriguez AD, Shi JG, Huang SD. J Nat Prod. 1999;62:1228–1237. doi: 10.1021/np990064r.Rodríguez AD, Shi YPJ. Nat Prod. 2000;63:1548–1550. doi: 10.1021/np000224v.
- 3.(a) Culver P, Jacobs RS. Toxicon. 1981;19:825–830. doi: 10.1016/0041-0101(81)90078-7. [DOI] [PubMed] [Google Scholar]; (b) Langdon RB, Jacobs RS. Life Sciences. 1983;32:1223–1228. doi: 10.1016/0024-3205(83)90191-1. [DOI] [PubMed] [Google Scholar]; (c) Sorenson EM, Culver P, Chiappinelli VA. Neuroscience. 1987;20:875–884. doi: 10.1016/0306-4522(87)90248-x. [DOI] [PubMed] [Google Scholar]; (d) Groebe DR, Abramson SN. J Biol Chem. 1995;270:281–286. doi: 10.1074/jbc.270.1.281. [DOI] [PubMed] [Google Scholar]; (e) Hyde EG, Boyer A, Tang P, Xu Y, Abramson SNJ. Med Chem. 1995;38:2231–2238. doi: 10.1021/jm00012a023. [DOI] [PubMed] [Google Scholar]; (f) Tornoe C, Holden-Dye L, Garland C, Abramson SN, Fleming JT, Sattelle DB. J Exper Biol. 1996;799:2161–2168. doi: 10.1242/jeb.199.10.2161. [DOI] [PubMed] [Google Scholar]
- 4.Waizumi N, Stankovic AR, Rawal VH. J Am Chem Soc. 2003;125:13022–13023. doi: 10.1021/ja035898h. [DOI] [PubMed] [Google Scholar]
- 5.For isolation of bipinnatin J, see: ref. 2g.
- 6.For the synthetic studies towards bipinnatin J, see: Tsubuki M, Takahashi K, Sakata K, Honda T. Heterocycles. 2005;65:531–540.While this manuscript was being readied for publication, we became aware of a completed synthesis of bipinnatin J: Trauner, D., private communication. See: Roethle PA, Trauner D. Org Lett. 2005 doi: 10.1021/ol052922i. ASAP.
- 7.For total synthesis of furanocembranolides, see for example: Rayner CM, Astles PC, Paquette LA. J Am Chem Soc. 1992;114:3926–3936.Paquette LA, Astles PC. J Org Chem. 1993;58:165–169.
- 8.For total synthesis of pseudopterolides, see: Marshall JA, Hartley GS, Wallace EM. J Org Chem. 1996;61:5729–5735.Marshall JA, Liao J. J Org Chem. 1998;63:5962–5970. doi: 10.1021/jo980603h.Marshall JA, Van Devender EA. J Org Chem. 2001;66:8037–8041. doi: 10.1021/jo016048s.
- 9.Other synthetic studies: Marshall JA, Wang XJ. J Org Chem. 1992;57:3387–3396.Marshall JA, McNulty LM, Zou D. J Org Chem. 1999;64:5193–5200. doi: 10.1021/jo990376z.Tsubuki M, Takahashi K, Honda T. J Org Chem. 2003;68:10183–10186. doi: 10.1021/jo035244r.
- 10.For isolation, see: Marrero J, Rodríguez AD, Barnes CL. Org Lett. 2005;7:1877–1880. doi: 10.1021/ol0505961.
- 11.Reviews of [5+2] cycloaddition: Mascarenas JL. Adv Cycloadd. 1999;6:1–54.Harmata M, Rashatasakhon P. Tetrahedron. 2003;59:2371–2395.
- 12.Andresen G, Eriksen A, Dalhus AB, Gundersen LL, Rise F. J Chem Soc, Perkin Trans 1. 2001:1662–1672. [Google Scholar]
- 13.Jefford CW, Sledeski AW, Boukouvalas J. J Chem Soc, Chem Commun. 1988:364–365. [Google Scholar]
- 14.Preparation of 3-bromo-1-iodo-2-methylpropene: Larock RC, Han X. J Org Chem. 1999;64:1875–1887. doi: 10.1021/jo981876f.
- 15.For examples of palladium promoted reductive coupling of allylic alcohols with aldehydes, see: Takahara JP, Masuyama Y, Kurusu Y. J Am Chem Soc. 1992;114:2577–2586.Kimura M, Tomizawa T, Horino Y, Tanaka S, Tamaru Y. Tetrahedron Lett. 2000;41:3627–3629.
- 16.Fürstner A. Chem Rev. 1999;99:991–1045. doi: 10.1021/cr9703360. [DOI] [PubMed] [Google Scholar]
- 17.For the Cr-promoted macrocyclization of allylic bromides, see: Still WC, Mobilio D. J Org Chem. 1983;48:4785–4786.Wender PA, Mckinney JA, Muka C. J Am Chem Soc. 1990;112:5369–5370.Wender PA, Grissom JW, Hoffmann U, Mah R. Tetrahedron Lett. 1990;31:6605–6608.
- 18.In Paquette’s synthesis of gorgiacerone, the Nozaki-Hiyama reaction was utilized to produce the pseudopterane skeleton (25% yield, see ref. 7a). Paquette has also utilized this process for construction of the furanocembrane skeleton (20% yield, see ref. 7b).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental procedures and characterization data for all key intermediates and bipinnatin J. This material is available free of charge via the Internet at http://pubs.acs.org.