

Abstract
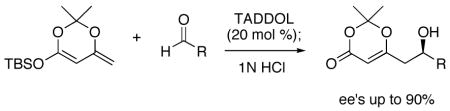
The concept of hydrogen bonding catalysis was extended to the vinylogous Mukaiyama aldol reaction, which gives rapid access to polyketide derivatives. The reaction of the silyldienol ether shown and a range of aldehydes catalyzed by TADDOL proceeds regiospecifically to produce the addition products in good yields and enantiomeric excesses.
Nature uses hydrogen bonding as a central force for the promotion of enzyme-catalyzed reactions. Yet, only recently have organic chemists begun to use this non-covalent interaction to catalyze chemical reactions of interest.1,2,3 We have previously demonstrated the successful use of hydrogen bond activation for the promotion of highly enantioselective Diels-Alder and Hetero-Diels-Alder reactions.4 Motivated by these results, we sought to expand this metal-free activation concept to other broadly useful classes of reactions. Herein we describe the first examples of hydrogen bond catalyzed enantioselective vinylogous Mukaiyama aldol (VMA) reactions.
The VMA reaction has proven to be a powerful method for complex molecule synthesis, as it provides rapid access to polyketide derivatives such as δ-hydroxy-β-ketoesters and α,β-unsaturated δ-hydroxy carbonyl compounds (Scheme 1).5 Further transformation of these compounds can be accomplished with good diastereoselectivity6 and paves the way to polyol units, a motif common to many natural products. Given the usefulness of the VMA reactions, considerable effort has been expended on the development of chiral catalysts for these reactions. Most of the reported catalysts are based on Lewis acidic metals, such as boron-,7 titanium-,8 copper-,9 chromium-,10 and promote the reaction by activating the electrophilic carbonyl group. Catalysts that promote the reaction by activating the nucleophilic component have also been shown to be effective.9c,11,12 Our objective was to promote the VMA reaction by activating the electrophile under metal-free conditions.
Scheme 1.
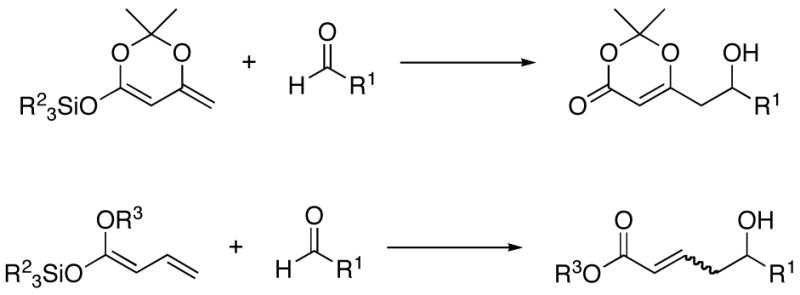
Vinylogous Mukaiyama Aldol Reactions
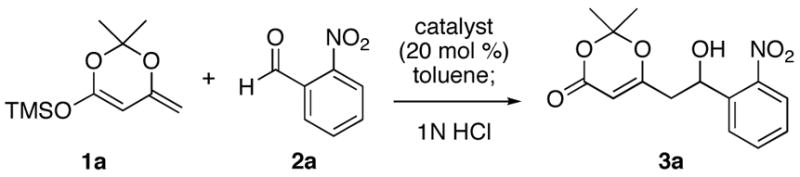
To the extent that hydrogen bonding represents a subset of Lewis acid activation, then a variety of chiral hydrogen bond donors should be capable of promoting asymmetric reactions. For the present study, we examined the effectiveness of several common chiral alkaloids and diols (Figure 1), as catalysts for the VMA reaction of trimethylsilyldienol ether of 2,2,6-trimethyl-4H-1,3-dioxin-4-one (1a) and 2-nitrobenzaldehyde (Table 1). These studies revealed that although many compounds promote the VMA reaction with varying levels of enantioselectivity, the best results were obtained using the TADDOL (α,α,α′,α′-tetraaryl-1,3-dioxolane-4,5-dimethanol) family of diols (Table 1, entries 13–15).
Figure 1.
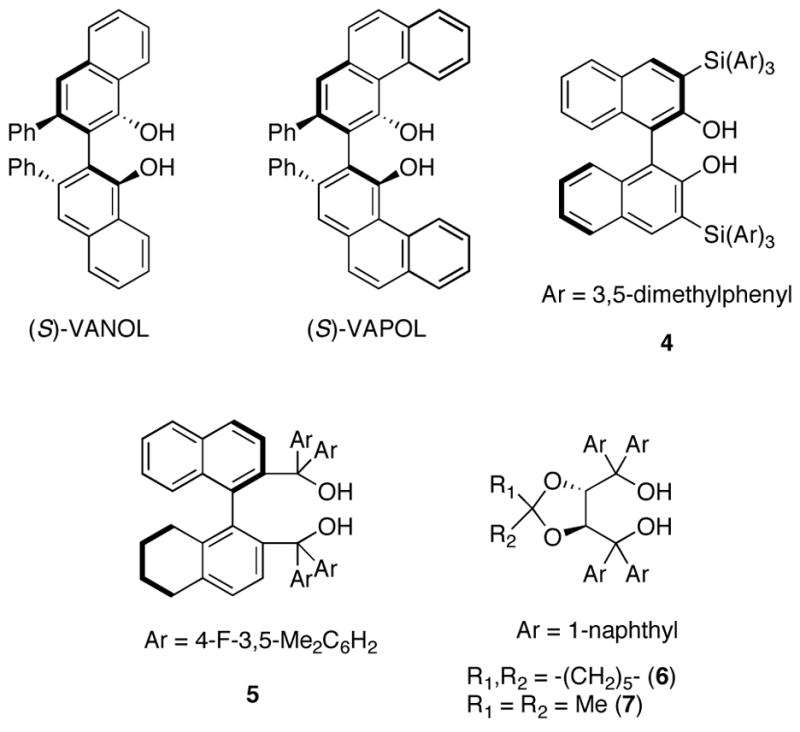
Diol catalysts used for the hydrogen bond catalyzed VMA reaction
Table 1.
Exploration of Various Hydrogen Bond Donors as Catalysts for the VMA Reaction
 | |||||
|---|---|---|---|---|---|
| entry | catalyst | temp. (°C) | time (h) | conversion (%)a | ee (%)b |
| 1 | (−)-ephedrine | −80 | 13 | 10 | 7 |
| 2 | (+)-N-methylephedrine | −80 | 13 | 9 | 20 |
| 3 | cinchonidine | −80 | 13 | 7 | 33 |
| 4 | quinine | −80 | 13 | 17 | 35 |
| 5 | Quinine·TFA | −80 | 10 | 0 | - |
| 6 | Quinine·TfOH | −80 | 10 | 0 | - |
| 7 | quinine·AcOH | −80 | 10 | 27 | 24 |
| 8 | (S)-BINOL | 20 | 3 | (50) | 4 |
| 9 | (S)-VANOL | −40 | 114 | (30) | 3 |
| 10 | (S)-VAPOL | −40 | 114 | (24) | 2 |
| 11 | 4 | −40 | 114 | (42) | 30 |
| 12 | 5 | 20 | 14 | (46) | 19 |
| 13 | 6 | 20 | 6 | (72) | 37 |
| 14 | 6 | −40 | 60 | (48) | 63 |
| 15 | 7 | −40 | 60 | (60) | 59 |
All the reactions were run at a 0.2 M concentration of the limiting reagent. For entries 1 to 7, 2 equiv of aldehyde was used. For entries 8 to 14, 3 equiv of silyldienol ether was used.
Determined by 1H NMR of the crude reaction mixture. Numbers in parentheses indicate the isolated yield.
Detemined by chiral HPLC analysis.
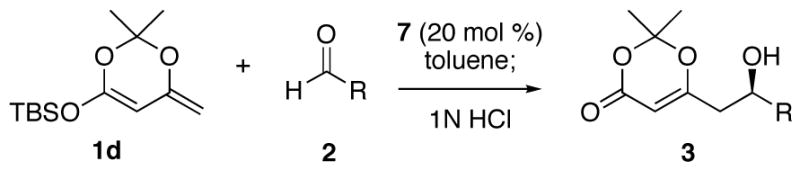
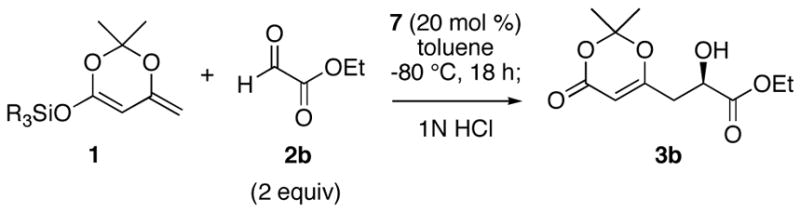
The optimization of reaction conditions was accomplished using the commercially available 1-naphthyl-TADDOL (7) as the catalyst and ethyl glyoxalate as the aldehyde partner (Table 2). When the reaction was carried out at −40 °C with 20 mol % of the catalyst, the VMA product was formed in 61% ee (Table 2, entry 1). The ee improved significantly upon lowering the reaction temperature to −80 °C (entry 2). In order to scavenge adventitious water or acidic species that might enhance the racemic background reaction, various additives were investigated, with Hunig’s base providing the greatest improvement in enantioselectivity (entries 3–6). The silyl substituent on the nucleophile was found to have a small but measurable effect on enantioselectivity, and optimum results were obtained with the TBS group (entry 9). It is noteworthy that the reactions between silyldienol ethers and glyoxalate 2b are generally very rapid, showing >90% conversion within 10 minutes.
Table 2.
Optimization of the Hydrogen Bond Catalyzed VMA Reaction
 | ||||
|---|---|---|---|---|
| entry | SiR3 | additive (10 mol%) | conv. (%)a | ee (%)b |
| 1c | TMS (1a) | - | 98 | 61 |
| 2 | TMS (1a) | - | 82 | 81 |
| 3 | TMS (1a) | 4 Å MSd | 95 | 63 |
| 4 | TMS (1a) | HC(OMe)3 | 98 | 84 |
| 5 | TMS (1a) | 2,6-Di-t-butylpyridine | 99 | 83 |
| 6 | TMS (1a) | (i-Pr)2NEt | 95 | 85 |
| 7 | TES (1b) | (i-Pr)2NEt | 90 | 85 |
| 8 | TIPS (1c) | (i-Pr)2NEt | 95 | 85 |
| 9 | TBS (1d) | (i-Pr)2NEt | 93 | 87 |
| 10 | TBDPS (1e) | (i-Pr)2NEt | 74 | 54 |
All the reactions were run at a 0.2 M concentration of silyldienol ether.
Determined by 1H NMR of the crude reaction mixture.
Detemined by chiral HPLC analysis.
Reaction run at −40 °C (bath).
100 mg used (0.2 mmolof 1).
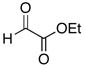
The hydrogen bond catalyzed VMA reaction was applied to a variety of reactive aldehydes (Table 3). The use of Hunig’s base, which decreases the rate of the reaction, was omitted for the reaction with aldehydes other than glyoxalates. In order to facilitate purification and analysis, the products were isolated as the free alcohols following a 1N HCl quench. As can be seen from these results, glyoxalate esters, α,β-unsaturated aldehydes, oxazole aldehydes, thiazole aldehydes, as well as electron-poor aromatic aldehydes can be used in this process, providing vinylogous aldol products in moderate to good ee’s (entries 1–9). Surprisingly, 4-nitrobenzaldehyde (entry 10) proved much less reactive than 2-nitrobenzaldehyde (entry 6) under otherwise identical conditions. Aliphatic aldehydes were less reactive and yielded products with low ee’s (entries 11 and 12). It should be noted that all these reactions showed excellent γ-selectivity, as the regioisomer resulting from attack at the α position of the silyldienol ether was not detected.
Table 3.
Hydrogen Bond Catalyzed VMA Reaction with Various Aldehydes
 | ||||||
|---|---|---|---|---|---|---|
| entry | aldehyde | temp. (°C) | time (h) | product | yield (%) | ee (%)a |
| 1b |
|
−80 | 1 | 3b | 60 | 87 |
| 2b |
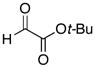
|
−80 | 1 | 3c | 54 | 84 |
| 3 |
|
−60 | 120 | 3d | 66 | 71 |
| 4 |
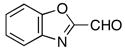
|
−80 | 66 | 3e | 55 | 83 |
| 5 |
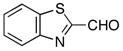
|
−80 | 98 | 3f | 73 | 90 |
| 6 |
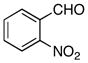
|
−60 | 130 | 3a | 58 | 75 |
| 7 |
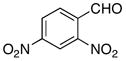
|
−60 | 96 | 3g | 40 | 62 |
| 8 |
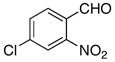
|
−60 | 120 | 3h | 25 | 62 |
| 9 |
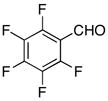
|
−60 | 120 | 3i | 57 | 67 |
| 10 |
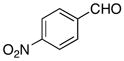
|
−60 | 96 | 3j | 0 | - |
| 11 |
|
−40 | 72 | 3k | 54 | 22 |
| 12 |
|
−40 | 72 | 3l | 23 | 27 |
See Supporting Information for experimental details.
Determined by chiral HPLC analysis.
10 mol % of (i-Pr)2NEt was added.
In addition to providing access to polyketide derivatives, the products can be easily transformed into the formal products of an aldol reaction with acetone, as shown in Scheme 2. Refluxing the VMA adducts 3b and 3d in a toluene/water mixture cleanly provides the desired product 8b and 8d respectively.8a The absolute configuration of VMA products 3k and 31 were determined by comparison of their optical rotations with the reported values,9a,13 whereas that of 8b was assigned by correlation with a known derivative (see Supporting Information).14
Scheme 2.
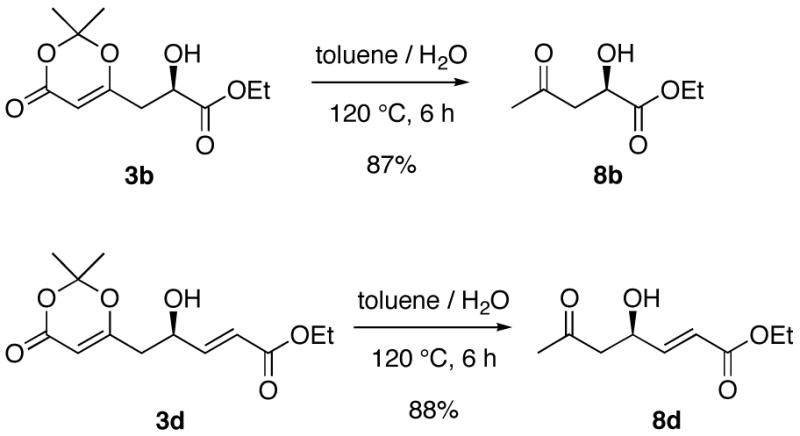
Further Transformations of VMA Products
The asymmetric induction observed in the formation of 3b can be rationalized using the general model (Figure 2) that we had developed for other TADDOL-catalyzed reactions.4 The TADDOL catalyst is expected to exist in a well-defined, internally hydrogen-bonded arrangement.15 The free hydrogen atom on the catalyst, which is expected to be more acidic than a normal OH, can hydrogen bond with the aldehyde oxygen, thereby lowering its lowest unoccupied molecular orbital energy. Stabilization of the hydrogen-bonded aldehyde through a postulated π-π* donor-acceptor interaction between the electron-rich proximal equatorial 1-naphthyl ring and the electron-deficient aldehyde carbonyl should further stabilize the hydrogen-bonded carbonyl, leaving its Re-face accessible to attack by the nucleophile.4b The above model correctly predicts the observed absolute stereochemistry in the product.
Figure 2.
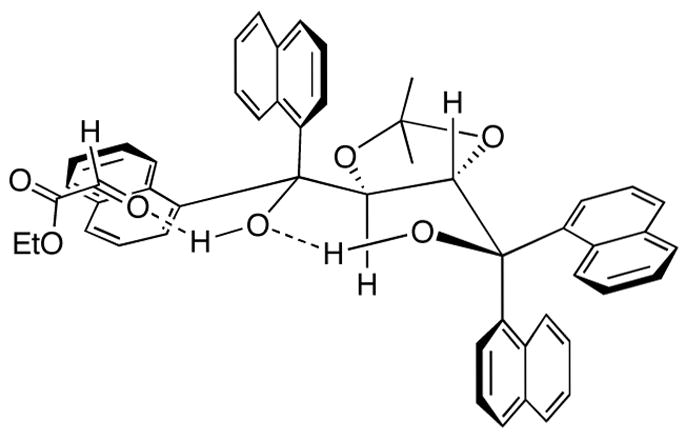
Proposed model for the hydrogen bond catalyzed VMA reaction.
In conclusion, we have demonstrated the successful use of hydrogen bonding catalysis in enantioselective vinylogous Mukaiyama aldol reactions. The reactions are most effective with highly reactive aldehydes and give the expected products in good to excellent yields and ee’s as high as 90%. We are currently investigating the use of other types of nucleophiles in order to expand the scope of this reaction. We are also examining a wider range of hydrogen bond donors, particularly those in which the proton acidity can be readily tuned, so as to catalyze a broader range of transformations.
Supplementary Material
Experimental procedures and characterization of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank the National Science Foundation and the National Institutes of Health for financial support. M.G. gratefully acknowledges the Natural Sciences and Engineering Research Council of Canada for a postdoctoral fellowship. We thank Professor William D. Wulff (Michigan State University) for samples of (S)-VANOL and (S)-VAPOL, and Professor Hisashi Yamamoto (University of Chicago) for a sample of catalyst 4.
References
- 1.Jeffrey GA, editor. An Introduction to Hydrogen Bonding. Oxford University Press; New York: 1997. p. 272. [Google Scholar]
- 2.For recent reviews, see: Pihko PM. Angew Chem, Int Ed. 2004;43:2062. doi: 10.1002/anie.200301732.Dalko PI, Moisan L. Angew Chem, Int Ed. 2004;43:5138. doi: 10.1002/anie.200400650.
- 3.Key contributions include: Sigman MS, Vachal P, Jacobsen EN. Angew Chem, Int Ed. 2000;39:1279. doi: 10.1002/(sici)1521-3773(20000403)39:7<1279::aid-anie1279>3.0.co;2-u.Schuster T, Bauch M, Dürner G, Göbel MW. Org Lett. 2000;2:179. doi: 10.1021/ol991276i.McDougal NT, Schaus SE. J Am Chem Soc. 2003;125:12094. doi: 10.1021/ja037705w.Okino T, Hoashi Y, Takemoto Y. J Am Chem Soc. 2003;125:12672. doi: 10.1021/ja036972z.Nugent BM, Yoder RA, Johnston JN. J Am Chem Soc. 2004;126:3418. doi: 10.1021/ja031906i.Akiyama T, Itoh J, Yokota K, Fuchibe K. Angew Chem, Int Ed. 2004;43:1566. doi: 10.1002/anie.200353240.Uraguchi D, Terada M. J Am Chem Soc. 2004;126:5356. doi: 10.1021/ja0491533.Momiyama N, Yamamoto H. J Am Chem Soc. 2005;127:1080. doi: 10.1021/ja0444637. and references cited therein.
- 4.(a) Huang Y, Unni AK, Thadani AN, Rawal VH. Nature. 2003;424:146. doi: 10.1038/424146a. [DOI] [PubMed] [Google Scholar]; (b) Thadani AN, Stankovic AR, Rawal VH. Proc Natl Acad Sci USA. 2004;101:5846. doi: 10.1073/pnas.0308545101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Unni AK, Takenaka N, Yamamoto H, Rawal VH. J Am Chem Soc. 2005;127:1336. doi: 10.1021/ja044076x. [DOI] [PubMed] [Google Scholar]
- 5.Mukaiyama T, Ishida A. Chem Lett. 1975:319.For recent reviews, see: Denmark SE, Heemstra JR, Beutner GL. Angew Chem, Int Ed. 2005;44:4682. doi: 10.1002/anie.200462338.Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem Rev. 2000;100:1929. doi: 10.1021/cr990247i.
- 6.For example, see: Mori Y, Kuhara M, Takeuchi A, Suzuki M. Tetrahedron Lett. 1988;29:5419.Evans DA, Hoveyda AH. J Am Chem Soc. 1990;112:6447.
- 7.Sato M, Sunami S, Sugita Y, Kaneko C. Chem Pharm Bull. 1994;42:839. [Google Scholar]
- 8.(a) Sato M, Sunami S, Sugita Y, Kaneko C. Heterocycles. 1995;41:1435. [Google Scholar]; (b) Singer RA, Carreira EM. J Am Chem Soc. 1995;117:12360. [Google Scholar]; (c) De Rosa M, Acocella MR, Villano R, Soriente A, Scettri A. Tetrahedron: Asymmetry. 2003;14:2499. and references therein. [Google Scholar]
- 9.(a) Evans DA, Murry JA, Kozlowski MC. J Am Chem Soc. 1996;118:5814. [Google Scholar]; (b) Evans DA, Kozlowski MC, Murry JA, Burgey CS, Campos KR, Connell BT, Staples RJ. J Am Chem Soc. 1999;121:669. [Google Scholar]; (c) Krüger J, Carreka EM. J Am Chem Soc. 1998;120:837. [Google Scholar]; d) Bluet G, Campagne JM. Tetrahedron Lett. 1999;40:221. [Google Scholar]
- 10.Shimada Y, Matsuoka Y, We R, Katsuki T. Synlett. 2004:57. [Google Scholar]
- 11.(a) Denmark SE, Beutner GL. J Am Chem Soc. 2003;125:7800. doi: 10.1021/ja035448p. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Beutner GL, Wynn T, Eastgate MD. J Am Chem Soc. 2005;127:3774. doi: 10.1021/ja047339w. [DOI] [PubMed] [Google Scholar]
- 12.Bluet G, Campagne JM. J Org Chem. 2001;66:4293. doi: 10.1021/jo015567s. [DOI] [PubMed] [Google Scholar]
- 13.Sugita Y, Sakaki J-i, Sato M, Kaneko C. J Chem Soc, Perkin Trans 1. 1992:2855. [Google Scholar]
- 14.Takano S, Iwabuchi Y, Ogasawara K. Tetrahedron Lett. 1991;32:3527. [Google Scholar]
- 15.(a) Beck AK, Bastani B, Planner DA, Fetter W, Seebach D, Braunschweiger H, Gysi P, La Vecchia L. Chimia. 1991;45:238. [Google Scholar]; (b) Seebach D, Dahinden R, Marti RE, Beck AK, Plattner DA, Kühnle FNM. J Org Chem. 1995;60:1788. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.