

Abstract
We determined the macrolide resistance phenotypes of 241 clinical isolates of erythromycin-resistant enterococci (MICs, ≥1 μg/ml), including 147 Enterococcus faecalis strains and 94 Enterococcus faecium strains, collected from a hospital in Seoul, Korea, between 1999 and 2000. By the erythromycin (40 μg)-josamycin (100 μg) double-disk test, 93 strains were assigned to the constitutive macrolide, lincosamide, and streptogramin B (MLSB) resistance (cMLSB) phenotype, and the remaining 148 strains were assigned to the inducible MLSB resistance (iMLSB) phenotype. Of the strains with the iMLSB phenotype, 36 exhibited a reversibly inducible MLSB (riMLSB) phenotype, i.e., blunting of the erythromycin zone of inhibition, which indicates that the 16-membered-ring macrolide josamycin is a more effective inducer than the 14-membered-ring macrolide erythromycin. Sequence analysis of the regulatory regions of the erm(B) genes from all of the strains exhibiting the riMLSB phenotype revealed not only erm(Bv) [where v represents variant; previously erm(AMR)] (n = 13), as reported previously, but also three kinds of erm(B) variants, which were designated erm(Bv1) (n = 17), erm(Bv2) (n = 3), and erm(Bv3) (n = 3), respectively. In lacZ reporter gene assays of these variants, the 16-membered-ring macrolide tylosin had stronger inducibility than erythromycin at ≥0.1 μg/ml. These findings highlight the versatility of erm(B) in induction specificity.
Cross-resistance to macrolide, lincosamide, and streptogramin B (MLSB) antibiotics is mediated by 23S rRNA mutations or the erm genes, which encode 23S rRNA methylases. The expression of MLSB resistance by the erm genes can be either constitutive or inducible, depending on the regulatory region located upstream of the structural gene. In inducible resistance, the specificity of induction differs according to the nature of the regulatory region. The erm(A) and erm(C) genes, the predominant determinants of MLSB resistance in staphylococci, are most often induced in the presence of 14-membered-ring macrolides and the lincosamide celesticetin, but not the 16-membered-ring macrolides and the lincosamide lincomycin (1, 8, 23, 24). Mutants of Staphylococcus aureus selected in the laboratory were reported to be induced by lincomycin and the 16-membered-ring macrolide carbomycin (22); and a recently found clinical strain of S. aureus with erm(A) had inducible cross-resistance to the 14-membered-ring macrolide erythromycin, the lincosamides clindamycin and lincomycin, and the streptogramin B quinupristin (5). The erm(D) gene is induced by the 14-membered-ring macrolides erythromycin and oleandomycin but is not induced by either the 16-membered-ring macrolide tylosin or the lincosamides clindamycin and lincomycin (6). Transcription of the erm(S) gene in the tylosin producer Streptomyces fradiae is autoinducible by tylosin (10). The erm(V) gene from Streptomyces viridochromogenes is induced by the 14- and 16-membered-ring macrolides and celesticetin (8). Interestingly, all MLSB antibiotics induce erm(B) (11), the most widely distributed erm determinant in Streptococcus pneumoniae (11), Streptococcus pyogenes (9), and Enterococcus (18). We previously reported that erm(Bv) [where v represents variant; previously erm(AMR)], the variant of erm(B) from a clinical isolate of Enterococcus faecalis, was induced more strongly by the 16-membered-ring macrolides josamycin, tylosin, and kitasamycin than by erythromycin (16, 17). This alteration of the induction specificity was due to a mutation encoding an arginine-to-cysteine change in the seventh codon of the putative leader peptide preceding the methylase gene.
In this work, we looked at the unexpected rate of the MLSB resistance phenotype in which the 16-membered-ring macrolides are more effective inducers than the 14-membered-ring macrolides in enterococcal clinical isolates. We also investigated the molecular basis of the alteration of induction specificity in these strains by DNA sequence analyses of the regulatory regions of erm(B) and lacZ reporter gene assays.
MATERIALS AND METHODS
Bacterial strains.
A total of 241 clinical isolates of erythromycin-resistant enterococci, including 147 Enterococcus faecalis isolates and 94 Enterococcus faecium isolates, were collected from the Severance Hospital in Seoul, Korea, between May 1999 and January 2000. Multiple isolates from the same patient were avoided. Erythromycin resistance (MIC, ≥1 μg/ml) was identified by the agar dilution method (see below).
Antimicrobials and MIC testing.
The following antimicrobial agents were used in this study: clarithromycin (Abbott Laboratories, Abbott Park, Ill.), erythromycin and tylosin (Sigma, St. Louis, Mo.), josamycin and kitasamycin (ICN Biomedicals, Aurora, Ohio), and rokitamycin (Asahi Kasei Co., Ltd., Tokyo, Japan). The MICs were determined by the agar dilution method in Mueller-Hinton agar (Difco, Detroit, Mich.) by the procedure recommended by the National Committee for Clinical Laboratory Standards (15).
Determination of resistance phenotype.
Erythromycin (40 μg) and josamycin (100 μg) disks were placed 5 to 10 mm apart on brain heart infusion (BHI) agar (Difco), and the plates were incubated aerobically at 37°C for 18 h. The absence of a significant zone of inhibition around the two disks was taken to indicate the constitutive type of MLSB resistance (cMLSB phenotype). Blunting of the josamycin zone of inhibition proximal to the erythromycin disk or blunting of the erythromycin zone of inhibition proximal to the josamycin disk was taken to indicate the inducible type of MLSB resistance (iMLSB phenotype). The iMLSB phenotype with blunting of the erythromycin zone of inhibition proximal to the josamycin disk was taken to indicate the reversibly inducible type of MLSB resistance (riMLSB phenotype).
Growth curves.
Bacterial cultures grown overnight in BHI broth without antibiotic were diluted to an optical density at 590 nm (OD590) of approximately 0.2 in BHI broth with 0.1 μg of tylosin or erythromycin per ml. After induction by incubation for various times at 37°C, the cultures were diluted to an OD590 of approximately 0.05 in BHI broth with 500 μg of erythromycin per ml. Growth at 37°C was monitored by measuring the turbidity of the culture at OD590 for up to 6 h.
Sequence analysis of the erm(B) gene regulatory regions and construction of erm(B)-lacZ reporter plasmids.
The regulatory regions of the erm(B) genes from 36 strains with an riMLSB phenotype were amplified with Vent DNA polymerase (New England Biolabs) and primers SR3 (5′-CTTAGAAGCAAACTTAAGAGTGTGT-3′) (19) and SR71 (5′-AATTTCGTAAACGGTATCGGTTTCT-3′). The 430-bp PCR products were purified with a QIAquick PCR purification kit (Qiagen) and were sequenced by using primer SR71 in a DNA sequencer (ABI PRISM 310; Applied Biosystems). After a comparison of the sequences, we selected three kinds of PCR products with novel sequences from E. faecalis C6608, Enterococcus faecium 3327, and E. faecalis U326, respectively. To construct in-phase fusions of erm(B) with lacZ, the PCR products from each selected strain were ligated to the SmaI site of pMM156 containing a promoterless lacZ gene (4). The resultant ligation products were introduced by electroporation into Escherichia coli CSH26, and transformants were selected on Luria-Bertani (LB) agar plates containing chloramphenicol (10 μg/ml). The transformants on each plate were pooled, and the plasmid DNAs were isolated. These pooled plasmid DNAs were introduced into Bacillus subtilis BR151 as described previously (2), and the transformants were selected on LB agar containing an inducing concentration of josamycin (0.1 μg/ml), 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (40 μg/ml), and chloramphenicol (10 μg/ml). A single blue colony of each strain was selected for the β-galactosidase induction assay. The sequences of all recombinant plasmids were confirmed by DNA sequencing.
β-Galactosidase induction assays.
Measurements of induction were performed for pEZ1, pEZ3, and pEZ4, which were constructed previously (16), as well as for the three plasmids constructed as described above. Plasmids pEZ1 and pEZ3 contain the upstream portion, including the regulatory regions, of erm(Bv) and erm(B) from Tn917, respectively, fused in frame with the lacZ gene in pMM156. Plasmid pEZ4 is identical to pEZ3 except for the lack of duplication of the TAAA sequence in the leader peptide. Cultures of B. subtilis BR151 harboring each erm(B)-lacZ fusion plasmid were grown to early log phase at 37°C in SPII medium (2). Cultures were induced for 120 min with either erythromycin or tylosin at concentrations from 1 ng/ml to 10 μg/ml, as indicated in Fig. 4. β-Galactosidase assays were carried out as described previously (14). Specific activities were expressed in Miller units and were averaged from at least three independent assays.
FIG. 4.
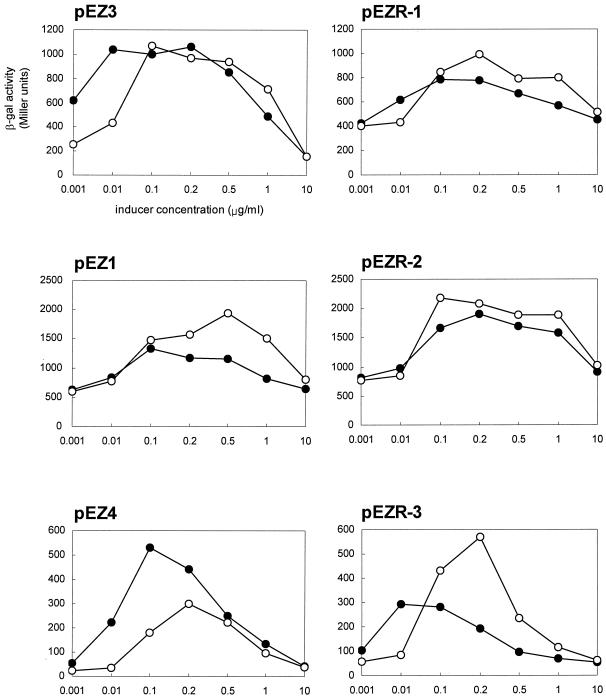
β-Galactosidase (β-gal) activity for B. subtilis BR151 carrying plasmids with erm(B)-lacZ fusions after 120 min of induction with various concentrations of erythromycin (solid circles) and tylosin (open circles).
RESULTS AND DISCUSSION
Identification of macrolide resistance phenotypes.
Since E. faecalis is intrinsically resistant to clindamycin (21), an erythromycin (40 μg) and josamycin (100 μg) double-disk test instead of a triple-disk test with erythromycin, josamycin, and clindamycin disks was performed to determine the resistance phenotypes of the enterococci. Of the 241 strains of erythromycin-resistant enterococci tested, 93 (39%) were assigned to the cMLSB phenotype and 148 (61%) were assigned to the iMLSB phenotype; among the latter strains, 36 (15%) showed the riMLSB phenotype. The rate of occurrence of the iMLSB phenotype might increase by the growth curve test. Isolates with the riMLSB phenotype exhibited blunting of the erythromycin zone of inhibition proximal to the josamycin disk (Fig. 1). The unexpected rate of enterococcal isolates with the riMLSB phenotype indicates that this novel macrolide resistance pattern is indeed disseminated among the enterococci in a Korean hospital.
FIG. 1.

riMLSB phenotype of erythromycin-resistant enterococcal isolates. The disks contained 40 μg of erythromycin (E) and 100 μg of josamycin (J).
When disks with lower amounts of drug (erythromycin, 15 μg; josamycin, 20 μg) were used, only 5 isolates presented the riMLSB phenotype, whereas the other 31 isolates exhibited a false cMLSB phenotype. Considering that these 31 isolates exhibited the riMLSB phenotype when they were tested with disks containing large amounts of drugs, the amounts of drug in the disks might be a critical factor in finding isolates with an riMLSB phenotype. Also, these results indicate that most of the 36 isolates with the riMLSB phenotype are highly resistant to both drugs.
The resistance inducibilities of erythromycin and tylosin were tested with 3 strains selected from among the 36 strains with the riMLSB phenotype by measuring their growth rates in broth. After exposure to a subinhibitory concentration (0.1 μg/ml) of either erythromycin or tylosin for various times, the cells were challenged with a high concentration (500 μg/ml) of erythromycin. E. faecalis R703 acquired resistance to the macrolide more rapidly when resistance was induced with tylosin than when it was induced with erythromycin; i.e., tylosin was a more potent inducer than erythromycin (Fig. 2). Similar results were obtained with the remaining strains, E. faecalis U326 and E. faecalis C6593 (data not shown). However, when the challenge concentration was decreased to 100 μg/ml, these kinetics of induction were not observed because the growth of the bacteria in which resistance was not induced was scarcely inhibited.
FIG. 2.
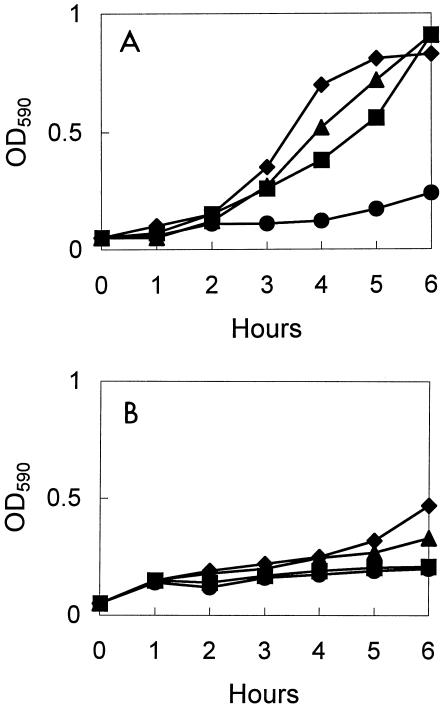
Growth curves for E. faecalis R703 with erm(Bv). The strain was induced with 0.1 μg of tylosin per ml (A) and 0.1 μg of erythromycin per ml (B) for 0 (solid circles), 15 (solid squares), 60 (solid triangles), and 120 (solid diamonds) min. The cultures were then challenged with 500 μg of erythromycin per ml for 6 h.
Resistance to macrolide antibiotics.
In a previous report (17), the MICs of the 16-membered-ring macrolides josamycin, tylosin, kitasamycin, and rokitamycin for E. faecalis 373, the first clinical isolate identified to contain erm(Bv), were at least 1,024 μg/ml, while the MICs of the 14-membered-ring macrolides erythromycin and clarithromycin were 16 μg/ml. In contrast, all 36 strains with the riMLSB phenotype tested in this study were highly resistant (MICs, ≥512 μg/ml) to erythromycin and clarithromycin as well as to josamycin, tylosin, kitasamycin, and rokitamycin. Thus, the high level of resistance of the isolates with the riMLSB phenotype might explain why this phenotype was most easily identified when disks with high doses in double-disk tests or high challenge concentrations in growth kinetic assays were used.
Sequence analysis of regulatory regions of erm(B) genes.
The 36 isolates with the riMLSB phenotype were analyzed for the presence of erm(B) by PCR with erm(B)-specific primers SR3 and SR71. For all isolates, an amplicon with the expected molecular size of 430 bp was obtained. The PCR product contains the putative promoter, the sequence encoding the leader peptide, and the first 108 nucleotides of the methylase gene. The nucleotide sequences of the PCR products were determined and are compared with that of erm(B) from Tn917 (GenBank accession no. M11180) in Fig. 3. Four groups of variations were detected. In the first variation, the sequences of the fragments amplified from 13 isolates were identical to that of erm(Bv), which had a C-to-T transition that resulted in an amino acid change from Arg to Cys at the seventh codon of the putative leader peptide. The second variation, observed for 17 isolates and designated erm(Bv1), had a duplication of CAAATGCGTTAT in the leader peptide, resulting in the duplication of four amino acids (Gln, Met, Arg, Tyr). This 12-nucleotide duplication was also observed in an erm(B) variant [previously erm(BC) from E. coli] (3). In addition, erm(Bv1) had two base substitutions in the sequence encoding the leader peptide, which led to changes in the corresponding amino acids. The third variation, observed for three isolates and designated erm(Bv2), was similar to erm(Bv1). In erm(Bv2), the 25th nucleotide of the sequence encoding the erm(Bv1) leader peptide, cytosine, was changed to adenine, and only one of the two point mutations found in erm(Bv1) was observed. The fourth variation, detected in the three remaining isolates and designated erm(Bv3), had a C-to-T transition that led to a single amino acid change from Thr to Ile at the 14th codon of the leader peptide and a point mutation located downstream from the leader peptide.
FIG. 3.

Sequences of erm(Bv), erm(Bv1), erm(Bv2), and erm(Bv3) in comparison with the sequences of Tn917 and pAM77. Only differences from the sequence of Tn917 are indicated. Gaps are indicated by dashes.
Meanwhile, we sequenced the structural erm(B) genes from seven riMLSB strains: two with erm(Bv), three with erm(Bv1), one with erm(Bv2), and one with erm(Bv3). In all erm(Bv) and erm(Bv3) sequences that were sequenced, the coding sequences of the methylase genes were identical to that of Tn917. All erm(Bv1) sequences had a substitution of Thr (ACT) for Ile (ATT) at codon 75 and three silent mutations at codons 6, 74, and 667 (AAA to AAG, AAC to AAT, and ACT to ACC, respectively). In erm(Bv2), two mutations at codons 74 and 75 found in erm(Bv1) were observed. Although it was not examined whether the amino acid substitution at codon 75 of the methylase gene has a role in the expression of various resistance phenotypes, it is likely that the changes in the regulatory region, including the leader peptide, are major causes of the alteration of induction specificity, on the basis of the following data from erm(B)-lacZ fusion experiments.
A translational attenuation model was proposed for the regulation mechanism of erm(B) (11). The expression of erm(C) is controlled by translational attenuation, in which alterations in the critical sequence of the leader peptide can affect the induction efficiency of inducer antibiotics (12, 13). This notion might be applied to the expression of erm(B). The replacement of Val with Ile at position 19 of the erm(Bv1) and erm(Bv2) leader peptides was also observed in pAM77 (7), and thus, it is not thought to have much of an effect on the specificity of induction. Therefore, the 4-amino-acid duplication in the leader peptides of erm(Bv1) and erm(Bv2) is likely to be responsible for the switch in induction specificity. The codon change at residue 14 of the leader peptide of erm(Bv3) is probably associated with the efficiency of induction by macrolides because it is the sole mutation in the leader peptide. Taken together, the sequence analysis results suggest that the critical region of erm(B) for induction contains at least the codons for residues 7 to 14 of the leader peptide.
β-Galactosidase induction assays.
To compare the induction of erm(B) from Tn917 by the 14- and 16-membered-ring macrolides with those of the four variants, we performed β-galactosidase assays with reporter constructs in which the methylase genes were translationally fused with E. coli lacZ. The β-galactosidase activities were measured after 120 min of induction with various concentrations of either erythromycin or tylosin (Fig. 4). For plasmid pEZ3 (16), which has the erm(B) sequence from Tn917, although erythromycin was a stronger inducer than tylosin at 1 and 10 ng/ml, the inducibilities of erythromycin and tylosin were similar at ≥0.1 μg/ml. At the optimum concentrations (0.1 and 0.2 μg/ml), both drugs were effective inducers for pEZ3, while erythromycin was a stronger inducer than tylosin for pEZ4 (16), which differs from pEZ3 by the lack of duplication of the TAAA motif in the leader peptide. In contrast to erm(B) from pAM77, that from Tn917 has a duplication of TAAA within the leader peptide-coding sequence, resulting in a peptide 9 amino acids shorter. Consistent with a previous report (16), the level of induction obtained with the erm(B)-lacZ fusion, which has the TAAA duplication (pEZ3), was higher than that obtained with the erm(B)-lacZ fusion, which has the single TAAA motif (pEZ4). Thus, the TAAA duplication appears to affect the induction specificity as well as the level of expression of erm(B). For pEZ1 (16), the erm(Bv)-lacZ fusion construct, the rates of β-galactosidase expression were similar at low concentrations of both drugs (1 and 10 ng/ml). At ≥0.1 μg/ml, however, tylosin had a higher induction efficiency than erythromycin. Similar induction patterns were also shown for pEZR-1, pEZR-2, and pEZR-3, which carry the erm(Bv1)-lacZ, erm(Bv2)-lacZ, and erm(Bv3)-lacZ fusions, respectively. In general, when the concentration of the inducer was low (1 and 10 ng/ml), erythromycin was a more potent inducer of these erm(B) variants than tylosin or had an induction efficiency equivalent to that of tylosin. In contrast, at inducer concentrations 0.1 μg/ml or higher, the expression of the erm(B) variants was more strongly induced by tylosin than by erythromycin. These data show the correlation between the inducer concentration and the induction specificity in erm(B) and the versatility of erm(B) in induction. We also assessed induction as a function of time (0 to 120 min), but there was no significant time-dependent effect on the inducibility of erythromycin and tylosin for any of the constructs (data not shown).
There were drastic differences in the induced levels of expression among the erm(B) variants (Fig. 4). The basal level of erm(B) expression could be influenced by mutations in the putative promoter or the region forming a stem-loop structure, which might sequester the ribosome binding site and the start codon for the methylase. However, such mutations were not found in the erm(B) sequences of the 36 strains analyzed in this study in comparison with the erm(B) sequence in Tn917 (Fig. 3). Thus, it is likely that a mutation in the leader peptide-coding sequence affects the secondary structure of putative upstream attenuators and, consequently, the downstream structure, which sequesters the initiation sequence for translation of the methylase gene. Also, this mutation could alter the efficiency or location of ribosome stalling, and therefore, the secondary structure of the putative attenuator would change.
All of the 36 isolates with the riMLSB phenotype exhibited inducible resistance to macrolide antibiotics and were highly resistant to these drugs (MICs, ≥512 μg/ml). It is possible that the high level of resistance results from a high basal level of expression. However, the level of expression of erm(Bv3) was quite low compared with those of the other erm(B) variants (see the result for pEZR-3 in Fig. 4). Also, as mentioned above, none of the erm(B) variants had a mutation in the putative promoter or the attenuator at the 3′ end. Moreover, the 13 isolates harboring erm(Bv) found in this study had a very high level of resistance to the 14-membered-ring macrolide, in contrast to the levels of resistance of a strain that carries the same erm(Bv) found previously, E. faecalis 373 (17). This discrepancy may be accounted for by differences in the genetic backgrounds of the hosts or the copy number of the erm(B) gene. It is conceivable that the expression of erm(B) can be changed by mutations not only of the regulatory region encompassing the leader peptide but also of a possible regulator, for example, open reading frame ORF3 followed by the methylase gene in Tn917 (20).
Acknowledgments
This work was supported by grant R01-1999-00110 from the Basic Research Program of the Korea Science & Engineering Foundation and was partially supported by grant HMP-00-CH-15-0014 from the Korean Ministry of Health & Welfare and Project BK21 for Medicine, Dentistry, and Pharmacy.
REFERENCES
- 1.Allen, N. E. 1977. Macrolide resistance in Staphylococcus aureus: inducers of macrolide resistance. Antimicrob. Agents Chemother. 11:669-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anagnostopoulus, C., and J. Spizizen. 1961. Requirements for transformation in Bacillus subtilis. J. Bacteriol. 81:741-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brisson-Noël, A., M. Arthur, and P. Courvalin. 1988. Evidence for natural gene transfer from gram-positive cocci to Escherichia coli. J. Bacteriol. 170:1739-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byeon, W. H., and B. Weisblum. 1990. Replication genes of plasmid pE194-cop and repF: transcripts and encoded proteins. J. Bacteriol. 172:5892-5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarebout, G., E. Nativelle, and R. Leclercq. 2001. Unusual inducible cross resistance to macrolides, lincosamides, and streptogramins B by methylase production in clinical isolates of Staphylococcus aureus. Microb. Drug Resist. 7:317-322. [DOI] [PubMed] [Google Scholar]
- 6.Docherty, A., G. Grandi, R. Grandi, T. J. Gryczan, A. G. Shivakumar, and D. Dubnau. 1981. Naturally occurring macrolide-lincosamide-streptogramin B resistance in Bacillus licheniformis. J. Bacteriol. 145:129-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Horinouchi, S., W. H. Byeon, and B. Weisblum. 1983. A complex attenuator regulates inducible resistance to macrolides, lincosamides, and streptogramin type B antibiotics in Streptococcus sanguis. J. Bacteriol. 154:1252-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamimiya, S., and B. Weisblum. 1997. Induction of ermSV by 16-membered-ring macrolide antibiotics. Antimicrob. Agents Chemother. 41:530-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kataja, J., P. Huovinen, M. Skurnik, the Finnish Study Group for Antimicrobial Resistance, and H. Seppälä. 1999. Erythromycin resistance genes in group A streptococci in Finland. Antimicrob. Agents Chemother. 43:48-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelemen, G., M. Zalacain, E. Culebras, E. Seno, and E. Cundliffe. 1994. Transcriptional attenuation control of the tylosin-resistance gene tlrA in Streptomyces fradiae. Mol. Microbiol. 14:833-842. [DOI] [PubMed] [Google Scholar]
- 11.Leclercq, R., and P. Courvalin. 2002. Resistance to macrolides and related antibiotics in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 46:2727-2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayford, M., and B. Weisblum. 1989. ermC leader peptide. Amino acid sequence critical for induction by translational attenuation. J. Mol. Biol. 206:69-79. [DOI] [PubMed] [Google Scholar]
- 13.Mayford, M., and B. Weisblum. 1990. The ermC leader peptide: amino acid alterations leading to differential efficiency of induction by macrolide-lincosamide-streptogramin B antibiotics. J. Bacteriol. 172:3772-3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 15.National Committee for Clinical Laboratory Standards. 1998. Performance standard for antimicrobial susceptibility testing: eighth informational supplement, M100-S8. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 16.Oh, T.-G., A.-R. Kwon, and E.-C. Choi. 1998. Induction of ermAMR from a clinical strain of Enterococcus faecalis by 16-membered-ring macrolide antibiotics. J. Bacteriol. 180:5788-5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oh, T.-G., M.-J. Lee, M.-C. Baek, B.-K. Kim, and E.-C. Choi. 1998. Resistance to macrolide-lincosamide-streptogramin B antibiotics is induced by 16 membered-ring macrolide antibiotics in Enterococcus faecalis 373. Arch. Pharm. Res. 21:76-78. [DOI] [PubMed] [Google Scholar]
- 18.Portillo, A., F. Ruiz-Larrea, M. Zarazaga, A. Alonso, J. L. Martinez, and C. Torres. 2000. Macrolide resistance genes in Enterococcus spp. Antimicrob. Agents Chemother. 44:967-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosato, A., H. Vicarini, and R. Leclercq. 1999. Inducible or constitutive expression of resistance in clinical isolates of streptococci and enterococci cross-resistant to erythromycin and lincomycin. J. Antimicrob. Chemother. 43:559-562. [DOI] [PubMed] [Google Scholar]
- 20.Shaw, J., and D. B. Clewell. 1985. Complete nucleotide sequence of macrolide-lincosamide-streptogramin B-resistance transposon Tn917 in Streptococcus faecalis. J. Bacteriol. 164:782-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh, K. V., G. M. Weinstock, and B. E. Murray. 2002. An Enterococcus faecalis ABC homologue (Lsa) is required for the resistance of this species to clindamycin and quinupristin-dalfopristin. Antimicrob. Agents Chemother. 46:1845-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka, T., and B. Weisblum. 1974. Mutant of Staphylococcus aureus with lincomycin- and carbomycin-inducible resistance to erythromycin. Antimicrob. Agents Chemother. 5:538-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weisblum, B. 1995. Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 39:577-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weisblum, B., C. Siddhikol, C. J. Lai, and V. Demohn. 1971. Erythromycin-inducible resistance in Staphylococcus aureus: requirements for induction. J. Bacteriol. 106:835-847. [DOI] [PMC free article] [PubMed] [Google Scholar]