

Abstract
The inhibitory effect of chloramphenicol on human cytochrome P450 (CYP) isoforms was evaluated with human liver microsomes and cDNA-expressed CYPs. Chloramphenicol had a potent inhibitory effect on CYP2C19-catalyzed S-mephytoin 4′-hydroxylation and CYP3A4-catalyzed midazolam 1-hydroxylation, with apparent 50% inhibitory concentrations (inhibitory constant [Ki] values are shown in parentheses) of 32.0 (7.7) and 48.1 (10.6) μM, respectively. Chloramphenicol also weakly inhibited CYP2D6, with an apparent 50% inhibitory concentration (Ki) of 375.9 (75.8) μM. The mechanism of the drug interaction reported between chloramphenicol and phenytoin, which results in the elevation of plasma phenytoin concentrations, is clinically assumed to result from the inhibition of CYP2C9 by chloramphenicol. However, using human liver microsomes and cDNA-expressed CYPs, we showed this interaction arises from the inhibition of CYP2C19- not CYP2C9-catalyzed phenytoin metabolism. In conclusion, inhibition of CYP2C19 and CYP3A4 is the probable mechanism by which chloramphenicol decreases the clearance of coadministered drugs, which manifests as a drug interaction with chloramphenicol.
Chloramphenicol, a broad-spectrum antibiotic, has been used to treat severe infections for several decades. Its use in contemporary medical practice has fallen out of favor due to dose-dependent and idiosyncratic bone marrow suppression (9). However, chloramphenicol showed good in vitro activity against many vancomycin-resistant enterococcal infections and is useful for certain life-threatening infections (22, 30).
Chloramphenicol increased the plasma drug concentrations or effects of several drugs, such as cyclosporine, tacrolimus, and phenytoin (4, 10, 14, 17, 20, 25, 29, 32, 36, 38).
Cytochrome P450s (CYPs) play a key role in the oxidation of numerous endogenous and exogenous compounds (24, 38). In previous animal studies, in vivo treatment with chloramphenicol has been shown to inhibit CYP-catalyzed reactions in microsomal preparations in rats, suggesting that the drug interactions of chloramphenicol possibly involve the CYPs (2, 8, 15).
However, there are no published studies investigating the effect of chloramphenicol on different CYP isoforms in human liver microsomes. Therefore, we studied the inhibitory effect of chloramphenicol on the activities of the major CYP isoforms in human liver microsomes and cDNA-expressed CYPs, using selective marker reactions to clarify the mechanism underlying the drug-drug interactions of chloramphenicol.
MATERIALS AND METHODS
Materials.
Chloramphenicol, phenytoin, 5-(p-hydroxyphenyl)-5-phenylhydantoin (HPPH), phenacetin, acetaminophen, chlorzoxazone, paclitaxel, dextromethorphan, dextrorphan, furafylline, NADP, EDTA, MgCl2, glucose-6-phosphate (G-6-P), and G-6-P dehydrogenase (G-6-PDH) were purchased from Sigma-Aldrich (St. Louis, Mo.). 1-Hydroxymidazolam, S-warfarin, 7-hydroxywarfarin, S-mephenytoin, 6-hydroxypaclitaxel, and 4′-hydroxymephenytoin were obtained from Ultrafine Chemical Co. (Manchester, United Kingdom). Acetonitrile and methanol were acquired from Fisher Scientific Co. (Pittsburgh, Pa.). Midazolam was kindly provided by Bukwang Pharmaceutical Co. (Seoul, Korea). All other reagents and chemicals used were of analytical or high-performance liquid chromatography (HPLC) grade. Human liver microsomes containing specific content of CYPs from donors (HG-43, HG-56, and HG-89) were obtained from Gentest Corp. (Woburn, Mass.). Human cDNA-expressed recombinant CYPs (1A2, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4) were also purchased from Gentest Corp.
Enzyme assay studies.
All incubations were performed in duplicate, and the mean values were used. We measured the activities of CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 by using specific reaction probe drugs. The Km and Vmax values were determined in human liver microsomes for the CYP isoform-specific reactions used, for which the incubation conditions and method references are listed in Table 1.
TABLE 1.
CYP isoform-specific probe drug reactions
| CYP | Index reaction | Final microsomal concn (mg/ml) | Incubation time (min) | Km (μM)a | Vmax (pmol/min/mg protein)b | Reference |
|---|---|---|---|---|---|---|
| CYP1A2 | Phenacetin O-deethylation | 2 | 25 | 35 | 879 | 27 |
| CYP2C9 | S-Warfarin 7-hydroxylation | 0.25 | 15 | 9.7 | 14.5 | 41 |
| CYP2C19 | S-Mephenytoin 4′-hydroxylation | 1 | 30 | 87 | 295 | 13 |
| CYP2D6 | Dextromethorphan O-demethylation | 0.5 | 30 | 11 | 255 | 1 |
| CYP2C8 | Paclitaxel 6α-hydroxylation | 1 | 20 | 25 | 400 | 40 |
| CYP3A4 | Midazolam 1-hydroxylation | 0.25 | 5 | 12 | 550 | 23 |
| CYP2E1 | Chlorzoxazone 6-hydroxylation | 0.4 | 25 | 85 | 960 | 19 |
Vmax, the maximum velocity of metabolite formation.
Km, Michaelis-Menten constant.
We used here the free base form of chloramphenicol, which is microbiologically active. A 25 mM chloramphenicol stock solution was prepared by dissolving chloramphenicol in distilled water, and this solution was serially diluted in distilled water. The reaction metabolite of the probe drug for each CYP was quantified by interpolating the peak area ratios of the respective metabolite and the internal standard from a standard curve of known metabolite concentrations.
Each incubation was performed with cDNA-expressed CYPs or human liver microsomes in a final incubation volume of 0.25 ml, after they were diluted from their original stock concentrations (final concentrations, 0.25 to 1 mg/ml for microsomes and 20 to 40 pmol/liter for cDNA-expressed CYPs). The incubation medium contained 100 mM phosphate buffer (pH 7.4) containing an NADPH-generating system (including 1.3 mM NADP, 3.3 mM G-6-P, 3.3 mM MgCl2, and 1.0 U of G-6-PDH per ml). For each substrate (probe drug), preliminary experiments were performed to determine whether metabolite formation was linear with respect to incubation time and microsomal protein concentrations. After incubation at 37°C with the probe drugs and microsomes in the absence (control) and presence of the NADPH-generating system, reactions were stopped by placing the reactants on ice and adding the appropriate stop solution (described in the corresponding reference in Table 1).
CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation was a modification of the reaction described by Ko et al. (13). After S-mephenytoin (25 to 100 μM) was incubated with human liver microsomes (1 mg/ml) for 30 min at 37°C in the presence of the NADPH-generating system, the reaction was stopped by placing it on ice and adding 70 μl of perchloric acid. Carbamazepine was added as the internal standard. The mixture was centrifuged at 10,000 × g for 5 min at 4°C, and aliquots of the supernatant were injected onto an HPLC system (Lichrosorb RP18 [4.6 by 250 mm]; UV, 225 nm; 320:680 acetonitrile-1% acetic acid [pH 4.0] at 0.7 ml/min).
CYP3A4-catalyzed midazolam 1-hydroxylation was performed by a modification of the method of Perloff et al. (23). After microsomes (0.25 mg/ml) were incubated for 30 min with midazolam (5 to 50 μM), the reaction was stopped with acetonitrile. Ketoconazole was added as the internal standard. Aliquots of the supernatant were injected onto an HPLC system for analysis (Capcell Pak C18 [1.5 by 250 mm]; UV, 245 nm; 265:45:690 acetonitrile-methanol-0.4 N acetate buffer [pH 4.0] at 0.12 ml/min).
CYP isoform-specific reactions for CYP1A2 (27), CYP2C8 (40), CYP2C9 (41), CYP2E1 (19), and CYP2D6 (1) are described in the corresponding references listed in Table 1.
To determine whether the inhibition of CYP isoform(s) by chloramphenicol is mechanism based, chloramphenicol was preincubated with the incubation medium at 37°C for 0 to 15 min, in either the presence or absence of the NADPH-generating system. After preincubation, the probe substrates were added either with or without the NADPH-generating system.
The HPLC system consisted of a Shiseido 2001 pump (Shiseido Co., Tokyo, Japan), a Shiseido model 2023 autosampler (Shiseido Co.), a dsChrom2000 integrator (Donam Instruments, Suwon, Korea), a Shiseido 2002 UV detector (Shiseido Co.), and a Jasco FP-2020 Plus fluorescence detector (Jasco Co., Tokyo, Japan).
Data analysis.
Fifty percent inhibitory concentrations (IC50s [concentration of the inhibitor required to cause 50% inhibition of the original enzyme activity]) were determined graphically by using curves of mean enzyme activity versus inhibitor concentrations. The apparent inhibitory constant (Ki) values were calculated by graphical analysis from secondary plots of slopes taken from double-reciprocal plots of chloramphenicol concentrations versus metabolite formation.
RESULTS
Inhibitory effect of chloramphenicol on CYP-catalyzed reactions.
Chloramphenicol strongly inhibited CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation and CYP3A4-catalyzed midazolam 1-hydroxylation in human liver microsomes, with apparent IC50s of 32.0 and 48.1 μM, respectively (Fig. 1). The Lineweaver-Burk plot, Dixon plots, and secondary reciprocal plots indicated that chloramphenicol inhibited CYP2C19 activity with a mixed-type inhibition pattern, with an apparent Ki value of 7.7 μM (Fig. 2 and Table 2). In the case of the CYP3A4-catalyzed reaction, chloramphenicol exhibited competitive inhibition with an apparent Ki value of 10.6 μM (Fig. 3 and Table 2). Chloramphenicol also showed competitive and moderate inhibition of the CYP2D6-catalyzed reaction with an IC50 (Ki) of 375.9 (75.8) μM, but did not inhibit or only minimally inhibited the other CYP-catalyzed reactions tested (Fig. 1). To evaluate whether chloramphenicol acts as a mechanism-based inhibitor of CYP-catalyzed reactions, we preincubated chloramphenicol with NADPH for 15 min prior to the addition of the CYP-specific substrates, but this did not increase the degree of inhibition (data not shown).
FIG. 1.
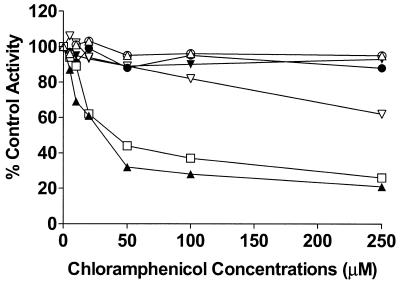
Inhibitory effect of chloramphenicol on CYP-catalyzed reactions in human liver microsomes. Effects of chloramphenicol (0 to 250 μM) on CYP1A2-catalyzed phenacetin O-deethylation (○), CYP2C8-catalyzed paclitaxel 6α-hydroxylation (•), CYP2C9-catalyzed S-warfarin 7-hydroxylation (▵), CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation (▴), CYP2D6-catalyzed dextromethorphan O-demethylation (▿), CYP2E1-catalyzed chlorzoxazone 6-hydroxylation (▾), and CYP3A4-catalyzed midazolam 1-hydroxylation (□). Each data point indicates the average value calculated from three different liver microsomal preparations.
FIG. 2.
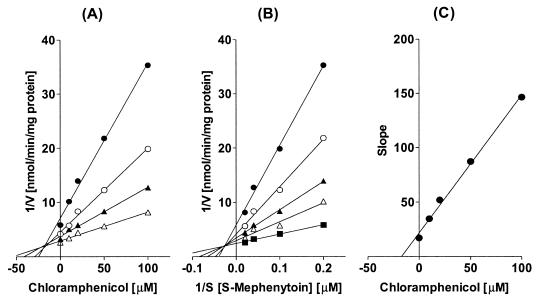
(A) Representative Dixon plot for the inhibition by chloramphenicol (0 to 100 μM) of CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation in human liver microsomes with 25 (•), 50 (○), 75 (▴), or 100 (▵) μM S-mephenytoin. (B) Lineweaver-Burk plot of CYP2C19-catalyzed S-mephenytoin 4′-hydroxylation in the absence (▪) or presence of 10 (▵), 20 (▴), 50 (○), or 100 (•) μM chloramphenicol. (C) Secondary plot of slopes taken from Lineweaver-Burk plots versus chloramphenicol concentration. Each data point represents the average of duplicate measurements.
TABLE 2.
Inhibition of CYP2C19, CYP2D6, and CYP3A4 isoforms by chloramphenicol and predicted in vivo inhibition of the metabolism of coadministered CYP2C19, CYP2D6, and CYP3A4 substrates by chloramphenicol, respectively, from in vitro data
| CYP isoform | IC50 (Ki) (μM)a | Type of inhibition | Predicted in vivo inhibition [I/(I+Ki)]b |
|---|---|---|---|
| CYP2C19 | 32.0 (7.7) | Mixed (α = 4.2) | 0.62-0.80 |
| CYP2D6 | 375.9 (75.8) | Competitive | 0.14-0.29 |
| CYP3A4 | 48.1 (10.6) | Competitive | 0.54-0.74 |
Ki values are derived from secondary plots of slopes taken from double-reciprocal plots versus chloramphenicol concentrations, and the IC50s were calculated graphically.
The inhibitor concentration (I) of chloramphenicol (therapeutic concentrations, 10 to 25 μg/ml [31 to 77 μM] and the fraction of drug unbound to protein are about 40% (see Discussion for details).
FIG. 3.
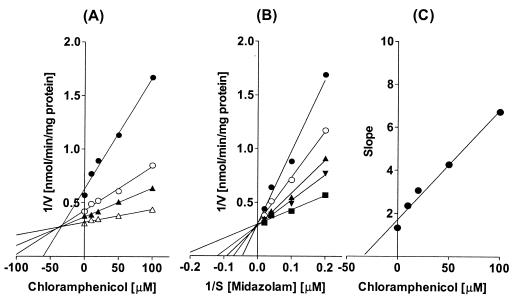
(A) Representative Dixon plot for the inhibition by chloramphenicol (0 to 100 μM) of CYP3A4-catalyzed midazolam 1-hydroxylation in human liver microsomes with 5 (•), 10 (○), 25 (▴), or 50 (▵) μM midazolam. (B) Lineweaver-Burk plot of CYP3A4-catalyzed midazolam 1-hydroxylation in the absence (▪) or presence of 10 (▵), 20 (▴), 50 (○), or 100 (•) μM chloramphenicol. (C) Secondary plot of slopes taken from Lineweaver-Burk plots versus chloramphenicol concentration. Each data point represents the average of duplicate measurements.
The mechanism of chloramphenicol-induced inhibition of phenytoin metabolism.
The inhibitory effect of chloramphenicol predominantly affected CYP2C19 and CYP3A4, but affected CYP2D6 only moderately. Therefore, we investigated the mechanism by which chloramphenicol interacts with phenytoin, which is metabolized mainly by CYP2C9 and to a lesser extent by CYP2C19 (3, 7, 35), leading to an increase in phenytoin levels in reported cases of the drug interaction. Chloramphenicol showed a weak inhibitory effect on phenytoin p-hydroxylation to HPPH, with an apparent IC50 of 619.7 μM (Fig. 4). Using cDNA-expressed CYPs, we confirmed that CYP2C9 and CYP2C19 resulted in HPPH metabolite formation at velocities of 29.5 ± 3.4 and 31.2 ± 5.2 pmol/min/pmol of CYP, respectively. However, little or no HPPH metabolite was formed from the other cDNA-expressed CYPs tested. When the inhibitory effect of chloramphenicol on CYP2C9- and CYP2C19-catalyzed phenytoin p-hydroxylation was evaluated, HPPH formation catalyzed by CYP2C9 was not inhibited, but the CYP2C19-catalyzed reaction was strongly and dose-dependently inhibited by chloramphenicol, suggesting that the drug interaction between phenytoin and chloramphenicol involves the inhibition of CYP2C19 (Fig. 4).
FIG. 4.
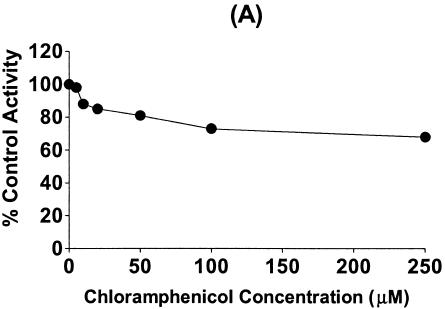
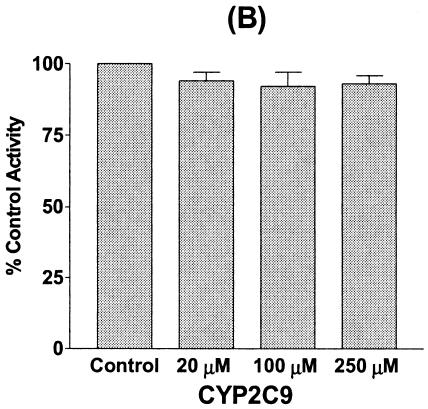
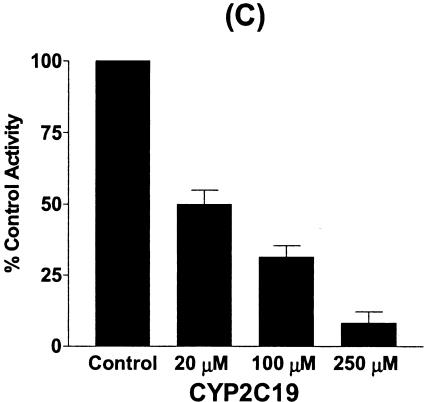
Inhibitory effect of chloramphenicol on phenytoin p-hydroxylation in human liver microsomes (A) and the inhibitory effect of chloramphenicol on HPPH formation from phenytoin (25 μM) in incubations with cDNA-expressed CYP2C9 (B) and CYP2C19 (C). Data are the averages of duplicate determinations.
DISCUSSION
Studies using rats or mice have shown that chloramphenicol is an inhibitor of CYP in liver microsomes (2, 24, 26). It has therefore been presumed a priori that chloramphenicol is an inhibitor of CYPs in humans. Furthermore, several drug interactions between chloramphenicol and coadministered drugs have supported this hypothesis (4, 10, 14, 17, 20, 25, 29, 32, 36, 38). However, little has been known of the inhibitory effect of chloramphenicol on human CYPs until now. In the present study, we confirmed that chloramphenicol has an inhibitory effect on CYP2C19, CYP3A4, and, to a lesser extent, CYP2D6 in human liver microsomes.
Chloramphenicol has been suggested to be effective in the treatment of vancomycin-resistant enterococcal infections in transplantation patients (22, 30). Calcineurin inhibitors, including cyclosporine and tacrolimus, interact with chloramphenicol when used in a combination therapy, causing an increase in their plasma drug concentrations and/or their toxicity (4, 20, 32, 36). Because both tacrolimus and cyclosporine are metabolized via CYP3A4 (16, 31, 37), the results of the present study indicate that the inhibition of CYP3A4-catalyzed reactions by chloramphenicol is the underlying mechanism by which the concentrations of both cyclosporine and tacrolimus in plasma are elevated by the administration of chloramphenicol.
In contrast, chloramphenicol is purported to be a CYP2C9 inhibitor, because chloramphenicol interacts with several drugs, such as phenytoin, warfarin, and phenobarbital (10, 14, 17, 25, 28, 38), which are mainly metabolized via CYP2C9 (7, 18, 21, 41). Interestingly, we observed no inhibitory effect of chloramphenicol on CYP2C9 in human liver microsomal preparations.
The CYP2C subfamily is the second-most-abundant CYP protein in the human liver following CYP3A (30%), representing about 20% of the total CYPs (34). CYP2C9 is clearly the most abundant CYP2C protein expressed, followed by CYP2C19 and CYP2C8 (28). Among the CYP2C isoforms tested, chloramphenicol only inhibited CYP2C19-catalyzed reaction. Phenytoin is metabolized by CYP2C9 and, to a lesser extent, by CYP2C19 (3, 7, 18, 35). Although CYP2C9 is mainly responsible for phenytoin metabolism, several drugs, such as tricyclic antidepressants, ticlopidine, omeprazole, and cimetidine, also influence concentrations of phenytoin in plasma through the inhibition of CYP2C19, but not through CYP2C9 inhibition (5, 6, 11). In the present study, chloramphenicol selectively inhibited CYP2C19-catalyzed reaction and not CYP2C9 in microsomal preparations. Furthermore, only CYP2C19-catalyzed phenytoin p-hydroxylation was inhibited by chloramphenicol. Taken together, these results indicate that chloramphenicol is an inhibitor of CYP2C19, and not of CYP2C9.
We found several reports of drug interactions between chloramphenicol and warfarin, but chloramphenicol did not inhibit S-warfarin 7-hydroxylation catalyzed by CYP2C9 in the present study. However, we cannot rule out the possibility that another isoform of warfarin (R-warfarin) interacts with chloramphenicol, because warfarin is a chiral drug composed of S and R forms (11). S-Warfarin is metabolized by CYP2C9, whereas R-warfarin is metabolized mainly by CYP1A2 and CYP3A4 (7, 11). Moreover, when we incubated a racemate of warfarin with chloramphenicol in human liver microsomes, we found chloramphenicol inhibited R-warfarin metabolism, whereas S-warfarin was not inhibited (data not shown). We have also found numerous case reports of interactions between warfarin and CYP3A4 inhibitors (12, 33, 42). Taken together, these data suggest that CYP3A4 inhibition by chloramphenicol may be the mechanism underlying the inhibition of warfarin metabolism, especially the inhibition of R-warfarin.
Theoretically, drug interactions based on the inhibition of hepatic metabolism, i, can be predicted by both the Ki value and the concentration of the inhibitor, [I], around the metabolic enzyme in the liver by using the following predictive model: i = [I]/([I] + Ki), assuming that the substrate concentration is much lower than its Km value (39). Based on the hypothesis that only the unbound forms of a drug in plasma are available for diffusion to intrahepatic regions of metabolic activity, the unbound concentration of an inhibitor in plasma has been used to predict in vivo drug interactions. Considering that therapeutic concentrations of chloramphenicol in plasma are 31 to 77 μmol/liter and protein binding is around 60%, we would expect approximately 54 to 80% inhibition of the clearance of CYP3A4 and CYP2C19 substrates by chloramphenicol (Table 2), suggesting that chloramphenicol is a potent inhibitor of CYP3A4 and CYP2C19 in vivo. Even though CYP2D6 was not potently inhibited by chloramphenicol, it also showed a 14 to 29% inhibition of clearance, suggesting that circumstances such as hypoalbuminemia and elevated concentrations of chloramphenicol and environmental factors (i.e., genetic polymorphism of CYP2D6), may affect the clearance of coadministered CYP2D6 substrates.
In conclusion, our in vitro study demonstrates that chloramphenicol at clinically relevant concentrations has a potent inhibitory effect on CYP2C19, CYP3A4, and, to a lesser extent, CYP2D6. The inhibition of CYP2C19 and CYP3A4 by chloramphenicol may explain the mechanism of the interactions between chloramphenicol and coadministered drugs.
REFERENCES
- 1.Abdel-Rahman, S. M., K. Marcucci, T. Boge, R. R. Gotschall, G. L. Kearns, and J. S. Leeder. 1999. Potent inhibition of cytochrome P-450 2D6-mediated dextromethorphan O-demethylation by terbinafine. Drug Metab. Dispos. 27:770-775. [PubMed] [Google Scholar]
- 2.Adams, H. R., E. L. Isaacson, and B. S. Masters. 1977. Inhibition of hepatic microsomal enzymes by chloramphenicol. J. Pharmacol. Exp. Ther. 203:388-396. [PubMed] [Google Scholar]
- 3.Bajpai, M., L. K. Roskos, D. D. Shen, and R. H. Levy. 1996. Roles of cytochrome P4502C9 and cytochrome P4502C19 in the stereoselective metabolism of phenytoin to its major metabolite. Drug Metab. Dispos. 24:1401-1403. [PubMed] [Google Scholar]
- 4.Bui, L., and D. D. Huang. 1999. Possible interaction between cyclosporine and chloramphenicol. Ann. Pharmacother. 33:252-253. [DOI] [PubMed] [Google Scholar]
- 5.Desta, Z., X. Zhao, J. G. Shin, and D. A. Flockhart. 2002. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin. Pharmacokinet. 41:913-958. [DOI] [PubMed] [Google Scholar]
- 6.Donahue, S., D. A. Flockhart, and D. R. Abernethy. 1999. Ticlopidine inhibits phenytoin clearance. Clin. Pharmacol. Ther. 66:563-568. [DOI] [PubMed] [Google Scholar]
- 7.Giancarlo, G. M., K. Venkatakrishnan, B. W. Granda, L. L. von Moltke, and D. J. Greenblatt. 2001. Relative contributions of CYP2C9 and 2C19 to phenytoin 4-hydroxylation in vitro: inhibition by sulfaphenazole, omeprazole, and ticlopidine. Eur. J. Clin. Pharmacol. 57:31-36. [DOI] [PubMed] [Google Scholar]
- 8.Halpert, J., B. Naslund, and I. Betner. 1983. Suicide inactivation of rat liver cytochrome P-450 by chloramphenicol in vivo and in vitro. Mol. Pharmacol. 23:445-452. [PubMed] [Google Scholar]
- 9.Holt, D., D. Harvey, and R. Hurley. 1993. Chloramphenicol toxicity. Adverse Drug React. Toxicol. Rev. 12:83-95. [PubMed] [Google Scholar]
- 10.Houghton, G. W., and A. Richens. 1975. Inhibition of phenytoin metabolism by other drugs used in epilepsy. Int. J. Clin. Pharmacol. Biopharm. 12:210-216. [PubMed] [Google Scholar]
- 11.Kaminsky, L. S., and Z. Y. Zhang. 1997. Human P450 metabolism of warfarin. Pharmacol. Ther. 73:67-74. [DOI] [PubMed] [Google Scholar]
- 12.Knoell, K. R., T. M. Young, and E. S. Cousins. 1998. Potential interaction involving warfarin and ritonavir. Ann. Pharmacother. 32:1299-1302. [DOI] [PubMed] [Google Scholar]
- 13.Ko, J. W., Z. Desta, N. V. Soukhova, T. Tracy, and D. A. Flockhart. 2000. In vitro inhibition of the cytochrome P450 (CYP450) system by the antiplatelet drug ticlopidine: potent effect on CYP2C19 and CYP2D6. Br. J. Clin. Pharmacol. 49:343-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koup, J. R., M. Gibaldi, P. McNamara, D. M. Hilligoss, W. A. Colburn, and E. Bruck. 1978. Interaction of chloramphenicol with phenytoin and phenobarbital. Case report. Clin. Pharmacol. Ther. 24:571-575. [DOI] [PubMed] [Google Scholar]
- 15.Kraner, J. C., E. T. Morgan, and J. R. Halpert. 1994. Selective suppression of rat hepatic cytochrome P450 2C11 by chloramphenicol. J. Pharmacol. Exp. Ther. 270:1367-1372. [PubMed] [Google Scholar]
- 16.Kronbach, T., V. Fischer, and U. A. Meyer. 1988. Cyclosporine metabolism in human liver: identification of a cytochrome P-450III gene family as the major cyclosporine-metabolizing enzyme explains interactions of cyclosporine with other drugs. Clin. Pharmacol. Ther. 43:630-635. [DOI] [PubMed] [Google Scholar]
- 17.Leone, R., E. Ghiotto, A. Conforti, and G. Velo. 1999. Potential interaction between warfarin and ocular chloramphenicol. Ann. Pharmacother. 33:114. [DOI] [PubMed] [Google Scholar]
- 18.Levy, R. H. 1995. Cytochrome P450 isozymes and antiepileptic drug interactions. Epilepsia 36:S8-S13. [DOI] [PubMed] [Google Scholar]
- 19.Lucas, D., J. F. Menez, and F. Berthou. 1996. Chlorzoxazone: an in vitro and in vivo substrate probe for liver CYP2E1. Methods Enzymol. 272:115-123. [DOI] [PubMed] [Google Scholar]
- 20.Mathis, A. S., N. Shah, G. T. Knipp, and G. S. Friedman. 2002. Interaction of chloramphenicol and the calcineurin inhibitors in renal transplant recipients. Transplant. Infect. Dis. 4:169-174. [DOI] [PubMed] [Google Scholar]
- 21.Miners, J. O., and D. J. Birkett. 1998. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. Br. J. Clin. Pharmacol. 45:525-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papanicolaou, G. A., B. R. Meyers, J. Meyers, M. H. Mendelson, W. Lou, S. Emre, P. Sheiner, and C. Miller. 1996. Nosocomial infections with vancomycin-resistant Enterococcus faecium in liver transplant recipients: risk factors for acquisition and mortality. Clin. Infect. Dis. 23:760-766. [DOI] [PubMed] [Google Scholar]
- 23.Perloff, M. D., L. L. von Moltke, M. H. Court, T. Kotegawa, R. I. Shader, and D. J. Greenblatt. 2000. Midazolam and triazolam biotransformation in mouse and human liver microsomes: relative contribution of CYP3A and CYP2C isoforms. J. Pharmacol. Exp. Ther. 292:618-628. [PubMed] [Google Scholar]
- 24.Porter, T. D., and M. J. Coon. 1991. Cytochrome P450. Multiplicity of isoforms, substrates, and catalytic regulatory mechanisms. J. Biol. Chem. 266:13469-13472. [PubMed] [Google Scholar]
- 25.Powell, D. A., M. C. Nahata, D. C. Durrell, J. P. Glazer, and M. D. Hilty. 1981. Interactions among chloramphenicol, phenytoin, and phenobarbital in a pediatric patient. J. Pediatr. 98:1001-1003. [DOI] [PubMed] [Google Scholar]
- 26.Rendic, S., and F. J. Di Carlo. 1997. Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 29:413-580. [DOI] [PubMed] [Google Scholar]
- 27.Rodrigues, A. D., B. W. Surber, Y. Yao, S. L. Wong, and E. M. Roberts. 1997. [O-Ethyl-14C]phenacetin O-deethylase activity in human liver microsomes. Drug Metab. Dispos. 25:1097-1100. [PubMed] [Google Scholar]
- 28.Rodrigues, A. D. 1999. Integrated cytochrome P450 reaction phenotyping: attempting to bridge the gap between cDNA-expressed cytochromes P450 and native human liver microsomes. Biochem. Pharmacol. 57:465-480. [DOI] [PubMed] [Google Scholar]
- 29.Rose, J. Q., H. K. Choi, J. J. Schentag, W. R. Kinkel, and W. J. Jusko. 1977. Intoxication caused by interaction of chloramphenicol and phenytoin. JAMA 237:2630-2631. [PubMed] [Google Scholar]
- 30.Sastry, V., P. J. Brennan, M. M. Levy, N. Fishman, A. L. Friedman, A. Naji, C. F. Barker, and K. L. Brayman. 1995. Vancomycin-resistant enterococci: an emerging pathogen in immunosuppressed transplant recipients. Transplant. Proc. 27:954-955. [PubMed] [Google Scholar]
- 31.Sattler, M., F. P. Guengerich, C. H. Yun, U. Christians, and K. F. Sewing. 1992. Cytochrome P-450 3A enzymes are responsible for biotransformation of FK506 and rapamycin in man and rat. Drug Metab. Dispos. 20:753-761. [PubMed] [Google Scholar]
- 32.Schulman, S. L., L. M. Shaw, K. Jabs, M. B. Leonard, and K. L. Brayman. 1998. Interaction between tacrolimus and chloramphenicol in a renal transplant recipient. Transplantation 65:1397-1398. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz, J., K. Bachmann, and E. Perrigo. 1983. Interaction between warfarin and erythromycin. South. Med. J. 76:91-93. [DOI] [PubMed] [Google Scholar]
- 34.Shimada, T., H. Yamazaki, M. Mimura, Y. Inui, and F. P. Guengerich. 1994. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 270:414-423. [PubMed] [Google Scholar]
- 35.Shin, J. G., J. Y. Park, M. J. Kim, J. H. Shon, Y. R. Yoon, I. J. Cha, S. S. Lee, S. W. Oh, S. W. Kim, and D. A. Flockhart. 2002. Inhibitory effects of tricyclic antidepressants (TCAs) on human cytochrome P450 enzymes in vitro: mechanism of drug interaction between TCAs and phenytoin. Drug Metab. Dispos. 30:1102-1107. [DOI] [PubMed] [Google Scholar]
- 36.Taber, D. J., R. E. Dupuis, K. D. Hollar, A. L. Strzalka, and M. W. Johnson. 2000. Drug-drug interaction between chloramphenicol and tacrolimus in a liver transplant recipient. Transplant. Proc. 32:660-662. [DOI] [PubMed] [Google Scholar]
- 37.Villeneuve, J. P., L. L'Ecuyer, S. De Maeght, and P. Bannon. 2000. Prediction of cyclosporine clearance in liver transplant recipients by the use of midazolam as a cytochrome P450 3A probe. Clin. Pharmacol. Ther. 67:242-248. [DOI] [PubMed] [Google Scholar]
- 38.Vincent, F. M., L. Mills, and J. K. Sullivan. 1978. Chloramphenicol-induced phenytoin intoxication. Ann. Neurol. 3:469. [DOI] [PubMed] [Google Scholar]
- 39.von Moltke, L. L., D. J. Greenblatt, J. Schmider, C. E. Wright, J. S. Harmatz, and R. I. Shader. 1998. In vitro approaches to predicting drug interactions in vivo. Biochem. Pharmacol. 55:113-122. [DOI] [PubMed] [Google Scholar]
- 40.Walle, T. 1996. Assays of CYP2C8- and CYP3A4-mediated metabolism of taxol in vivo and in vitro. Methods Enzymol. 272:145-151. [DOI] [PubMed] [Google Scholar]
- 41.Yamazaki, H., and T. Shimada. 1997. Human liver cytochrome P450 enzymes involved in the 7-hydroxylation of R- and S-warfarin enantiomers. Biochem. Pharmacol. 54:1195-1203. [DOI] [PubMed] [Google Scholar]
- 42.Yeh, J., S. C. Soo, C. Summerton, and C. Richardson. 1990. Potentiation of action of warfarin by itraconazole. Br. Med. J. 301:669. [DOI] [PMC free article] [PubMed] [Google Scholar]