

Abstract
Human immunodeficiency viruses in 321 samples from tenofovir-naïve patients were retrospectively evaluated for resistance to this nucleotide analogue. All virus strains with insertions between amino acids 67 and 70 of the reverse transcriptase (n = 6) were highly resistant. Virus strains with the Q151M mutation were divided into susceptible (n = 12) and highly resistant (n = 8) viruses. This difference was due to the absence or presence of the K65R mutation, which was confirmed by site-directed mutagenesis. Viral clones with various combinations of the mutations M41L, K70R, L210W, and T215F or T215Y were analyzed for cross-resistance induced by thymidine analogue mutations (TAMs). The levels of increased resistance induced by single, double, and triple mutations at the indicated positions could be ranked as follows: for mutants with single mutations, mutations at positions 41 > 215 > 70; for mutants with double mutations, mutations at positions 41 and 215 > 70 and 215 = 210 and 215 > 41 and 70; for mutants with triple mutations, mutations at positions 41, 210, and 215 > 41, 70, and 215. Viral clones with M184V or M184I exhibited slightly increased susceptibilities to tenofovir (0.7-fold). Almost all clones with TAM-induced resistance were resensitized when M184V was present (P < 0.001). Among the viruses in the clinical samples, the rate of tenofovir resistance significantly increased with the number of TAMs both in the samples with 184M and in those with 184V (P = 0.005 and P = 0.003, respectively). A resensitizing effect of M184V was confirmed for all samples exhibiting at least one TAM (P = 0.03). However, accumulation of at least two TAMs resulted in more than 2.0-fold reduced susceptibility to tenofovir, irrespective of the presence of M184V. Decision tree building, a classical machine learning technique, was used to generate models for the interpretation of mutations with respect to tenofovir resistance. The application of previously proposed cutoffs for a reduced response to therapy and treatment failure demonstrated the central roles of positions 215 and 65 for 1.5- and 4.0-fold reduced susceptibilities, respectively. Thus, clinically relevant resistance may be conferred by the accumulation of TAMs, and the resensitizing effect of M184V should be considered only minor.
The arsenal of antiretroviral therapy has recently been enlarged by tenofovir, an acyclic nucleotide analogue, which has proven to be active against human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) in vitro (1, 14, 16), in SIV-infected animals (22, 24), and in clinical studies (8, 19). Its oral prodrug, tenofovir disoproxil fumarate, is rapidly converted to tenofovir after cell membrane penetration (13), and its long intracellular half-life makes once-daily dosing possible (15). Only two phosphorylation steps by cellular enzymes are required, after which the drug acts as a chain termination inhibitor of the reverse transcriptase (RT). Since this mechanism is similar to those of nucleoside RT inhibitors (NRTIs), cross-resistance to these drugs has been suspected. However, the data which have been generated so far do not argue for broad cross-resistance.
In vitro experiments (20, 25) and the administration of tenofovir to SIV-infected rhesus macaques (24) have shown the emergence of the K65R mutation, which confers three- to fourfold reduced susceptibility to tenofovir. Furthermore, tenofovir resistance has been observed for multiple dideoxynucleoside-resistant viruses with insertions between RT amino acids 67 and 70, while tenofovir was still active in the presence of Q151M (10). K65R as well as insertions have only rarely been observed in clinical isolates (5). Likewise, resistance to tenofovir was detected at only a low frequency in isolates from treatment-experienced patients, with the greatest degree of cross-resistance observed in the context of reduced susceptibility to thymidine analogues (5). Furthermore, the M184V mutation was described to confer increased susceptibility to tenofovir (25) and to reduce the level of K65R-induced resistance (28). In the presence of thymidine analogue mutations (TAMs), M184V was shown to increase the susceptibility of viruses to tenofovir, at least in part (5); however, the clinical relevance of this effect is still unclear (8). Recently, 4.0-fold reduced susceptibility to tenofovir has been defined as the cutoff for the failure of therapy (8). More recent data suggest that the efficacy of tenofovir can be reduced at even lower levels of resistance (7).
This study was performed to focus on three aspects of tenofovir resistance: (i) to characterize the resistance profile of multiple dideoxynucleoside-resistant viral strains, (ii) to analyze TAM-induced cross-resistance and the degree of resensitization by M184V, and (iii) to develop computational models to predict tenofovir resistance from the genotypes by using the recently suggested clinically relevant cutoffs for a bioinformatics approach which has previously been described for other antiretroviral drugs (2, 3).
(This study was presented in part at the 11th International HIV Drug Resistance Workshop Basic Principles and Clinical Implications, Seville, Spain, July 2002 [K. Wolf, H. Walter, T. Schnell, W. Keulen, N. Beerenwinkel, J. Selbig, A.-M. Vandamme, K. Korn, and B. Schmidt, Abstr. 11th Int. HIV Drug Resist. Workshop Basic Principles Clin. Implications, abstr. 20, 2002].)
MATERIALS AND METHODS
Selection of samples.
HIV strains in 321 samples from 294 patients were retrospectively analyzed for resistance to tenofovir. These were selected from among the viruses in samples which had been sent to the German National Reference Centre for Retroviruses for drug resistance testing between January 1998 and July 2002. Virus selection was based only on the availability of genotypic data and a corresponding recombinant virus stock, although there was a special focus on samples with multiple dideoxynucleoside-resistant strains. Samples were obtained from patients treated at more than 20 outpatient centers and clinical practices in Germany. Treatment histories were available for 133 patients. Except for four seroconverters, all patients had been pretreated with an NRTI: 1, 40, 15, 27, 35, and 11 patients had received one, two, three, four, five, and six NRTIs, respectively. None of them had previously received tenofovir.
Resistance testing.
Phenotypic resistance testing was performed by a recombinant virus assay as described previously (26). In brief, the complete protease gene and the first 900 bp of the RT gene were amplified from patient plasma and cloned into a matched deletion mutant of pNL4-3. Transient transfection of 293T cells resulted in recombinant viral stocks, which were analyzed on a CEMx174-derived cell line containing the gene for the secreted alkaline phosphatase (SEAP) under the control of the SIV long terminal repeat (9). Tenofovir resistance was determined in triplicate by measuring secreted alkaline phosphatase activity after 3 days by using six tenofovir concentrations ranging from 0.45 to 150 μM. The fold change in susceptibility was calculated by dividing the 50% inhibitory concentration (IC50) for the respective recombinant virus by the IC50 for nonresistant reference strain NL4-3, which was included in each independent assay. The fold reductions in the susceptibilities of viruses resistant to multiple dideoxynucleosides as well as the viral clones were determined in at least three independent runs. Genotypic analysis was performed by population sequencing of the amplification product described above by using dye terminators (Amersham, Cleveland, Ohio). The sequences were aligned by using the Wisconsin package (version 10.0; Genetics Computer Group, Madison, Wis.). The detection limit for minority species was about 30%. Resistance-associated mutations were analyzed as described by Schinazi et al. (17).
Mutagenesis.
Viral clones were constructed by site-directed mutagenesis (6). Viral clones were constructed by insertion of the following mutations into the HXB2 backbone: M41L; K70R; M184I; M184V; T215F; T215Y; M41L and K70R; M41L and M184V; M41L and T215F; M41L and T215Y; K70R and M184V; K70R and T215Y; M184V and T215Y; M41L, K70R, and T215Y; M41L, M184V, and T215Y; K70R, M184V, and T215Y; and M41L, K70R, M184V, and T215Y. Construction of viral clones with mutations within the NL4-3 backbone (mutations M184V; M184V and T215Y; L210W and T215Y; M184V, L210W, and T215Y; M41L, L210W, R211K, and T215Y; and M41L, M184V, L210W, R211K, and T215Y) was described previously (27). For the mutagenesis of positions 62 and 65 of the RT, the following sense (s) and antisense (as) primers were used: A65A-K65K-s (5′-CCAGTATTTGCCATAAAGAAAAAAAATA-3′), A62A-K65K-as (5′-ATTTTTTTTCTTTATGGCAAATACTGGA-3′), A62V-K65K-s (5′-CCAGTATTTGTAATAAAGAAAAAAAA-3′), A62V-K65K-as (5′-TTTTTTTCTTTATTACAAATACTGGA-3′), A62A-K65R-s (5′-CCAGTATTTGCCATAAAGAGAAAAAA-3′), and A62A-K65R-as (5′-TTTTTCTCTTTATGGCAAATACTGGA-3′).
Bioinformatics analysis.
The DNA sequences were aligned with the pol gene of reference strain HXB2CG (GenBank accession number K03455) and translated into amino acids. One attribute was defined for each of the first 250 amino acids of the RT, with the attributes classified as an amino acid, a deletion, or unknown for ambiguous sequence information or a lack of sequence information. Additionally, one binary attribute was defined for the occurrence of an insertion. The viruses in the sample set were divided into susceptible and resistant subgroups according to the two phenotypic cutoffs which have recently been proposed as clinically relevant for a reduced response to therapy and treatment failure (1.5- and 4.0-fold, respectively) (7, 8). Mutual information profiles were created. These profiles quantify the information content (in bits) of each sequence position for the discrimination between tenofovir-susceptible and -resistant strains. Decision trees were generated by recursively splitting the data set according to the amino acid position with the largest amount of normalized mutual information plus the ratio between the amount of mutual information and its average amount over all RT sequence positions and by repeating this splitting for the respective subsets. Pruning was used to avoid overfitting.
Statistical analysis.
For statistical evaluation of the data, Fisher's exact test and the Wilcoxon rank-sum test were applied as appropriate. Linear regression analysis was used to investigate the relationship between the number of TAMs and the median fold change in susceptibility. Significant values in the mutual information profiles were calculated by using 5,000 permutations for each position, which allowed the detection of significant differences from background at a 95% confidence level with Bonferroni's correction for multiple comparisons (see the legend to Fig. 2). The expected prediction error of the decision trees for unseen sequences was estimated by the leave-one-out method, a common cross-validation technique (3).
FIG. 2.
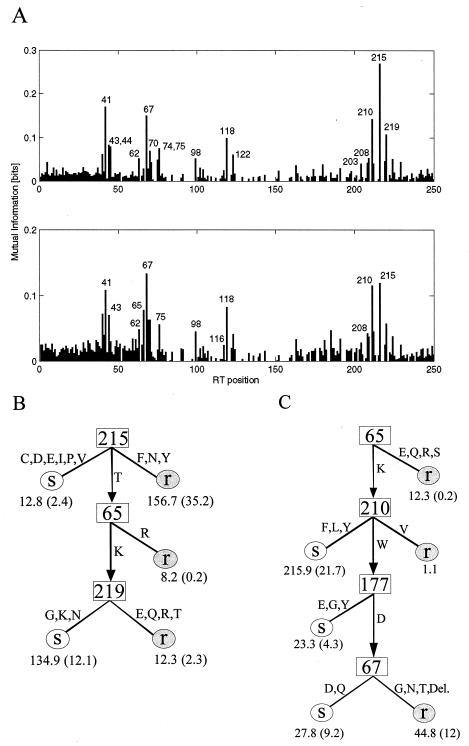
Mutual information profiles and decision tree models for tenofovir resistance obtained by using the two cutoffs for which clinical significance has been suggested (7, 8). (A) Mutual information profiles for 1.5- and 4.0-fold reduced susceptibility (upper and lower panels, respectively). The leftmost bar (position 0) on the x axis corresponds to the attribute indicating an insertion; positions 1 through 250 represent the first 250 amino acid positions of the HIV type 1 RT. The y axis gives the mutual information (in bits), which represents the information content of each sequence position that can be used to discriminate between tenofovir-susceptible and -resistant viruses. All amino acid positions with an information content significantly different from the background level are labeled. (B) Decision tree for 1.5-fold reduced susceptibility, which requires two branches with at least four viruses, resulting in a leave-one-out error of 19.1%. (C) Decision tree for 4.0-fold reduced susceptibility, which requires two branches with at least seven viruses, resulting in a leave-one-out error of 22.2%. In panels B and C, each number below an “S” (susceptible) or “r” (resistant) indicates the number of viruses classified along this path and the number in parentheses indicates the number of misclassifications. Fractional numbers are due to sequence heterogeneities resulting in more than one amino acid at the respective position.
RESULTS
Determination of drug susceptibility of reference wild-type virus.
The mean tenofovir IC50 for susceptible reference clone NL4-3 was determined to be 4.75 ± 2.2 μM in 15 consecutive independent runs of the phenotypic assay. This value is very similar to those reported previously (12, 25, 28).
Tenofovir resistance in multiple-dideoxynucleoside-resistant viruses.
Among the 321 clinical samples analyzed, the viruses in 27 displayed a pattern of multiple dideoxynucleoside resistance. Of these, six samples contained viruses with an insertion of two amino acids (SV and SS in three viruses each) between RT positions 67 and 70, one sample contained a virus with a deletion of one amino acid in this region (M41L, deletion of amino acid 67, T69G, K70R, L74I, A98G, V108I, M184V, L214F, T215F, K219E), and 19 isolates displayed the Q151M mutation pattern (18). The virus in one sample contained a deletion, in addition to the Q151M complex (A62V, S68G, deletion of amino acid 69, mixture of 75V and 75I, F77L, Q151M, M184V, R211K, L214F). All viruses with insertions were resistant to tenofovir, with a median reduction in susceptibility of 16.8-fold (range, 10.0- to 19.3-fold). The isolate exhibiting a deletion in the context of the Q151M complex was susceptible to tenofovir (1.7-fold), whereas the isolate displaying the deletion without Q151M showed a 6.3-fold reduced susceptibility to tenofovir. The group of viruses with the Q151M complex could be divided into two subsets: the viruses from 12 samples showed a median reduction in susceptibility to tenofovir of 2.0-fold (range, 0.9- to 3.3-fold), whereas the viruses from 8 samples were highly resistant, with a median reduction in susceptibility of 13.5-fold (range, 9.3- to 17.0-fold). These subsets differed in the presence of K65R: none of the 12 susceptible viruses but 7 of 8 resistant viruses exhibited K65R (P < 0.0001, Fisher's exact test). Since the second subset contained five and two consecutive samples from two patients, respectively, statistics were recalculated by including the data for only one sample from each patient. The difference between the subsets was still significant (P < 0.05, Fisher's exact test).
Clonal analysis of K65R in the presence of Q151M.
Since all tenofovir-resistant viruses carrying the Q151M mutation also exhibited mutations at position 62 (V in viruses from six samples, F in the virus from one sample, and P in the virus from one sample), in addition to K65R, we performed a clonal analysis to confirm the role of K65R and to verify the contribution of a mutation at position 62. An individual clone was isolated from the plasmid preparation of one of the five consecutive samples described above which had been generated during phenotypic resistance testing (the RT profile was A62V, K65R, D67N, S68K, T69N, V75I, F77L, K103N, V108I, Y115F, F116Y, Q151M, I178L, M184V, R211K, L214F, K219E). Mutations K65R and A62V were consecutively removed from this clone by site-directed mutagenesis (Fig. 1). The clones with K65R were highly resistant to tenofovir, irrespective of the presence of A62V, whereas the clones with the wild-type sequence at position 65 were susceptible. Resistance could thus be attributed to the presence of K65R.
FIG. 1.
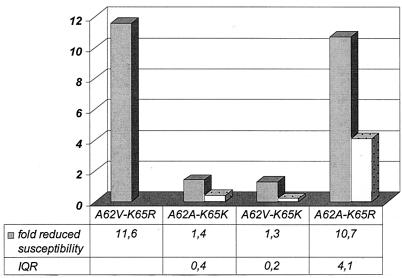
Tenofovir resistance of a viral clone derived from a patient sample (with RT mutations A62V, K65R, D67N, S68K, T69N, V75I, F77L, K103N, V108I, Y115F, F116Y, Q151 M, I178L, M184V, R211K, L214F, and K219E) and three clones whose sequences are identical at all positions except positions 62 and 65. Data are given as the medians of at least three independent runs for each phenotypic resistance test. IQR, interquartile range (the interquartile range was not available for mutant A62V-K65R).
Clonal analysis of TAMs in the presence and absence of M184V or M184I.
To determine the role of TAMs in tenofovir resistance, clones that contained one, two, or three of TAMs were analyzed (Table 1). Among the mutants with single mutations, those with M41L displayed the highest levels of tenofovir resistance, followed by those with the T215Y or T215F and K70R mutations. The mutants with double mutations showed a similar ranking (M41L and T215Y > M41L and T215F > L210W and T215Y = K70R and T215Y > M41L and K70R). The mutant containing the triple mutations M41L, K70R, and T215Y showed a 3.2-fold resistance, whereas the clone containing the triple mutations M41L, L210W, and T215Y was highly resistant (6.0-fold). Viruses with the M184V or M184I mutation alone exhibited slightly increased levels of susceptibility to tenofovir (0.6- and 0.8-fold, respectively). If M184V was introduced into a TAM background, all but one clone with tenofovir resistance was resensitized (P < 0.0001, Wilcoxon rank sum test) (Table 1). It is noteworthy that the resistant virus with the M41L, L210W, and T215Y mutations was not completely resensitized when the M184V mutation was present (6.1- versus 1.9-fold). Next, the resensitizing effect of M184V on TAM-induced tenofovir resistance was evaluated by using the data derived from the viruses from clinical samples.
TABLE 1.
Tenofovir resistance of individual clones in the context of TAMs in the absence or presence of M184Va
| TAM(s) | Median fold reduced susceptibility to tenofovir (IQR)b | nc | Mutation or additional mutation | Median fold reduced susceptibility to tenofovir (IQR) | n |
|---|---|---|---|---|---|
| M184I | 0.8 (0.2) | 3 | |||
| M184V | 0.6 (0.4) | 8 | |||
| M41L | 2.8 (2.1) | 3 | +M184V | 0.8 (0.4) | 3 |
| K70R | 1.1 (0.5) | 3 | +M184V | 1.6 (1.1) | 3 |
| T215F | 1.9 (0.2) | 3 | |||
| T215Y | 1.7 (0.3) | 3 | +M184V | 1.0 (0.3) | 5 |
| M41L + K70R | 0.8 (0.2) | 3 | |||
| M41L + T215F | 2.3 (0.6) | 3 | |||
| M41L + T215Y | 2.9 (0.5) | 3 | +M184V | 1.3 (0.4) | 2d |
| K70R + T215Y | 1.8 (1.3) | 3 | +M184V | 0.8 (0.2) | 3 |
| L210W + T215Y | 1.8 (2.0) | 3 | +M184V | 0.5 (0.0) | 2d |
| M41L + K70R + T215Y | 3.2 (0.2) | 3 | +M184V | 1.0 (0.5) | 3 |
| M41L + L210W + T215Ye | 6.0 (4.1) | 11 | +M184V | 1.9 (1.4) | 10 |
| Any of thesef | 2.4 (1.8) | 41 | +M184V | 0.9 (0.8) | 42 |
Mutations were inserted into the pNL4-3 or HXB2 backbone (see Materials and Methods). Tenofovir susceptibilities were equivalent for two clones (clones with the M184V mutation and the M184V and T215Y mutations) which were available within the NL4-3 and HxB2 background.
Fold increase in the IC50 for the respective recombinant virus compared to the IC50 for susceptible reference virus NL4-3. IQR, interquartile range.
n, number of independent runs of phenotypic resistance testing.
Drug resistance was determined in only two independent runs due to poorly replicating recombinant viruses.
In the context of R211K.
P < 0.0001.
Analysis of TAMs in the presence and absence of M184V in viruses from clinical samples.
The viruses from the clinical samples were evaluated for the number of TAMs, defined as the mutations M41L; D67N; K70R; L210W; T215Y, T215F, or T215C; or K219Q, K219E, or K219R. First, the median fold reduced susceptibility to tenofovir significantly increased with the number of TAMs both in the group with 184M and in the group with 184V (P = 0.005 and 0.003, respectively) (Table 2). Next, the resensitizing effect of M184V was evaluated. When all viruses exhibiting at least one TAM were compared, the level of resistance to tenofovir was significantly lower for viruses with 184V than for viruses with 184M (P = 0.03). A reduction in the level of tenofovir resistance was present in almost all subsets of viruses with different numbers of TAMs and M184V; however, due to small differences and small sample numbers, the differences between the subsets were not statistically significant. The average number of TAMs (3.2 versus 3.3) as well as the frequency of individual TAMs and TAM combinations was comparable in viruses with and without M184V.
TABLE 2.
Tenofovir resistance of clinical isolates in the context of TAMs in the absence or presence of M184Va
| No. of TAM(s)b | Median fold reduced susceptibility to tenofovir (IQR)c | nd | Additional mutation | Median fold reduced susceptibility to tenofovir (IQR) | n |
|---|---|---|---|---|---|
| 0 | 0.9 (0.6) | 87 | +M184V | 0.9 (0.9) | 26 |
| 1 | 1.7 (3.7) | 6 | +M184V | 1.1 (0.7) | 7 |
| 2 | 2.4 (2.9) | 15 | +M184V | 2.5 (9.5) | 11 |
| 3 | 3.6 (3.5) | 23 | +M184V | 2.4 (2.2) | 15 |
| 4 | 3.8 (3.3) | 24 | +M184V | 3.6 (3.8) | 17 |
| 5 | 7.1 (5.7) | 8 | +M184V | 3.7 (3.6) | 12 |
| 6 | 12.0 (4.0) | 2 | +M184V | 3.4 (0) | 2 |
| 1-6e | 3.8 (3.6) | 78 | +M184V | 2.9 (3.9) | 64 |
Tenofovir resistance increased concomitantly with the number of TAMs, which proved to be significant both in the group with 184M and in the group with 184V (P = 0.005 and 0.003, respectively).
TAMs were considered M41L; D67N; K70R; L210W; T215Y, T215F, or T215C; and K219Q, K219E, or K219R. All samples with mixed populations at these sites and/or at position 184 were excluded from the analysis. IQR interquartile range.
Data are given as the medians for all samples displaying the respective number of TAMs.
n, number of samples in which the isolates displayed the respective number of TAMs.
P = 0.03.
Bioinformatics approach to prediction of tenofovir resistance from genotypes.
A total of 40.4, 34.3, and 25.3% of the viruses had less than 1.5-fold, 1.5- to 4.0-fold, and more than 4.0-fold reduced susceptibilities to tenofovir, respectively. In the mutual information profile for 1.5-fold reduced susceptibility, TAMs at positions 41, 67, 210, 215, and 219 as well as position 118 showed the highest peaks (Fig. 2A, upper panel). The relevance of position 65 increased for 4.0-fold reduced susceptibility (Fig. 2A, lower panel). As described in the Materials and Methods section, decision trees were generated from the mutual information profiles for these two cutoffs. By varying the number of samples which were required for splitting of the data set into different branches, two trees with the lowest leave-one-out error (19.1 and 22.2%, respectively) were selected (Fig. 2B and C). For the prediction of >1.5-fold reduced susceptibility, position 215 was the most important, followed by position 65 in those samples with the wild-type sequence at position 215 and then position 219 for samples with the wild-type sequence at position 65 as well. In the decision tree for the prediction of >4.0-fold reduced susceptibility, the first split is according to the amino acid at position 65, followed by those at positions 210, 177, and 67. Position 184 showed up in only one of the decision trees, requiring two branches with at least six viruses: viruses with 184M were classified as resistant, whereas those with 184V were predicted to be susceptible (data not shown). Interestingly, the tree for 4.0-fold reduced susceptibility picked up a new resensitizing effect, namely, reversal of L210W-induced resistance by 177E, 177G, or 177Y. Site-directed mutagenesis should be performed to further evaluate whether this is a true mechanism of resensitization or if the substitutions at position 177 are just compensatory changes associated with M184V. The decision tree models can be used for phenotype predictions as part of geno2pheno (version 2.1; http://www.genafor.org/). This service is freely available via the Internet and provides further services such as quantitative predictions of resistance.
DISCUSSION
The aim of this study was to characterize the tenofovir resistance profiles of NRTI-resistant viruses. One aspect focused on the susceptibility of viral strains resistant to multiple dideoxynucleosides. Previous data showing tenofovir resistance in viruses with insertions between RT amino acids 67 and 70 (10) could be confirmed. The same group reported that tenofovir resistance occurred only rarely in viruses carrying Q151M (5). We could clarify by statistical and clonal analyses that K65R also has a central role in this context, thus describing another mutational pattern leading to high-level tenofovir resistance. A similar observation was recently presented in another context (23). Interestingly, K65R was frequently found in viruses with the Q151M mutation (35%) but in less than 1% of the remaining clinical isolates. K65R has been reported to reduce replication capacity and RT processivity (28), but this may be different for viruses with Q151M. Furthermore, K65R seems more likely to develop with thymidine analogue-sparing regimens in the absence of TAMs (4, 21, 29); however, in our study three of four patients who harbored viruses with K65R and whose drug histories were available had initially been treated with zidovudine plus another NRTI and only one patient had received dideoxycytidine.
However, these highly resistant variants are rare even in populations of patients who have been heavily treated in the past. Therefore, frequent mutational patterns conferring lower levels of tenofovir resistance may have a much greater influence on the success of tenofovir therapy in patients previously treated with an NRTI. An analysis of more than 5,000 clinical viruses (5) revealed that only 12, 4, and 1% exhibited more than 3.0-, 5.0-, and 10.0-fold resistance to tenofovir, respectively; and thus, it was argued that the degree of cross-resistance to tenofovir is low. The corresponding values for our data set are higher (34, 21, and 7%, respectively). The high prevalence of viruses with >10.0-fold resistance is caused by the preselection of viruses resistant to multiple dideoxynucleosides. The higher frequency of viruses with 4.0- to 10.0-fold resistance may be explained by the extensive treatment with NRTIs that our clinical cohort had previously received; i.e., more than half of the patients had received four or more NRTIs. Furthermore, resistance factors may not be directly comparable between different phenotypic platforms, although the IC50 for our nonresistant reference virus was very similar to those in three previous studies (12, 25, 28), and clones with the M184V mutation showed the same extent of hypersusceptibility determined in our assay (10, 12, 25).
In addition, previous data and the present data should be interpreted in light of the clinically relevant cutoffs for tenofovir resistance. Thus, a durable treatment response with a decline in the plasma viral load of more than 0.5 log10 copies/ml was observed in a cohort of HIV-infected patients for more than 48 weeks if <4.0-fold resistance to tenofovir was present at the baseline, whereas tenofovir therapy failed if >4.0-fold resistance was present (8). However, another recent study showed that the response to tenofovir was impaired even with >1.5-fold resistance (7), a cutoff which interferes with the interassay variabilities of phenotypic assays. In consideration of these cutoffs, the level of clinically relevant cross-resistance to tenofovir may be broader than was originally thought, in particular in patients previously heavily treated with NRTIs. This may be a reason for the lower than expected level of reduction of the viral load with tenofovir treatment in clinical studies (19). However, other mechanisms that could contribute to the durability of the antiretroviral response should be considered, such as less efficient removal of tenofovir by pyrophosphorolysis and nucleotide-dependent chain terminator removal (11).
Our data allow some interesting conclusions about TAM-induced tenofovir resistance and the clinical role of M184V-induced resensitization to be made. First, the level of tenofovir resistance increased gradually with the number of TAMs. Without the M184V mutation, the median resistance of the clinical samples rose to >4.0-fold for viruses with five or six TAMs, whereas one clone with only three TAMs (M41L, L210W, and T215Y) also reached this threshold. Thus, not all TAMs are of equal importance for tenofovir resistance, which should be evaluated in further studies. Concerning the effect of M184V in NRTI-treated patients, recent data showed a more extensive viral load reduction in patients carrying viruses with M184V (8). After the study had been completed, it became obvious that the same effect was observed with tenofovir and placebo, so the authors concluded that the occurrence of M184V indicated good compliance rather than a resensitizing effect (8). Our study adds some information to this point of interest. First, data on the tenofovir hypersusceptibilities of viral clones carrying M184V could be confirmed. Second, data from clonal studies also support the fact that M184V resensitizes clones with TAM-induced resistance (5), although the resistance of viruses with the M41L, L210W, and T215Y mutations was only partially reversed to a level which may still be clinically relevant (Table 1). Third, stratified analysis of clinical isolates showed significantly lower levels of tenofovir resistance in viruses carrying TAMs with 184V than in viruses carrying TAMs with 184M (Table 2) but not in viruses with two TAMs. This subgroup contained four virus isolates from one patient with two TAMs (D67N and K219E) plus M184V, but also K65R and Q151M, which explains the higher median level of resistance. Overall, the resensitizing effect of M184V seemed to be less pronounced in the clinical isolates than in the clones. This was further supported by the fact that M184V did not play an important role in the decision trees. A possible explanation may be the accumulation of additional mutations such as those indicated in the mutual information profiles, e.g., V118I. Additionally, population sequencing of clinical isolates showed mutations from a pool of viruses, and this approach possibly failed to detect minorities of resistant variants.
The decision trees confirmed the central role of mutations at position 215 and 65 for 1.5- and 4.0-fold reduced susceptibilities to tenofovir, respectively. Importantly, these data were derived from viruses from tenofovir-naïve patients, thus describing cross-resistance rather than resistance to tenofovir in tenofovir-treated patients. Since the K65R mutation is rare in clinical samples, the bulk of cross-resistance to tenofovir appears to be due to TAMs. When the in vitro data and clinical data for tenofovir are combined, the resistance-response relationship appears to be more continuous, ranging from full response to an intermediate response to no response. The phenotypic cutoffs of 1.5- and 4.0-fold approximately define these response zones. A similar type of continuous response pattern has been observed for abacavir and will likely hold true for other antiretrovirals, which should be evaluated in further clinical studies.
Acknowledgments
We thank B. Fleckenstein for continuous support; K. van Laethem and M. D. Miller for helpful discussions; and G. Moschik, C. Paatz, T. Schnell, E. Schwingel, and C. Thein for excellent technical assistance. The indicator cell line was kindly provided by R. E. Means and R. C. Desrosiers. We are grateful to all physicians and their patients for providing samples for resistance testing. Tenofovir was kindly provided by Gilead Sciences.
Financial support for this study was obtained through grants from the Bayerische Staatsministerium für Kultus, Erziehung und Wissenschaft (to K. Korn), the Robert Koch-Institute, Berlin (National Reference Centre for Retroviruses), the Deutsche Forschungsgemeinschaft (to D. Hoffmann, R. Kaiser, and J. Selbig), and the Federal Ministry of Education and Research (HIV Competence Network grant AZ 01 KI 0211). This work was supported in part by the AIDS Reference Laboratory of Leuven, Belgium, which received funding from the Belgian Ministry of Social Affairs through the Health Insurance System.
REFERENCES
- 1.Balzarini, J., A. Holy, J. Jindrich, L. Naesens, R. Snoeck, D. Schols, and E. De Clercq. 1993. Differential antiherpesvirus and antiretrovirus effects of the (S) and (R) enantiomers of acyclic nucleoside phosphonates: potent and selective in vitro and in vivo antiretrovirus activities of (R)-9-(2-phosphonomethoxypropyl)-2,6-diaminopurine. Antimicrob. Agents Chemother. 37:332-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beerenwinkel, N., B. Schmidt, H. Walter, R. Kaiser, T. Lengauer, D. Hoffmann, K. Korn, and J. Selbig. 2001. Geno2pheno: interpreting genotypic HIV drug resistance tests. IEEE Intellig. Syst. 16:35-41. [Google Scholar]
- 3.Beerenwinkel, N., B. Schmidt, H. Walter, H., R. Kaiser, T. Lengauer, D. Hoffmann, K. Korn, and J. Selbig. 2002. Diversity and complexity of HIV-1 drug resistance: a bioinformatics approach to predicting phenotype from genotype. Proc. Natl. Acad. Sci. USA 99:8271-8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gu, Z., Q. Gao, H. Fang, H. Salomon, M. A. Parniak, E. Goldberg, J. Cameron, and M. A. Wainberg. 1994. Identification of a mutation at codon 65 in the IKKK motif of reverse transcriptase that encodes human immunodeficiency virus resistance to 2′,3′-dideoxycytidine and 2′,3′-dideoxy-3′-thiacytidine. Antimicrob. Agents Chemother. 38:275-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrigan, P. R., M. D. Miller, P. McKenna, Z. L. Brumme, and B. A. Larder. 2002. Phenotypic susceptibilities to tenofovir in a large panel of clinically derived human immunodeficiency virus type 1 isolates. Antimicrob. Agents Chemother. 46:1067-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho, S. N., H. D. Hunt, R. M. Horton, J. P. Pullen, and L. R. Pease. 1989. Site directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51-59. [DOI] [PubMed] [Google Scholar]
- 7.Lu, B., N. S. Hellmann, M. Bates, K. Dawson, J. Rooney, and M. D. Miller. 2002. Determination of clinical cut-offs for reduced response to tenofovir DF therapy in antiretroviral-experienced patients. Antivir. Ther. 7:S104. [Google Scholar]
- 8.Margot, N. A., E. Isaacson, I. McGowan, A. K. Cheng, R. T. Schooley, and M. D. Miller. 2002. Genotypic and phenotypic analyses of HIV-1 in antiretroviral-experienced patients treated with tenofovir DF. AIDS 16:1227-1235. [DOI] [PubMed] [Google Scholar]
- 9.Means, R. E., T. Greenough, and R. C. Desrosiers. 1997. Neutralization sensitivity of cell culture-passaged simian immunodeficiency virus. J. Virol. 69:5431-5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller, M. D., N. A. Margot, K. Hertogs, B. Larder, and V. Miller. 2001. Antiviral activity of tenofovir (PMPA) against nucleoside-resistant clinical HIV samples. Nucleosides Nucleotides Nucleic Acids 20:1025-1028. [DOI] [PubMed] [Google Scholar]
- 11.Naeger, L. K., N. A. Margot, and M. D. Miller. 2002. ATP-dependent removal of nucleoside reverse transcriptase inhibitors by human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 46:2179-2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naeger, L. K., N. A. Margot, and M. D. Miller. 2001. Increased drug susceptibility of HIV-1 reverse transcriptase mutants containing M184V and zidovudine-associated mutations: analysis of enzyme processivity, chain-terminator removal and viral replication. Antivir. Ther. 6:115-126. [PubMed] [Google Scholar]
- 13.Naesens, L., N. Bischofberger, P. Augustijns, P. Annaert, G. Van Den Mooter, M. N. Arimilli, C. U. Kim, and E. De Clercq. 1998. Antiretroviral efficacy and pharmacokinetics of oral bis(isopropyloxymethyl)-9(2-phosphonylmethoxypropyl) adenine in mice. Antimicrob. Agents Chemother. 42:1568-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer, S., N. Margot, H. Gilbert, N. Shaw, R. Buckheit, Jr., and M. Miller. 2001. Tenofovir, adefovir, and zidovudine susceptibilities of primary human immunodeficiency virus type 1 isolates with non-B subtypes or nucleoside resistance. AIDS Res. Hum. Retrovir. 17:1167-1173. [DOI] [PubMed] [Google Scholar]
- 15.Robbins, B. L., R. V. Srinivas, C. Kim, N. Bischofberger, and A. Fridland. 1998. Anti-human immunodeficiency virus activity and cellular metabolism of a potential prodrug of the acyclic nucleoside phosphonate 9-R-(2-phosphonomethoxypropyl) adenine (PMPA), bis(isopropyloxymethylcarboyl) PMPA. Antimicrob. Agents Chemother. 42:612-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenwirth, B., P. Ten Haaft, W. M. J. M. Bogers, I. G. Nieuwenhuis, H. Niphuis, E.-M. Kuhn, N. Bischofberger, J. L. Heeney, and K. Uberla. 2000. Antiretroviral therapy during primary immunodeficiency virus infection can induce persistent suppression of virus load and protection from heterologous challenge in rhesus macaques. J. Virol. 74:1704-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schinazi, R. F., B. Larder, and J. W. Mellors. 2000. Mutations in retroviral genes associated with drug resistance: 2000-2001 update. Int. Antivir. News 8:65-92. [Google Scholar]
- 18.Schmit, J. C., J. Cogniaux, P. Hermans, D. Van Vaeck, S. Sprecher, B. Van Remoortel, M. Witvrouw, J. Balzarini, J. Desmyter, E. De Clercq, and A. M. Vandamme. 1996. Multiple drug resistance to nucleoside analogues and nonnucleoside reverse transcriptase inhibitors in an efficiently replicating human immunodeficiency virus type 1 patient strain. J. Infect. Dis. 174:962-968. [DOI] [PubMed] [Google Scholar]
- 19.Schooley, R. T., R. Ruane, R. A. Myers, G. Beall, H. Lampiris, D. Berger, S. S. Chen, M. D. Miller, E. Isaacson, A. K. Cheng, and Study 902 Team. 2002. Tenofovir DF in highly antiretroviral-experienced patients: results from a 48-week, randomized, double-blind study. AIDS 16:1257-1263. [DOI] [PubMed] [Google Scholar]
- 20.Srinivas, R. V., and A. Fridland. 1998. Antiviral activities of 9-R-2-phosphonomethoxypropyl adenine (PMPA) and bis(isopropyloxymethylcarbonyl) PMPA against various drug-resistant human immunodeficiency virus strains. Antimicrob. Agents Chemother. 42:1484-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tisdale, M., T. Alnadaf, and C. Cousens. 1997. Combination of mutations in human immunodeficiency virus type 1 reverse transcriptase required for resistance to the carbocyclic nucleoside 1592U89. Antimicrob. Agents Chemother. 41:1094-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai, C. C., K. E. Follis, A. Sabo, T. W. Beck, R. F. Grant, N. Bischofberger, R. E. Benveniste, and R. Black. 1995. Prevention of SIV infection in macaques by (R)-9-(2-phosphonomethoxypropyl)adenine. Science 270:1197-1199. [DOI] [PubMed] [Google Scholar]
- 23.Van Laethem, K., M. Witvrouw, C. Pannecouque, B. Van Remoortel, R. Esnouf, J. Balzarini, J. Desmyter, E. De Clerq, and A.-M. Vandamme. 2000. Phenotypic multi-nucleoside resistance linked to the Q151M mutation is enhanced under the selective pressure of acyclic nucleotide analogues but reduced by foscarnet. AIDS 14(Suppl. 4):S118. [Google Scholar]
- 24.Van Rompay, K. K., J. M. Cherrington, M. L. Marthas, C. J. Berardi, A. S. Mulato, A. Spinner, R. P. Tarara, D. R. Canfield, S. Telm, N. Bischofberger, and N. C. Pedersen. 1996. 9-[2-(Phosphonomethoxy)propyl]adenine therapy of established simian immunodeficiency virus infection in infant rhesus macaques. Antimicrob. Agents Chemother. 40:2586-2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wainberg, M. A., M. D. Miller, Y. Quan, H. Salomon, A. S. Mulato, P. D. Lamy, N. A. Margot, K. E. Anton, and J. M. Cherrington. 1999. In vitro selection and characterization of HIV-1 with reduced susceptibility to PMPA. Antivir. Ther. 4:87-94. [DOI] [PubMed] [Google Scholar]
- 26.Walter, H., B. Schmidt, K. Korn, A. M. Vandamme, T. Harrer, and K. Überla. 1999. Rapid, phenotypic HIV-1 drug sensitivity assay for protease and reverse transcriptase inhibitors. J. Clin. Virol. 13:71-80. [DOI] [PubMed] [Google Scholar]
- 27.Walter, H., B. Schmidt, M. Werwein, E. Schwingel, and K. Korn. 2002. Prediction of abacavir resistance from genotypic data: impact of zidovudine and lamivudine resistance in vitro and in vivo. Antimicrob. Agents Chemother. 46:89-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White, K. L., N. A. Margot, T. W. Wrin, C. J. Petropoulos, M. D. Miller, and L. K. Naeger. 2002. Molecular mechanisms of resistance to human immunodeficiency virus type 1 with reverse transcriptase mutations K65R and K65R+M184V and their effects on enzyme function and viral replication capacity. Antimicrob. Agents Chemother. 46:3437-3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang, D., A. M. Caliendo, J. J. Eron, K. M. DeVore, J. C. Kaplan, M. S. Hirsch, and R. T. D'Aquila. 1994. Resistance to 2′,3′-dideoxycytidine conferred by a mutation in codon 65 of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 38:282-287. [DOI] [PMC free article] [PubMed] [Google Scholar]