

Abstract
A synthetic peptide containing amino acids 77 to 95 of the intracellular GTPase RhoA has previously been shown to inhibit replication of respiratory syncytial virus (RSV) in cultured cells. We show that residues 80 to 90 of RhoA are sufficient for this activity and that the cysteine residue at position 83 is critical. Further studies with an optimal peptide sequence containing amino acids 80 to 94 of RhoA revealed that the antiviral potency of the peptide is dependent on the oxidation of cysteine 83. Size-exclusion chromatography and sedimentation equilibrium studies of the peptide comprising residues 80 to 94 revealed that it is capable of forming aggregates in both reduced and oxidized states. A peptide (83A) in which the cysteine residue is replaced by an alanine does not form dimers or higher-order aggregates and did not inhibit RSV replication at any concentration tested. These data indicate that formation of peptide multimers is necessary for the antiviral activities of RhoA-derived peptides and suggest that the observed antiviral activities of these peptides may be unrelated to the biological functions of their parent molecule.
Respiratory syncytial virus (RSV) is an enveloped, negative-sense, single-stranded RNA virus of the Paramyxovirus family. It is the most important cause of lower respiratory tract infections in children worldwide and is a significant cause of morbidity and mortality among immunocompromised adults and the institutionalized elderly (4). RSV enters host cells by fusing its viral envelope with the host cell plasma membrane to allow penetration of the viral nucleocapsid. The processes of viral attachment and fusion are mediated by the viral glycoproteins, termed the fusion protein (F), glycoprotein (G), and the small hydrophobic protein (SH). While G and SH can increase the efficiency of the viral entry process, F alone is sufficient (10, 13, 27).
It has previously been observed that RSV F can interact with the small GTPase RhoA (20) and that a peptide derived from the F-binding region of RhoA is able to block the entry of RSV into susceptible host cells (21). This peptide, which we termed peptide 77-95, comprises the linear peptide sequence corresponding to amino acids (aa) 77 to 95 of RhoA. On the basis of the observation that peptide 77-95 can interfere with binding of F to RhoA in an in vitro enzyme-linked immunosorbent assay (ELISA), it was originally hypothesized that peptide 77-95 may inhibit an interaction between RSV F and RhoA essential to F-mediated membrane fusion (21). However, an in vivo interaction between F and RhoA at the time of viral entry has not been demonstrated. In addition, other agents that should be capable of inhibiting a RhoA-F interaction, such as anti-RhoA antibodies and exogenous, purified RhoA, have no inhibitory effect on RSV entry (unpublished data). Thus, the ability of the RhoA-derived peptide to inhibit RSV entry may be unrelated to its ability to disrupt an in vitro F-RhoA interaction.
The region from aa 77 to 95 of RhoA corresponds to an internal beta strand with a highly hydrophobic nature (residues 79 to 86), followed by a surface-exposed alpha helix (residues 88 to 95). There are acidic residues at positions 78 (Asp), 87 (Asp), 90 (Asp), and 93 (Glu) (11, 19, 25, 28). Thus, at neutral pH linear peptide 77-95 has a bipartite nature: a mainly hydrophobic N terminus (aa 77 to 86) and a negatively charged C terminus (aa 87 to 95). There are several reports of inhibition of RSV and other enveloped viruses by polyanionic compounds, including naturally occurring glycosaminoglycans as well as synthetic polyanionic molecules (for reviews, see references 18 and 22). For example, heparan sulfate binds to both F and G (5, 6), and soluble heparin or other iduronic acid-containing glycosaminoglycans can inhibit RSV infection of cultured cells (3, 5, 9, 14). Other reported inhibitors of RSV are largely of a hydrophobic and anionic character (2, 12, 22, 29). Thus, the inhibitory activity of the RhoA-derived peptide may be a result of the physical properties of the linear peptide itself that are independent of its ability to disrupt a specific protein-protein interaction. In the present study we have sought to define the structural properties of peptide 77-95 that are responsible for its antiviral effects.
MATERIALS AND METHODS
Viruses and cells.
HEp-2 cells were used in all experiments unless indicated otherwise. Cells were propagated in minimum essential medium supplemented with glutamine, amphotericin B, gentamicin, and 10% fetal bovine serum (MEM 10). The A2 strain of RSV was kindly provided by Robert Channock, and working stocks of virus were prepared as described previously (8).
Synthetic peptides.
The unpurified peptide preparations used for the initial screens (Table 1) were purchased from Research Genetics (now ResGen; Huntsville, Ala.) at a stated purity of approximately 70%. The crude preparation of peptide 78-94 was synthesized and fractionated by SynPep Corporation (Dublin, Calif.). All other peptides were synthesized by 9-fluorenylmethoxy carbonyl solid-phase chemistry (Chiron Technologies, San Diego, Calif.) by the Food and Drug Administration Facility for Biotechnology Resources (Bethesda, Md.). Peptide 80-94 is a linear peptide corresponding to amino acids 80 to 94 of RhoA: ILMCFSIDSPDSLEN. Peptide 83A is the same as peptide 80-94 except for an alteration by the substitution of an alanine residue for Cys83: ILMAFSIDSPDSLEN. The sequences of the other peptides tested are shown in Table 1 or Fig. 1B. In general, peptides were dissolved to 10 mg/ml in stock solutions and stored in aliquots at −70°C. The solvents varied on the basis of solubility properties, as indicated in the text or Table 1.
TABLE 1.
Effective antiviral concentrations of RhoA-derived peptidesa
| Peptide | Peptide | Sequence | IC50 (μM) |
|---|---|---|---|
| Wild type | 77-95 | TDVILMCFSIDSPDSLENI | 7.60 |
| Scrambled | 77-95 | CSIELSDIPLSVDFNTMIDb | >50 |
| Alanine substiutited | 77-95-77A | ADVILMCFSIDSPDSLENIb | 2.56 |
| 77-95-78A | TAVILMCFSIDSPDSLENI | 1.37 | |
| 77-95-79A | TDAILMCFSIDSPDSLENI | 6.60 | |
| 77-95-80A | TDVALMCFSIDSPDSLENI | 11.60 | |
| 77-95-81A | TDVIAMCFSIDSPDSLENIb | 5.42 | |
| 77-95-82A | TDVILACFSIDSPDSLENI | 1.43 | |
| 77-95-83A | TDVILMAFSIDSPDSLENIb | >50 | |
| 77-95-84A | TDVILMCASIDSPDSLENIb | 6.29 | |
| 77-95-85A | TDVILMCFAIDSPDSLENIb | 6.82 | |
| 77-95-86A | TDVILMCFSADSPDSLENI | 3.52 | |
| 77-95-87A | TDVILMCFSIASPDSLENIb | 4.36 | |
| 77-95-88A | TDVILMCFSIDAPDSLENI | 2.26 | |
| 77-95-89A | TDVILMCFSIDSADSLENI | 15.32 | |
| 77-95-90A | TDVILMCFSIDSPASLENIb | 2.61 | |
| 77-95-91A | TDVILMCFSIDSPDALENI | 1.19 | |
| 77-95-92A | TDVILMCFSIDSPDSAENIb | 2.27 | |
| 77-95-93A | TDVILMCFSIDSPDSLANI | 9.83 | |
| 77-95-94A | TDVILMCFSIDSPDSLEAIb | 18.47 | |
| 77-95-95A | TDVILMCFSIDSPDSLENAb | 4.89 | |
| Truncated | 77-86 | TDVILMCFSIb,c | >50 |
| 77-89 | TDVILMCFSIDSPb | >50 | |
| 77-92 | TDVILMCFSIDSPDSL | 10.86 | |
| 78-95 | DVILMCFSIDSPDSLENIb | 1.23 | |
| 79-95 | VILMCFSIDSPDSLENI | 16.95 | |
| 80-95 | ILMCFSIDSPDSLENIb | 7.17 | |
| 83-95 | CFSIDSPDSLENIb,c | >50 | |
| 80-94 | ILMCFSIDSPDSLEN | 1.75 | |
| 80-93 | ILMCFSIDSPDSLE | 3.50 | |
| 80-92 | ILMCFSIDSPDSL | 12.40 | |
| 80-91 | ILMCFSIDSPDSb | 6.36 | |
| 80-90 | ILMCFSIDSPD | 4.61 | |
| 80-89 | ILMCFSIDSPb,c | 35.77 | |
| 80-88 | ILMCFSIDSb,c | >50 | |
| 80-87 | ILMCFSIDb,c | >50 | |
| 80-86 | ILMCFSIb,c | >50 | |
| 80-85 | ILMCFSb,c | >50 |
The peptides shown were sythesized and tested for antiviral activity, and IC50 were calculated as described in the text. Combined data from two or more separate experiments were used to calculate each value. Unless indicated otherwise, the peptide stocks were dissolved in PBS or distilled, deionized water. Where applicable, the position of the alanine substitution is underscored. Boldface type indicates peptides which did not inhibit 50% viral replication at any concentration tested.
The peptide stocks were disolved in DMSO.
The peptide caused a visible CPE.
FIG. 1.
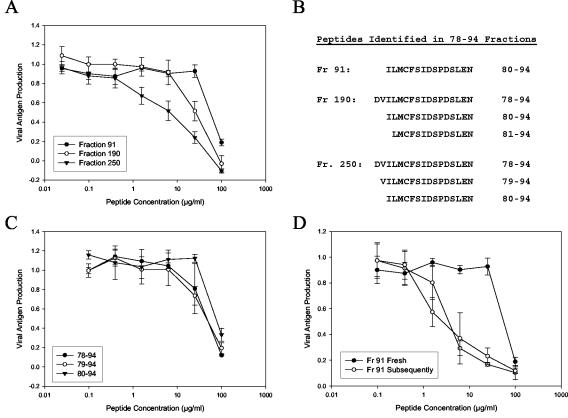
The antiviral activity of a RhoA-derived peptide preparation is highest in late-eluting aggregates and aged peptide aliquots. A crude peptide corresponding to the sequence from residues 78 to 94 of RhoA was synthesized and subjected to fractionation by reverse-phase HPLC. A total of 300 fractions were collected, and selected fractions were tested for antiviral activity. (A) Inhibition curves for the three fractions with the highest activities. (B) The fractions shown in panel A were analyzed by mass spectrometry to identify peptide products. Shown are the peptide sequences that likely correspond to the observed masses. For fraction 250, the validity of these assignments was verified by sequencing. (C) The peptides identified as components of fraction 250 were individually synthesized and tested for antiviral activity. (D) Compiled data from multiple assays of peptide 80-94 from fraction (Fr) 91. Closed circles, the first test of a peptide aliquot after it was dissolved; open circles, subsequent tests of the same aliquot. Results are representative of several tests with both crude and purified 80-94 peptides. Each datum point represents the mean for three replicate wells at each concentration, and error bars represent the standard deviations.
Peptide purification and analysis.
Fractionation of the crude preparation of peptide 78-94 was performed at SynPep Corporation. Crude peptide was loaded onto a C18 column and eluted with a gradient of 0 to 100% acetonitrile containing 0.075% trifluoroacetic acid over 150 min. A total of 300 fractions were collected, lyophilized, and stored at −20°C until testing. The molecular weight components of active fractions were determined by matrix-assisted laser desorption ionization-time of flight mass spectrometry analysis. The N-terminal sequence and purity of the peptide product were determined by Edman sequencing on a model 494A peptide/protein sequenator (Applied Biosystems, Foster City, Calif.) by using the software of the manufacturer. Peptides 80-94 and 83A were purified to ≥95% by reverse-phase high-pressure liquid chromatography (HPLC) at the Peptide Synthesis and Analysis Unit, National Institute of Allergy and Infectious Diseases (Rockville, Md.), and mass spectrometry analysis of purified peptides was performed by the Bio-Analytical Mass Spectrometry Laboratory, National Institute of Allergy and Infectious Diseases (Rockville, Md.).
Microplaque reduction assay.
Peptides were serially diluted fourfold in MEM 10 to give final test concentrations ranging from 0.10 to 100 μg/ml. An equal volume of MEM 10 containing RSV (calculated to give a final virus concentration of 30 to 50 PFU/well) was then added to the diluted peptides. A total of 90 μl of the virus-peptide mixture was then added in triplicate to HEp-2 cells in 96-well plates that had been seeded the previous day with 1.5 × 104 to 2.0 × 104 cells per well. At 2 days postinfection, the cells were fixed in methanol and the extent of viral replication was determined by immunohistochemical staining with a mixture of anti-F monoclonal antibodies (kindly provided by Judy Beeler through the World Health Organization Reagent Bank for RSV and PIV3, Center for Biologics Evaluation and Research, Food and Drug Administration, Rockville, Md.).
Viral antigen reduction assay.
To simplify the quantitation of virus replication, the microplaque assay was modified in later experiments to detect virus replication by an in situ ELISA method rather than by counting of plaques. The following adaptations to the microplaque assay were made: (i) a higher inoculum of virus (≥100 PFU/well) was used; (ii) the cells were fixed on days 2 to 3 postinfection, depending on the extent of a visible cytopathic effect (CPE); and (iii) 100 μl of 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid substrate solution (Kirkegaard & Perry Laboratories, Gaithersburg, Md.) rather than the diaminobutyric acid substrate used in the microplaque assay was added. After the substrate was allowed to develop, the absorbance of the wells was read at 405 nm with an MRX microplate reader (Dynex Technologies, Chantilly, Va.). The results were directly comparable to those obtained by the microplaque assay. Table 1 presents the data gathered by the microplaque assay. Figures 1 to 3 present the data collected by the antigen reduction assay.
FIG. 3.
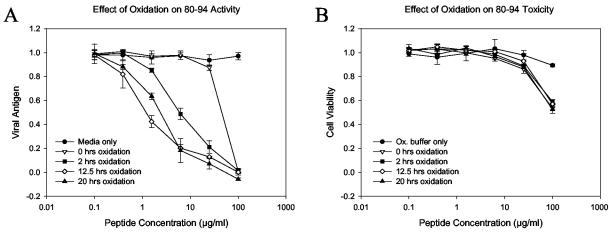

The antiviral potency of 80-94 is dependent on the extent of cysteine oxidation. Peptide 80-94 was purified to >95% purity by reverse-phase HPLC and was then dissolved in oxidation buffer (ammonium bicarbonate buffer [pH 8.0] containing 20% DMSO) to promote disulfide bond formation. At several time points aliquots of peptide solution were removed and assayed for free sulfhydryl content, diluted in PBS, and stored at −20°C. The peptides were later thawed and assayed for antiviral activity. (A) Inhibition curves for peptide aliquots drawn at the indicated time points; (B) cytotoxicity profile of the same aliquots shown in panel A; (C) free sulfhydryl (SH) concentrations of corresponding samples. This pattern of oxidation-dependent enhancement of peptide activity is representative of those from numerous experiments with both crude and highly purified peptides. In panels A and B each datum point represents the mean for three replicate wells, and error bars represent the standard deviations.
Calculation of IC50s.
Each peptide was tested in at least two separate experiments as described above. Data from each experiment were normalized to the level for the untreated control wells. All data for each peptide were then combined and fit to a sigmoid curve by using the “regression wizard” function of SigmaPlot 2001 (SPSS Inc., Chicago, Ill.). The 50% inhibitory concentrations (IC50s) were calculated by solving the resulting curve-fit equation for the concentration corresponding to a 50% reduction in plaque number or the level of production of viral antigen compared to the plaque numbers or antigen levels in the untreated control wells.
Cell viability assay.
For the initial testing of unpurified peptide preparations (data shown in Table 1), cellular cytotoxicity was assessed by visual examination of cell monolayers for CPE. In later experiments viability assays were performed by using the Cell Proliferation Assay kit from the American Type Culture Collection (Manassas, Va.). Assays were performed by using the same format used for the microplaque reduction assay or in situ ELISA, but in the absence of virus. Peptides were serially diluted in MEM 10 and were then transferred to HEp-2 cells in 96-well plates. After coincubation for 48 to 72 h, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent was added and the assay was performed according to the protocol of the manufacturer.
Ellman assay.
The free sulfhydryl content of peptide stocks was assessed by using Ellman's reagent (Pierce Biotechnology, Rockford, Ill.), according to the protocol of the manufacturer, adapted to a microwell format. Cysteine standards and peptide stocks were diluted 1:50 in 200 μl of reaction buffer (0.1 M sodium phosphate [pH 8.0], 1 mM EDTA) containing freshly dissolved Ellman's reagent (1 mM) in 96-well microtiter plates (Nunc International). Samples were mixed for ∼30 s and were incubated for 15 min at room temperature. The absorbance at 405 nM was then measured by using an MRX microplate reader (Dynex Technologies), and free sulfhydryl values were calculated on the basis of a standard curve with concentrations in the range of 0.078 to 10 mM. All R2 values for curve fitting were within the range of 0.9990 to 1.000.
Sedimentation equilibrium experiments.
Sedimentation equilibrium analysis was performed with a Beckman Optima XL-A/I analytical ultracentrifuge with absorbance optical scanning. The cells were loaded with 130- to 145-μl volumes of sample in the optical density range of 0.3 to 0.7 absorbance units, and the absorbance was measured at either 230 or 202 nm (depending on the absorbance spectrum of the peptide). An An-60Ti 4 cell rotor was used in order to achieve the necessary centrifugal speeds cited below. Data on the sedimentation equilibrium absorbance versus the radial position were obtained at radial increments of 0.001 cm with 10 repeats at 20°C. Rotor speeds between 52,000 and 58,000 rpm were used for each sample. For a single ideal component at sedimentation equilibrium, the total solute concentration at any radial position r (Cr) is given by the following expression:
 |
(1) |
where Cref is the concentration at a reference position, M is the molecular weight of the solute, νbar is the partial specific volume of the solute, ρ is the density of the sedimentation solvent, ω is the angular velocity, rref is the radius at a reference position, R is the gas constant, and T is the temperature (in Kelvin). The molar mass is readily determined by nonlinear regression analysis at a single centrifugal speed or global analysis of the data obtained with multiple speeds. Classically, equation 1 was converted to a linear form given by equation 2:
 |
(2) |
The weight-average molar mass is determined from the slope of the plot of ln C versus r2. For both equations 1 and 2 the absorbance can replace the concentration for molecular weight determinations. Both nonlinear regression fitting of the data to equation 1 and the slope determined by equation 2 were used in this study since the latter provides an easy comparison of the molecular weight of the peptide on the basis of differences in the slopes. Data analysis was performed with the software package (version 4.0) provided by Beckman-Coulter Instruments linked to Origin 4.1 (Microcal Software, Inc.). The partial specific volumes of peptides 83A and 80-94 were 0.735 and 0.728 ml/g, respectively, as determined by the public domain software program Sednterp (http://www.bbri.org/RASMB/rasmb.html) developed by D. B. Hayes, T. Laue, and J. Philo.
Gel filtration chromatography.
All chromatography was performed with an Äkta fast-protein liquid chromatography system from Amersham Biosciences (Piscataway, N.J.). Prepacked Superdex Peptide HR 10/30 columns, which have an effective separation range of 100 to 7,000 Da and a molecular mass cutoff of approximately 20,000 Da, were used for all analyses. Peptides samples were prepared as described above and were then diluted to 1 mg/ml in phosphate-buffered saline (PBS; pH 7.4) and stored at −20°C. The column was preequilibrated with two column volumes of PBS, and 50 μl of peptide solution was then loaded and eluted with PBS at a rate of 0.5 ml/min. The peptides that eluted were detected by use of UV absorbance at 214 nm. When desired, eluted peptide was collected for further analysis in 0.25-ml fractions by using an automated fraction collector (Amersham) with microplate adaptor.
Assignment of monomer and dimer elution profiles.
To verify the elution profile of monomeric peptide 80-94, purified peptide 80-94 was reacted with a fivefold molar excess of iodoacetamide (catalog no. A3221; Sigma). After 1 h of incubation at room temperature there was no detectable free sulfhydryl by the Ellman reaction. The mixture was then diluted in PBS, and the peptide was separated from unreacted iodoacetamide by size-exclusion chromatography. The peptide component eluted in single peak at a 12.01-ml elution volume and was collected for analysis by sedimentation equilibrium centrifugation. Purified peptide dimers were isolated by oxidation of peptide 80-94 in 20% dimethyl sulfoxide (DMSO), followed by elution from the size-exclusion column. The putative dimer fraction (centered at an elution volume of 10.91 ml) was collected and analyzed by sedimentation equilibrium centrifugation.
RESULTS
Cysteine 83 is essential for peptide activity.
In order to determine which structural features of peptide 77-95 are necessary for inhibition of RSV, we first asked which amino acids of the peptide were required for its antiviral activity. We tested a series of altered 77-95 peptides in which an alanine residue was singly substituted for the native amino acid at each position (Table 1). In these experiments the IC50s of the unaltered peptide for virus growth were 25 μg/ml or higher, with a calculated IC50 of 7.6 μM. Individual substitution of 18 of the 19 amino acids had little effect on the observed peptide potency (less than 10-fold). The substitution of cysteine 83, however, resulted in a reduction in antiviral activity to below the level of detection of the assay (100 μg/ml; approximately 50 μM for the 77-95 series of peptides).
Residues 80 to 90 are essential.
We next determined the minimal linear sequence necessary for peptide activity by testing peptides truncated from the N and/or the C terminus. Table 1 shows the sequence of each peptide tested and its calculated antiviral potency. These experiments indicated that the amino acids outside of residues 80 to 90 are dispensable for antiviral activity. The majority of peptides whose sequences excluded this region also caused a nonviral CPE in the cell monolayers. This cellular toxicity was not attributable to the DMSO used as a solvent for some of the peptide stocks. At a final concentration of 1% (the amount present in 100 μg of peptide per ml), DMSO had no inhibitory effect on RSV replication and no visible effects on cell morphology. By a more sensitive assay, 1% DMSO was shown to have minimal effects on cellular viability (<20% decrease in the metabolism of MTT) over 48 to 72 h, with no effects at concentrations below 1% (data not shown). Of all the truncated peptides tested, peptides 78-95 and 80-94 showed the most potent activities, with IC50s of 2.3 and 2.1 μg/ml (1.23 and 1.75 μM), respectively.
The activity of crude 77-95 is reduced by purification.
The experiments described above and previously published experiments (21) describing the antiviral activity of peptide 77-95 were done with synthetic peptides that were approximately 70% pure by HPLC. When we attempted to validate the previous results using 77-95 peptides purified to ≥90% by reverse-phase RP-HPLC, we observed a decrease in potency of approximately fivefold (data not shown). These observations suggested the presence of one or more minor components in the crude peptide preparations that were more active at inhibiting RSV than purified peptide 77-95.
Late-eluting peptide aggregates are enriched for antiviral activity.
In order to isolate the most active product from a crude peptide preparation, a large-scale synthesis was performed, and the products of this synthesis were fractionated by reverse-phase HPLC. A slightly truncated sequence, residues 78 to 94, was used, since deletion of residues 77 and 95 had been shown to slightly improve peptide activity. The crude peptide preparation was eluted from a C18 column in 300 fractions, corresponding to 11 distinguishable peaks. Fractions corresponding to each peak were tested for antiviral activity, and those that contained active peptide product were then analyzed by mass spectrometry. As shown in Fig. 1A, the highest activity was found in late-eluting fractions (fractions 240 to 280) that appeared to contain aggregates of coeluting peptides. One of these fractions was analyzed by N-terminal sequencing and was found to contain relatively equal amounts of peptides 78-94, 79-94, and 80-94 (Fig. 1B). Of the fractions analyzed by mass spectrometry, only fraction 91 contained a product of a single mass, which corresponded to the Mw of a peptide spanning residues 80 to 94 of RhoA. Interestingly, this relatively pure fraction was the least active of those tested, suggesting that preformed heterotypic peptide aggregates are more potent inhibitors than purified peptide 80-94.
To determine the relative activity of each of the peptide components of fraction 250, peptides corresponding to products of each of the observed masses were synthesized, verified by mass spectrometry to be free from contaminating truncation products, and then tested for antiviral activity (Fig. 1C). Each of the individual peptides showed some level of antiviral activity at the highest concentrations tested, yet none of the individual peptides was as potent alone as the fraction in which it had coeluted. This further suggested that the formation of peptide aggregates leads to increased antiviral potency.
This hypothesis is further supported by the subsequent discovery that peptide 80-94 gains antiviral potency with increasing time in DMSO or aqueous solution. While performing the experiments described above, we noted that aliquots of both fraction 91 (Fig. 1D) and the individually synthesized peptide 80-94 (data not shown) exhibited increased antiviral activities when the initial stock solutions were retested. Due to the hydrophobic and acidic nature of the peptide, we had generally used either DMSO or 1% (wt/vol) ammonium bicarbonate buffer (pH 8.0) as solvents when making peptide stock solutions. Both of these solvents are conducive to disulfide bond formation between cysteine residues (1). This suggested that the observed increase in peptide potency was possibly due to formation of peptide dimers via intermolecular disulfide bonds.
Improvement in peptide 80-94 activity is dependent on cysteine 83.
To determine whether the observed increase in peptide activity was due to disulfide bond formation, we compared the antiviral properties of highly purified (>95% by HPLC) peptide 80-94 and a control peptide (peptide 83A) that differs from 80-94 only by the substitution of an alanine residue for the native cysteine residue at position 83. Lyophilized peptides were dissolved in bicarbonate buffer and then split into two aliquots. One aliquot was immediately placed at −20°C to prevent oxidation, while the other was left at room temperature overnight to allow air oxidation. The next day these aliquots were tested for antiviral activity.
As seen in previous assays, peptide 80-94 showed limited activity when it was not allowed to oxidize. However, after 20 h of air oxidation the activity of peptide 80-94 had increased approximately 16-fold (Fig. 2A). In contrast, peptide 83A showed very little activity before incubation and no detectable activity afterwards (Fig. 2B). Evaluation of the amount of free sulfhydryl in the aliquots tested showed that the fresh sample was approximately 20% oxidized, based on comparison with a fresh cysteine standard, while the sample left to air oxidize overnight was approximately 50% oxidized (data not shown).
FIG. 2.
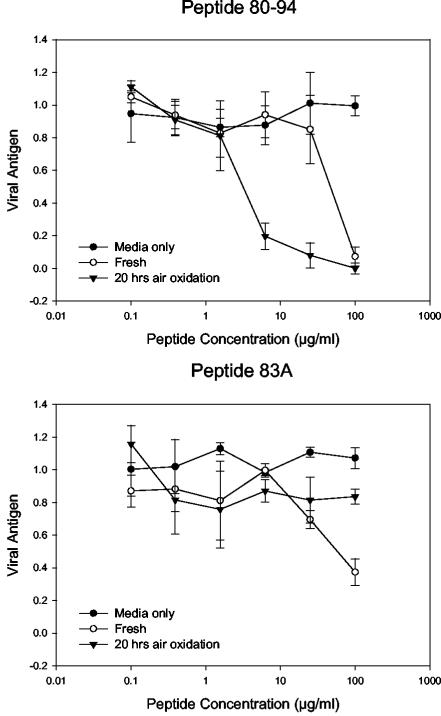
Improvement in the activity of peptide 80-94 is dependent on cysteine 83. Peptide 80-94 and a control peptide lacking a sulfhydryl moiety at position 83 (peptide 83A) were tested for antiviral activity under oxidizing and nonoxidizing conditions. The peptides were dissolved in bicarbonate buffer and either immediately diluted in PBS and frozen or left at room temperature to allow air oxidation, followed by dilution in PBS. Samples were then tested side by side for antiviral activity. Each datum point represents the mean for three replicate wells at each concentration, and error bars represent the standard deviations.
To obtain complete oxidation of peptide 80-94, we next tried incubation in 1% bicarbonate buffer (pH 8.0) containing 20% DMSO. Oxidation of cysteine-containing peptides by DMSO has been shown to occur over a wide pH range and to be very specific, with no tendency to induce oxidation of other nucleophilic amino acids, except under very acidic conditions (26). Incubation of peptide 80-94 in 20% DMSO resulted in oxidation of the peptide to >90% within 12.5 h of incubation at room temperature. As shown in Fig. 3, the extent of oxidation of the peptide correlated directly with its antiviral activity but had no effect on peptide toxicity. Peptide 83A, when incubated under the same conditions, showed no improvement in antiviral activity or increased toxicity (data not shown). These data indicate that the improved activity of peptide 80-94 after incubation in 20% DMSO is entirely dependent on the cysteine residue and is not due to chemical changes involving other residues of the peptide. Given the selectivity of DMSO in promoting disulfide bond formation, these data strongly suggest that the formation of peptide dimers is responsible for the observed increase in peptide potency. The viability data further indicate that the antiviral activity of the peptide is independent of its effect on cellular metabolism at high concentrations.
The multimeric state of peptide 80-94 varies with the extent of oxidation.
We next used gel filtration chromatography (GFC) and sedimentation equilibrium centrifugation to determine the molecular radii and molecular weights of peptides 80-94 and 83A (Fig. 4 and 5). GFC separates molecules on the basis of their shape and can be affected by properties of molecules that are independent of molecular weight, such as the three-dimensional structure of the molecule or its interaction with or repulsion from the gel matrix. Sedimentation equilibrium centrifugation, on the other hand, is affected only by the mass of the molecule studied, independent of its other properties (for a recent review, see reference 16). Figure 4 shows that the GFC profile for fresh or oxidized peptide 83A is predominantly homogeneous, with a very predominant peak at 11.7 to 11.8 ml and only very minor higher-molecular-weight peaks eluting at lower volumes. Sedimentation equilibrium measurement gave weight-average molar weights of 2,070 (fresh) and 1,954 (oxidation treatment) for peptide 83A (actual formula weight, 1,652). These results support a monomeric state for peptide 83A under reducing or oxidizing conditions. On the other hand, peptide 80-94 exists as a mixture of different multimeric states, depending on its oxidation state. The GFC pattern for freshly dissolved peptide 80-94 shows a monomeric elution peak unresolved from a leading shoulder presumed to represent peptide dimers (Fig. 4). Additional peaks in the 9-ml region can be seen, as can a flowthrough fraction. Upon oxidation, the monomer peak decreases and the presumptive dimer peak increases. This presumed dimer peak was purified by GFC and characterized by sedimentation equilibrium centrifugation. A weight-average molar weight of 3,510 was obtained (the actual formula weight of dimers should be 3,366), which supports the assignment of dimers to the GFC peak labeled as such in Fig. 4. Reduced and acetylated peptide 80-94 yielded a weight-average molar weight of 2,253, consistent with the stabilization of the monomeric state. In Fig. 5A we present the nonlinear regression fit of the absorbance versus the radial position for the dimer purified by GFC. Comparable nonlinear regression fits were also obtained for peptide 83A both fresh and under oxidizing conditions and acetylated peptide 80-94 (results not shown). We have provided a comparison of the plots of the natural log of the absorbance versus r2 (see Materials and Methods) for fresh peptide 83A, acetylated peptide 80-94, and oxidized peptide 80-94 (Fig. 5B). It is evident that the plots for fresh peptide 83A and acetylated peptide 80-94 have almost identical slopes and give values close to those for the monomer, whereas oxidized peptide 80-94 has a twofold greater slope and is dimeric.
FIG. 4.
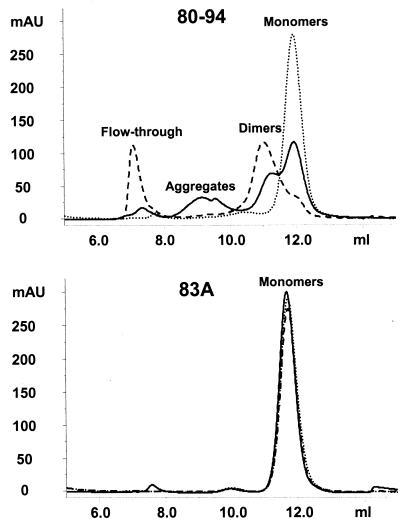
Size-exclusion chromatography of reduced and oxidized peptides. Peptides 80-94 and 83A were analyzed by size-exclusion chromatography with a prepacked Superdex Peptide HR 10/30 column (Amersham Biosciences). A total of 50 μl of peptide solution (0.5 mg/ml) was injected onto the column and eluted with PBS. Solid lines, peptide solutions with minimal oxidation times (diluted in PBS and frozen for <15 min after they were dissolved in oxidation buffer); dashed lines, peptides oxidized to ≥95% (<95% of the original free sulfhydryl content by the Ellman assay). Oxidized samples were also diluted in 50 mM DTT to reduce any disulfide bonds and were then applied to the column (dotted lines). Although some sample-to-sample variability in the amounts of peptides eluting at each point was seen, substantial dimer peaks were observed only in oxidized samples, and aggregates eluting at 9.0 ml were seen only in fresh, nonoxidized samples. mAU, milli-absorbance units
FIG. 5.
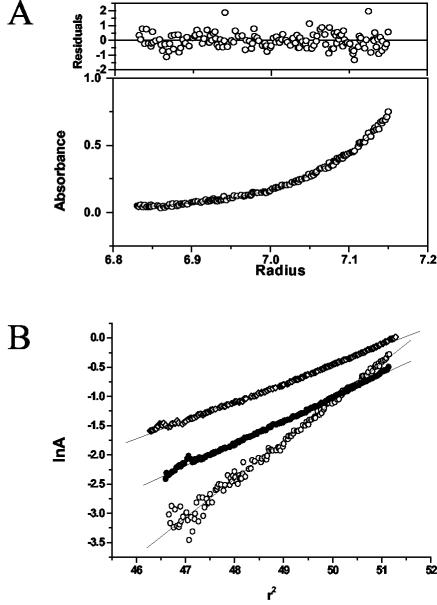
Analysis of peptides by sedimentation equilibrium centrifugation. (A) Sedimentation equilibrium absorbance data obtained at 202 nm versus radial position profile for the purified dimer fraction of peptide 80-94. The solid line shows the best fit obtained from weighted nonlinear regression modeling of the data. The weighted residuals of the fitted line to the experimental data are shown in the upper panel. (B) Plots of the natural log of the absorbance (lnA) versus r2 for fresh peptide 83A (open diamonds), acetylated peptide 80-94 (solid circles), and the purified dimer (open circles).
To further verify the interpretation of the oxidation state of peptide 80-94 described above, it was reduced with dithiothreitol (DTT) and reexamined (Fig. 4). Under these conditions the peptide eluted entirely as monomers. As expected, the DTT had no effect on the elution profile of 83A, confirming that the changes in molecular weight were dependent on the cysteine residue.
DISCUSSION
The inhibitory peptides studied here represent a novel class of entry inhibitors for paramyxoviruses. The mechanism of action of these peptides, however, remains to be elucidated. Previously described peptide inhibitors of RSV fall into two categories: heptad repeat peptides derived from F (15, 22) and heparin-binding peptides derived from G (6, 7). The latter are thought to inhibit binding of RSV to host cells by saturating cellular receptor sites, with the former blocking the conformational changes in F necessary to complete the entry process (24). It is unlikely that RhoA-derived peptides share a common mechanism with these peptides. The G peptides are predominantly basic (and the RhoA peptides are acidic), while the F heptad repeat peptides require a precise molecular fit (30) that the RhoA-derived peptides could not provide.
Because peptide 77-95 is derived from the region of RhoA with which F interacts and because the peptide can inhibit an in vitro interaction between F and RhoA (21), we expected that peptide 77-95 would mimic a surface-exposed region of RhoA. We were surprised to find that a single substitution at any of the positions of the peptide that correspond to surface-exposed residues of RhoA (11, 17, 19, 23, 25, 28) had little effect on the ability of the peptide to inhibit RSV (Fig. 6). Indeed, the only single substitution that caused more than a 10-fold loss in activity was the substitution of cysteine 83, a residue deeply buried in the RhoA structure. The fact that antiviral activity can be achieved by a peptide as short as 11 amino acids and the additional observation that intermolecular disulfide bond formation increases the activity of peptide 80-94 strongly suggest that the most active form of this peptide has little resemblance to the structure of the protein from which it was derived. In this light, it seems unlikely that the antiviral activities of peptides derived from the region from residues 77 to 95 of RhoA relate to a highly specific protein-protein interaction between F and RhoA. An alternate explanation for peptide inhibition of RSV entry would be the ability of polyanionic peptide dimers to disrupt F or G binding to cellular glycosaminoglycans or other receptors, based on charge-charge interactions. Experiments are under way to test this hypothesis.
FIG. 6.
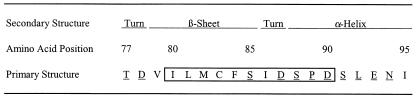
Structural properties of residues 77 to 95 of RhoA. The one-letter amino acid code of RhoA aa 77 to 95 is presented with secondary structure assignments based on several published crystal structures of RhoA (see text for references). Residues with side chains that are surface exposed in the crystal structures are underlined. The boxed region indicates the minimum peptide sequence necessary for RSV inhibition.
In many assays with both crude and highly purified peptides, dimers of peptide 80-94 consistently showed high levels of antiviral activity. The calculated IC50s of oxidized peptide 80-94 were in the range of 3.0 to 3.3 μg/ml. This would correspond to a dimer concentration of approximately 0.9 μM. Purified peptide 77-95, on the other hand, had a calculated IC50 in the range of 25 to 50 μM (data not shown). This value is considerably higher than those published previously (21). The reason for this discrepancy may be multifactorial. First, the peptides used previously were not purified by HPLC. Thus, both truncated synthesis products and preformed peptide aggregates may have been present. We have shown here that either of these impurities can increase the activity of a peptide solution. Second, the oxidation state of the peptide 77-95 preparation used in previous studies is unknown. In the present studies involving purified peptide 77-95, care was taken to avoid conditions conducive to oxidation (freezing-thawing or extended incubation at room temperature), which likely resulted in higher observed IC50s than would have been seen with oxidized peptide. Finally, differences in assay conditions and techniques may have been responsible for some of the observed variation.
Despite these differences, we show here that antiviral activity comparable to that reported previously (21) can be achieved by highly purified RhoA-derived peptides. This activity is dependent, however, on the formation of peptide dimers via disulfide bonds. It has been shown with polyanionic polysaccharides (29) and synthetic polyanionic inhibitors of enveloped viruses (2) that the antiviral activities of these compounds increase with increasing molecular weight and with an increasing number of negatively charged residues. The antiviral activities of RhoA-derived peptides appear to follow the same pattern. Peptide 80-94, which is capable of forming dimers and aggregates of higher molecular weight, can achieve relatively potent antiviral activity. However, a nearly identical peptide lacking only the sulfhydryl group necessary for disulfide bond formation does not readily form aggregates and does not inhibit RSV entry. These observations suggest that RhoA-derived peptides may act in a manner similar to those in which other polyanionic inhibitors of RSV and other enveloped viruses act (18, 22). Determination of whether the mechanism of RhoA-derived peptide inhibition of RSV is equivalent to or separate from the mechanisms of other polyanionic inhibitors will shed new light on the process of RSV entry and the ways in which it can be inhibited.
Acknowledgments
We thank Judy Beeler for the F-specific monoclonal antibodies and for much helpful advice and Lynne Crim, Susette Audet, and Manoj Pastey for discussion and review of the manuscript. We also thank Nga Ngyuen and her staff for helpful consultation and peptide synthesis, and we thank Jan Lukszo and Carl Hammer for service and advice regarding peptide chemistry and analysis.
This work was supported in part by NIH grant R01-AI-33933.
REFERENCES
- 1.Andreu, D., F. Albericio, N. A. Sole, M. C. Munson, M. Ferrer, and G. Barany. 1994. Formation of disulfide bonds in synthetic peptides and proteins. Methods Mol. Biol. 35:91-169. [DOI] [PubMed] [Google Scholar]
- 2.Baba, M., D. Schols, E. Declerco, R. Pauwels, M. Nagy, J. Gyorgyiedelenyi, M. Low, and S. Gorog. 1990. Novel sulfated polymers as highly potent and selective inhibitors of human immunodeficiency virus-replication and giant-cell formation. Antimicrob. Agents Chemother. 34:134-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bourgeois, C., J. B. Bour, K. Lidholt, C. Gauthray, and P. Pothier. 1998. Heparin-like structures on respiratory syncytial virus are involved in its infectivity in vitro. J. Virol. 72:7221-7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins, P. L., R. M. Chanock, and B. R. Murphy. 2001. Respiratory syncytial virus, p. 1443-1485. In P. M. Howley and D. M. Knipe (ed.), Fields virology, vol. 1, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 5.Feldman, S. A., S. Audet, and J. A. Beeler. 2000. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 74:6442-6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feldman, S. A., R. M. Hendry, and J. A. Beeler. 1999. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J. Virol. 73:6610-6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorman, J. J., J. L. McKimm-Breschkin, R. S. Norton, and K. J. Barnham. 2001. Antiviral activity and structural characteristics of the nonglycosylated central subdomain of human respiratory syncytial virus attachment (G) glycoprotein. J. Biol. Chem. 276:38988-38994. [DOI] [PubMed] [Google Scholar]
- 8.Graham, B. S., M. D. Perkins, P. F. Wright, and D. T. Karzon. 1988. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 26:153-162. [DOI] [PubMed] [Google Scholar]
- 9.Hallak, L. K., P. L. Collins, W. Knudson, and M. E. Peeples. 2000. Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology 271:264-275. [DOI] [PubMed] [Google Scholar]
- 10.Heminway, B. R., Y. Yu, Y. Tanaka, K. G. Perrine, E. Gustafson, J. M. Bernstein, and M. S. Galinski. 1994. Analysis of respiratory syncytial virus F, G, and SH proteins in cell fusion. Virology 200:801-805. [DOI] [PubMed] [Google Scholar]
- 11.Ihara, K., S. Muraguchi, M. Kato, T. Shimizu, M. Shirakawa, S. Kuroda, K. Kaibuchi, and T. Hakoshima. 1998. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J. Biol. Chem. 273:9656-9666. [DOI] [PubMed] [Google Scholar]
- 12.Ikeda, S., J. Neyts, S. Verma, A. Wickramasinghe, P. Mohan, and E. De Clercq. 1994. In vitro and in vivo inhibition of ortho- and paramyxovirus infections by a new class of sulfonic acid polymers interacting with virus-cell binding and/or fusion. Antimicrob. Agents Chemother. 38:256-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karron, R. A., D. A. Buonagurio, A. F. Georgiu, S. S. Whitehead, J. E. Adamus, M. L. Clements-Mann, D. O. Harris, V. B. Randolph, S. A. Udem, B. R. Murphy, and M. S. Sidhu. 1997. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. USA 94:13961-13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krusat, T., and H. J. Streckert. 1997. Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch. Virol. 142:1247-1254. [DOI] [PubMed] [Google Scholar]
- 15.Lambert, D. M., S. Barney, A. L. Lambert, K. Guthrie, R. Medinas, D. E. Davis, T. Bucy, J. Erickson, G. Merutka, and S. R. Petteway, Jr. 1996. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA 93:2186-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lebowitz, J., M. S. Lewis, and P. Schuck. 2002. Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11:2067-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longenecker, K., P. Read, U. Derewenda, Z. Dauter, X. Liu, S. Garrard, L. Walker, A. V. Somlyo, R. K. Nakamoto, A. P. Somlyo, and Z. S. Derewenda. 1999. How RhoGDI binds Rho. Acta Crystallogr. D Biol. Crystallogr. 55(Pt 9):1503-1515. [DOI] [PubMed] [Google Scholar]
- 18.Luscher-Mattli, M. 2000. Polyanions—a lost chance in the fight against HIV and other virus diseases? Antivir. Chem. Chemother. 11:249-259. [DOI] [PubMed] [Google Scholar]
- 19.Maesaki, R., K. Ihara, T. Shimizu, S. Kuroda, K. Kaibuchi, and T. Hakoshima. 1999. The structural basis of Rho effector recognition revealed by the crystal structure of human RhoA complexed with the effector domain of PKN/PRK1. Mol. Cell 4:793-803. [DOI] [PubMed] [Google Scholar]
- 20.Pastey, M. K., J. E. Crowe, Jr., and B. S. Graham. 1999. RhoA interacts with the fusion glycoprotein of respiratory syncytial virus and facilitates virus-induced syncytium formation. J. Virol. 73:7262-7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pastey, M. K., T. L. Gower, P. W. Spearman, J. E. Crowe, Jr., and B. S. Graham. 2000. A RhoA-derived peptide inhibits syncytium formation induced by respiratory syncytial virus and parainfluenza virus type 3. Nat. Med. 6:35-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prince, G. A. 2001. An update on respiratory syncytial virus antiviral agents. Expert Opin. Investig. Drugs 10:297-308. [DOI] [PubMed] [Google Scholar]
- 23.Rittinger, K., P. A. Walker, J. F. Eccleston, S. J. Smerdon, and S. J. Gamblin. 1997. Structure at 1.65 A of RhoA and its GTPase-activating protein in complex with a transition-state analogue. Nature 389:758-762. [DOI] [PubMed] [Google Scholar]
- 24.Russell, C. J., T. S. Jardetzky, and R. A. Lamb. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024-4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimizu, T., K. Ihara, R. Maesaki, S. Kuroda, K. Kaibuchi, and T. Hakoshima. 2000. An open conformation of switch I revealed by the crystal structure of a Mg2+-free form of RHOA complexed with GDP. Implications for the GDP/GTP exchange mechanism. J. Biol. Chem. 275:18311-18317. [DOI] [PubMed] [Google Scholar]
- 26.Tam, J. P., C. R. Wu, W. Liu, and J. W. Zhang. 1991. Disulfide bond formation in peptides by dimethyl-sulfoxide—scope and applications. J. Am. Chem. Soc. 113:6657-6662. [Google Scholar]
- 27.Techaarpornkul, S., N. Barretto, and M. E. Peeples. 2001. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J. Virol. 75:6825-6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei, Y., Y. Zhang, U. Derewenda, X. Liu, W. Minor, R. K. Nakamoto, A. V. Somlyo, A. P. Somlyo, and Z. S. Derewenda. 1997. Crystal structure of RhoA-GDP and its functional implications. Nat. Struct. Biol. 4:699-703. [DOI] [PubMed] [Google Scholar]
- 29.Witvrouw, M., and E. DeClercq. 1997. Sulfated polysaccharides extracted from sea algae as potential antiviral drugs. Gen. Pharmacol. 29:497-511. [DOI] [PubMed] [Google Scholar]
- 30.Zhao, X., M. Singh, V. N. Malashkevich, and P. S. Kim. 2000. Structural characterization of the human respiratory syncytial virus fusion protein core. Proc. Natl. Acad. Sci. USA 97:14172-14177. [DOI] [PMC free article] [PubMed] [Google Scholar]