

Abstract
The relationships between the pharmacokinetic properties of quinine during a 7-day treatment course and the therapeutic response were studied in 30 adult patients with uncomplicated falciparum malaria monitored for ≥28 days. All patients received a 7-day oral quinine regimen either alone (n = 22) or in combination with rifampin (n = 8). The median fever clearance time was 58.5 h, and the mean ± standard deviation parasite clearance time was 73 ± 24 h. After recovery, six patients had recrudescences of Plasmodium falciparum malaria and seven had delayed appearances of P. vivax infection between days 16 and 23. Between the patients with and without recrudescences, there were no significant differences either in fever clearance time or parasite clearance time or in the overall pharmacokinetics of quinine and 3-hydroxyquinine. Patients for whom the area under the concentration-time curve from 3 to 7 days for quinine in plasma was <20 μg · day/ml had a relative risk of 5.3 (95% confidence interval = 1.6 to 17.7) of having a subsequent recrudescence of infection (P = 0.016). Modeling of these data suggested an average minimum parasiticidal concentration of quinine in plasma of 3.4 μg/ml and an MIC of 0.7 μg/ml for uncomplicated falciparum malaria in Thailand. To ensure a cure, the minimum parasiticidal concentration must be exceeded during four asexual cycles (>6 days).
The cinchona alkaloids have been important antimalarial drugs for more than 350 years. The principal alkaloid, quinine, still remains effective against chloroquine-resistant falciparum malaria, and it is widely used. Development of quinine resistance in Plasmodium falciparum has been relatively slow and incomplete by comparison with those of the other principal antimalarial drugs, e.g., chloroquine, mefloquine, and sufadoxine-pyrimethamine. In areas with multidrug-resistant strains, 7-day regimens of quinine and tetracycline still provide cure rates well over 90% in patients with uncomplicated falciparum malaria (4, 5). To date there is no convincing evidence of high-grade quinine resistance in the treatment of severe malaria (9). The pharmacokinetic properties of and therapeutic responses to quinine vary with age, pregnancy, immunity, and disease severity (2, 8, 14). As patients recover from malaria, the total apparent volume of distribution of quinine expands and systemic clearance increases (13). As a result, concentrations in plasma fall. In areas with resistant strains of P. falciparum in order to achieve good therapeutic responses in children, the dose of quinine must be increased after the 3rd day of treatment to compensate for this decline in the concentration in plasma which occurs with recovery (2). These observations suggest that quinine concentrations must remain above levels which inhibit parasite multiplication throughout the course of treatment to eradicate the infection from the body (14). Knowledge of the in vivo minimum parasiticidal concentrations and MICs (14) of quinine in patients with malaria are necessary for optimization of dosing regimens, but they have not been determined previously and quinine has a relatively narrow therapeutic ratio. In this report the relationship of the plasma quinine concentration profile to the therapeutic response has been examined in adult patients with uncomplicated falciparum malaria who were included in larger prospective clinical treatment trials in which the standard 7-day course of oral quinine was used (6).
MATERIALS AND METHODS
Patients.
This pharmacokinetic-pharmacodynamic study was conducted with adult male patients with acute P. falciparum malaria admitted to the Bangkok Hospital for Tropical Diseases, Bangkok, Thailand. Informed consent was obtained from each subject. Patients with severe malaria (15) or patients with mixed malaria infections were excluded. Patients who gave a history of drug hypersensitivity, who had taken any antimalarial drugs within the previous 48 h, or whose urine was positive in screening tests for sulfonamides (lignin test) or 4-aminoquinolines (Wilson-Edeson test) were also excluded. All patients were monitored for at least 28 days outside malaria transmission areas. These studies were approved by the ethics committee of the Faculty of Tropical Medicine, Mahidol University, Bangkok. The clinical responses of 10 of these patients have been published previously (6).
Management.
After clinical assessment and confirmation of the diagnosis from thick and thin blood smears, baseline blood samples were taken for routine hematology and biochemistry. All patients were treated with the standard 7-day oral treatment regimen: quinine sulfate (10 mg of salt/kg of body weight three times a day; Thai Government Pharmaceutical Organization) either alone or in combination with rifampin (15 mg/kg/day for 7 days; Merrell Dow Pharmaceuticals Inc.). These patients were part of larger therapeutic studies (6).
Oral acetaminophen (0.5 to 1 g every 4 h) was given to patients with temperatures >38°C. Vital signs were recorded every 4 h until resolution of fever and thereafter every 6 to 12 h. Fever clearance time (FCT) was the time that it took for the body temperature to fall below 37.5°C and remain below this value for >48 h. Patients who were subsequently unable to stay in the hospital until clearance of both fever and parasites were excluded from the study. Reappearance of infection was assessed in patients who remained in Bangkok either in the hospital or at home (i.e., outside the malaria transmission area) for at least 28 days. Patients with recrudescences were retreated with a 7-day course of quinine (10 mg of salt/kg three times a day) combined with tetracycline (4 mg/kg four times a day; Thai Government Pharmaceutical Organization), and those who had late vivax malaria appearances were subsequently treated with the standard doses of chloroquine and primaquine.
Laboratory investigations.
Parasite counts in Giemsa-stained thin films or thick films were measured every 12 h until clearance and thereafter daily for 28 days. Patients were hospitalized away from the area of endemicity so that reinfection could be confidently excluded. Parasite density was expressed as the number of parasites per microliter of blood, derived from the numbers of parasites per 1,000 red blood cells in a thin film stained with Giemsa or Field’s stain or calculated from the white cell count and the numbers of parasites per 200 white blood cells in a thick film. Routine biochemical and hematological tests were repeated on days 7, 14, 21, and 28 after admission.
Quinine pharmacokinetics.
Serial venous blood samples were taken for quinine level determination before at 12 and 24 h during treatment and then daily until day 7. All blood samples were taken before quinine intake. Each sample (4 ml) was collected in a heparinized tube and was immediately centrifuged at 1,500 × g for 10 min. All plasma samples were stored at −20°C until analysis. Quinine and 3-hydroxyquinine (3OH-Q) concentrations in plasma were assayed by high-pressure liquid chromatography as described previously (7, 12). All drug measurements were carried out without the knowledge of the treatment regimens given to the patients.
Quinine pharmacokinetics were evaluated by noncompartmental modeling with the Win-NONLIN program (Statistical Consultants, Lexington, Ky.). The drug concentration profiles were expressed as the areas under the plasma drug concentration-time curves (AUCs) from the time of the start of treatment to day 7 (AUC0-7). The AUCs for quinine and 3OH-Q in plasma during the course of therapy were divided prospectively into those during the acute phase of illness (days 0 to 2) and those during recovery (days 3 to 7) (denoted AUC0-2 and AUC3-7, respectively). Maximum concentrations in plasma (Cmaxs) and the times to Cmax (Tmaxs) were calculated from the 7-day sequential quinine and 3OH-Q concentrations.
Pharmacokinetic-pharmacodynamic analysis.
Three assumptions were made: first, that the minimum parasiticidal concentrations (MPCs) were present throughout the treatment course in cured patients but fell below these values in patients with recrudescent infections; second, that the slope of the concentration-effect relationship was similar in vivo and in vitro; and third, that as rifampin did not shorten parasite clearance times (PCTs), it did not contribute significantly to parasite killing. The average MPC was therefore estimated as the concentration less than or equal to the minimum concentration in the average drug profiles of cured patients. Patients with recrudescences who presented with low drug levels were used to estimate the MIC, i.e., the concentration associated with a parasite multiplication rate of 1 per cycle.
Estimation of MIC.
The standard pharmacodynamic sigmoid Emax model was assumed, as follows:
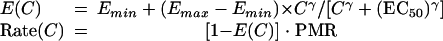 |
(1) |
where E represents the effect of treatment on the parasite multiplication rate (PMR), rate denotes the parasite multiplication rate corresponding to effect E, Emin is the effect on the parasite multiplication rate when no drug is present and is assumed to be equal to 0, Emax gives the lowest multiplication rate or the maximum parasite killing rate under the treatment (Emax is <1), γ is a slope parameter, C is the drug concentration in blood, and EC50 is the concentration giving 50% of the maximum effect.
The MIC is the concentration associated with a parasite multiplication rate of 1 per cycle, i.e., 1 = PMR · [1 − E(MIC)].
Therefore, if, in the absence of drug, the parasite multiplication rate is equal to 10, then 1 = 10[1 − E(MIC)] and, thus, E(MIC) is equal to 0.9.
After substitution into Equation 1, we get
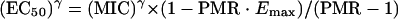 |
In our series of patients the lowest parasite multiplication rate, PMR(1 − Emax), ranges from 0.0005 to 0.003, so we can approximate the EC50 as follows:
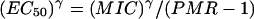 |
We have parasite counts available for the patients who recrudesced on day 2 (P2) and again on the day of recrudescence (PR). We assumed that after day 8 the treatment had no effect on the parasite multiplication rate. The relationship between these two parasite counts can then be expressed as
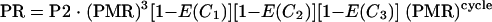 |
(2) |
where Ci is the geometric mean plasma quinine concentration over cycle i since day 2 (average concentration, assuming exponential decay for the drug concentration in blood), and cycle is the number of cycles since day 8 to recrudescence. As one parasite life cycle is 2 days, cycle · 2 is equal to the number of days.
Figure 1 depicts the sequence of events in the model.
FIG. 1.
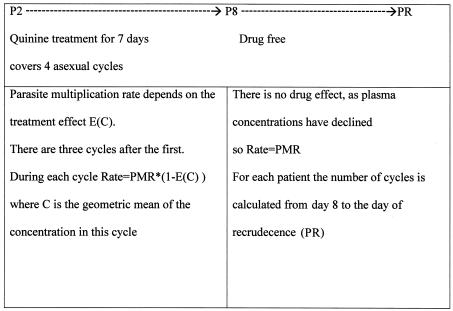
Sequence of events in the model.
As the MIC was estimated by using a number of assumptions, we also performed a sensitivity analysis with different values for γ (2.5, 3, and 3.5), the parasite multiplication rate (6, 10, and 20), and 1 − Emax (0.0001, 0.00001, and 0.000001).
Estimation of MPC.
MPC is the minimum concentration giving Emax. As a sigmoid function never really reaches Emax, Emax is its asymptote, and we calculate MPC as a concentration reaching Emax with precision ɛ, that is, [Emax − E(MPC)]/Emax = ɛ, and we get MPC = MIC[(1 − ɛ)/(PMR − 1)ɛ]1/γ.
Statistical analysis.
Normally distributed data were compared by unpaired t tests, and data not conforming to a normal distribution were compared by the Mann-Whitney U test. The cumulative cure rate was calculated by Kaplan-Meier survival analysis, and the cure rates were compared by the log-rank test. Correlations were assessed by the method of Spearman. All statistical analyses were performed with the SPSS statistical computing package (version 10.0 for Windows; SSPS Inc.).
RESULTS
Clinical responses.
The study included 30 adult male patients with uncomplicated falciparum malaria and a mean ± standard deviation (SD) age of 24.9 ± 8.9 years. The majority of these patients (70%) came from the western border of Thailand, where multidrug-resistant P. falciparum is most prevalent, and more than half of the patients (60%) had a previous history of malaria. The mean ± SD number of previous malaria infections was 1.7 ± 1.1 (range, 1 to 5). All patients were treated with oral quinine (10 mg/kg 3 times/day for 7 days) either alone (n = 22) or in combination with rifampin (n = 8). All patients recovered following the treatment. The overall median FCT was 58.5 h (range, 4 to 152 h), and the mean ± SD PCT was 73 ± 24 h. PCTs were similar in those who received rifampin and those who did not. One patient had a prolonged FCT of 152 h; this was ascribed to a simultaneous respiratory tract infection. His parasitemia cleared normally in 29 h. There was a significant correlation between the overall FCT and PCT (r = 0.48, P = 0.007). During the 28-day monitoring period, six patients had recrudescent infections (two were treated with quinine alone and four were treated with quinine plus rifampin). These occurred at a mean ± SD time of 20 ± 2.5 days (range, 17 to 23 days). Seven patients had delayed appearances of P. vivax infections at 19 ± 2 days (range, 16 to 21 days). Between patients with and without recrudescences, there were no significant differences in either FCT (median, 62 versus 56 h [P = 0.27]) or PCT (80 ± 32 versus 72 ± 23 h [P = 0.17]). As shown in Table 1, the demographic data and baseline laboratory data were also similar between patients with and without recrudescences.
TABLE 1.
Laboratory findings on admission for patients with P. falciparum malariaa
| Characteristic | Cure (n = 24) | Recrudescence (n = 6) | Total (n = 30) | P value |
|---|---|---|---|---|
| No. of patients | 24 | 6 | 30 | |
| Age (yr)a | 26.9 ± 10.9 | 24.2 ± 3.9 | 25.9 ± 9.9 | 0.63 |
| No. (%) of patients with previous malaria | 15 (62.5) | 3 (50) | 18 (60) | 0.67 |
| No. (%) of patients from the west | 17 (71) | 4 (67) | 21 (70) | 0.52 |
| Parasite count (no. of parasites/μl)b | 50,925 | 8,125 | 12,039 | 0.07 |
| Hematocrit (%)a | 34.7 ± 8.5 | 38.8 ± 5.2 | 35.7 ± 8.0 | 0.28 |
| White blood cell count (109/liter)a | 6.1 ± 2.1 | 7.1 ± 0.9 | 6.3 ± 1.9 | 0.29 |
| Platelet count (109/liter)a | 104.3 ± 61.2 | 63.5 ± 16.2 | 95.3 ± 56.8 | 0.12 |
| Serum creatinine level (mg/dl)a | 1.0 ± 0.1 | 1.2 ± 0.3 | 1.1 ± 0.2 | 0.01 |
| Total bilirubin level (mg/dl)a | 1.8 ± 1.2 | 2.4 ± 1.7 | 1.9 ± 1.3 | 0.35 |
| SGPTc (U/liter)a | 37.7 ± 28.7 | 52.7 ± 10.1 | 41.0 ± 34.0 | 0.35 |
Data are presented as means ± SDs.
Data are presented as the geometric mean.
SGPT, serum glutamic pyruvic transaminase.
Quinine pharmacokinetics.
Following quinine treatment, the quinine levels increased to an average maximum level within 1.5 days. The overall median peak concentration was 11.4 μg/ml. The concentrations of the active metabolite, 3OH-Q, increased after the first dose of quinine and reached maximum values at 2.5 days, or 1 day after the Tmax of quinine (Table 2). The AUC0-7 for quinine was approximately 10-fold higher than that for 3OH-Q, but the AUC0-7s for the drugs were not correlated (r = 0.23, P = 0.22). The AUCs during and after the first 48 h (AUC0-2 and AUC3-7) correlated significantly for both quinine (r = 0.73, P < 0.001) and 3OH-Q (r = 0.87, P < 0.001). The median ratios of AUCs during and after the first 48 h (AUC0-2/AUC3-7) were 0.45 (range, 0.27 to 1.46) for quinine and 0.30 (range, 0.07 to 0.98) for 3OH-Q. The overall AUC0-7 for 3OH-Q correlated significantly with the AUC0-2 of quinine (r = 0.38, P = 0.041) but not with the AUC3-7 (r = 0.23, P = 0.21).
TABLE 2.
Pharmacokinetic parameters for quinine and 3OH-Q following oral administration of quinine or quinine plus rifampin in patients with P. falciparum malariaa
| Patient group | Quinine
|
|||||
|---|---|---|---|---|---|---|
| AUC0-2 (μg · day/ml) | AUC3-7 (μg · day/ml) | AUC0-7 (μg · day/ml) | AUC0-2/ AUC3-7 | Tmax (days) | Cmax (μg/ml) | |
| Cured (n = 24) | 17.40 (8.81-26.60) | 35.37 (10.15-75.94) | 54.01 (20.17-97.62) | 0.45 (0.29-1.42) | 1.50 (0.50-5.00) | 11.43 (5.54-17.62) |
| Recrudesced (n = 6) | 16.80 (11.67-19.58) | 13.26 (8.17-70.68) | 29.88 (19.84-89.83) | 1.11 (0.27-1.46) | 0.50 (0.50-5.00) | 11.53 (8.36-16.50) |
| Total (n = 30) | 17.4 (8.81-26.60) | 34.67 (8.17-75.94) | 51.66 (19.84-97.62) | 0.46 (0.27-1.46) | 1.50 (0.50-5.00) | 11.43 (5.54-17.62) |
Data are presented as medians (ranges).
Quinine pharmacokinetic parameters and clinical responses.
Both FCT and PCT correlated positively with the Tmax of quinine (r > 0.41, P < 0.024) and with the overall AUC0-7 for quinine (r > 0.46, P < 0.011) (Fig. 2) and inversely with the ratio of quinine increments during and after the first 48 h (AUC0-2/AUC3-7) (r = −0.35 and P = 0.054 for FCT; r = −0.49 and P = 0.007 for PCT). The overall ratios of quinine and 3OH-Q AUCs (AUC0-7 quinine/AUC0-7 3OH-Q) also correlated significantly with both FCT and PCT (r = 0.42 and P = 0.022 and r = 0.39 and P = 0.034, respectively). FCT and PCT were not related to the AUC0-2 but significantly correlated with the AUC3-7 for quinine (r > 0.47, P < 0.008) and correlated inversely with the first 3OH-Q increment (r > −0.37, P < 0.046). PCT also correlated inversely with the AUC0-2/AUC3-7 ratio for 3OH-Q (r = −0.39, P = 0.036) and directly with the Tmax of 3OH-Q (r = 0.42, P = 0.020).
FIG. 2.
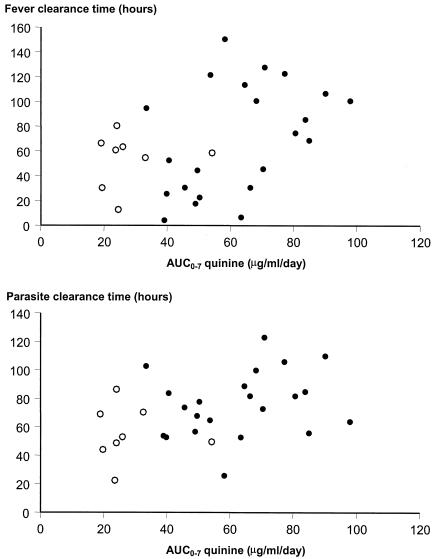
Relationship between the AUC0-7 for quinine and FCT and PCT in patients treated with quinine alone (closed circles) and quinine plus rifampin (open circles).
Quinine pharmacokinetics and clinical outcome.
During the first 48 h of treatment, the profiles of quinine concentrations in plasma were similar for patients with or without subsequent recrudescences but were lower thereafter for patients with recrudescences (Fig. 3). The serial concentrations of 3OH-Q were similar in both groups (Fig. 3). Of the six patients with subsequent recrudescences, four had low AUC3-7s for quinine (<20 μg · day/ml), low AUC0-7s for quinine (<35 μg · day/ml), and high AUC0-2/AUC3-7 ratios for quinine (>1.0). The proportions of patients with these pharmacokinetic characteristics were significantly higher among patients with recrudescences than among those with cures (P < 0.029). Patients with AUC3-7s for quinine less than 20 μg · day/ml had a relative risk of a subsequent recrudescence of 5.3 (95% confidence interval [CI] = 1.6 to 17.7) (P = 0.016). The cumulative cure rate for these patients was therefore significantly lower than that for patients with higher AUC3-7s for quinine (P = 0.005) (Fig. 4). There were no significant differences in the other pharmacokinetic parameters for quinine or 3OH-Q between patients with and without recrudescences (Table 2).
FIG. 3.
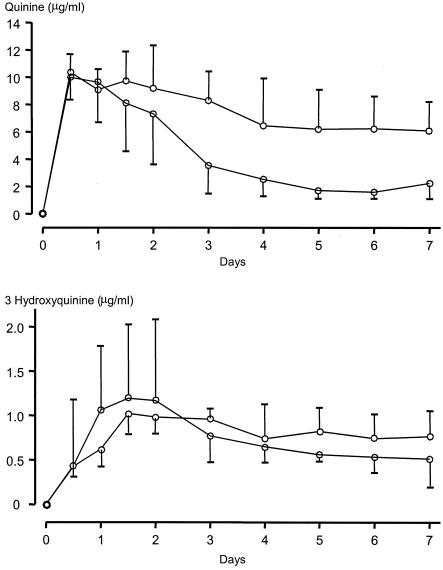
Concentrations of quinine and 3OH-Q in the plasma of patients with P. falciparum malaria with (upper graph) and without (lower graph) subsequent recrudescences. The data are shown as medians and the 95% CI of the median.
FIG. 4.
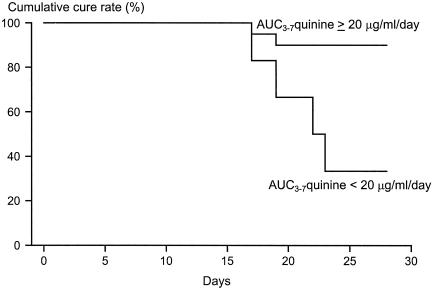
Cumulative cure rates for patients with P. falciparum malaria for whom the AUC3-7 for quinine was ≥20 or <20 μg · day/ml.
In vivo pharmacodynamics of quinine.
Estimation of the total parasite burden in individual patients with falciparum malaria is confounded by sequestration, but it can be approximated from the changes in peripheral parasitemia over time (14). Parasite killing is a first-order process, while the concentrations in plasma exceed the MPC. The MIC results in a parasite multiplication rate of 1, and with an unrestrained approximate parasite multiplication rate of 10 per cycle, this is similar to the 90% inhibitory concentration (IC90) value in vitro (11). A model of in vivo parasite population dynamics for an average patient in this series is shown in Fig. 4. By using data for the patients whose infections recrudesced, MICs were estimated by using different values of the three parameters: slope, Emax, and parasite multiplication rate. These results are presented in Table 3. The mean MIC was estimated to be 0.68 μg/ml (95% CI, 0.65 to 0.71 μg/ml) and the mean MPC was estimated to be 7.25 μg/ml (95% CI, 3.12 to 3.41 μg/ml) assuming a slope of 3 (1), Emax equal to 0.9999, which is a 3-log reduction in the parasite count over a cycle, and ɛ equal to 0.0001. With ɛ equal to 0.001, the MPC estimate falls to 3.36 μg/ml (Fig. 5).
TABLE 3.
Estimated quinine MICs for different values of the parameters parasite multiplication rate, Emax, and slope γ
| PMRa and γ values | Estimated MIC (μg/ml) (95% CI) for 1 − Emax values of:
|
||
|---|---|---|---|
| 0.0001 | 0.00001 | 0.000001 | |
| PMR = 6 | |||
| γ = 2.5 | 0.88 (0.84-0.92) | 0.89 (0.84-0.93) | 0.89 (0.85-0.93) |
| γ = 3.0 | 1.04 (1.00-1.08) | 1.05 (1.01-1.08) | 1.05 (1.01-1.08) |
| γ = 3.5 | 1.16 (1.12-1.20) | 1.17 (1.13-1.21) | 1.18 (1.14-1.22) |
| PMR = 10 | |||
| γ = 2.5 | 0.54 (0.51-0.57) | 0.55 (0.53-0.59) | 0.56 (0.53-0.59) |
| γ = 3.0 | 0.68 (0.65-0.71) | 0.71 (0.68-0.74) | 0.71 (0.68-0.74) |
| γ = 3.5 | 0.80 (0.76-0.83) | 0.84 (0.81-0.86) | 0.84 (0.81-0.87) |
| PMR = 20 | |||
| γ = 2.5 | 0.22 (0.19-0.24) | 0.29 (0.27-0.31) | 0.30 (0.28-0.31) |
| γ = 3.0 | 0.30 (0.27-0.33) | 0.40 (0.38-0.42) | 0.42 (0.40-0.44) |
| γ = 3.5 | 0.38 (0.35-0.41) | 0.51 (0.48-0.53) | 0.53 (0.51-0.55) |
PMR, parasite multiplication rate.
FIG. 5.
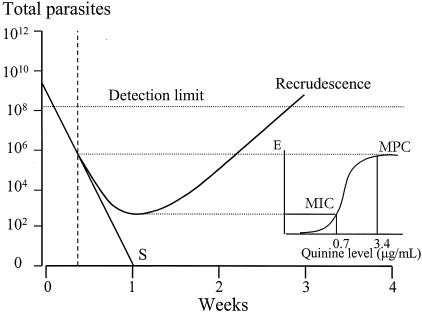
In vivo pharmacodynamics of quinine in falciparum malaria. The mean profile of the parasite burden in successfully treated patients declines logarithmically until total eradication(s) because the concentrations in plasma remain above the MPC (approximately 3.3 μg/ml), whereas in patients with recrudescent infections, parasite killing (E) falls below maximal values after 48 h of treatment. A parasite multiplication rate of 1 per cycle results from average MICs of 0.7 μg/ml at the end of the first week, and thereafter, the parasite multiplication rate (10 per cycle) is unrestrained (11).
DISCUSSION
Despite the relative ease of quantitating the burden of infection in patients with malaria compared with that in patients with bacterial and viral infections, there is very little information on the relationship between the concentrations of antimalarial drugs in plasma in vivo and the therapeutic response. The concentration-effect relationship has been studied extensively in vitro, but direct extrapolation from the in vitro to the in vivo situation cannot be assumed. In the artificial setting of ex vivo culture, malaria parasites are grown in nutrient medium in the absence of host cells or binding proteins, while the concentrations of the antimalarial drugs remain constant. In this in vivo pharmacodynamic assessment, it is assumed that whereas the absolute concentrations differ, the slope of the concentration-effect relationship is the same in vivo or ex vivo. Estimates of the concentrations of cinchona alkaloids required in vivo for a therapeutic response in patients with malaria were first made in the classic series of studies conducted in the United States immediately following the Second World War (3). These early studies relied on an imprecise spectrophotometric assay for quinine and quinidine (which included all fluorescent metabolites and which therefore overestimated the true concentration) and were based on daily blood sampling. The infections were artificially induced by a single strain (3). Since then there have been no systematic attempts to relate the blood quinine concentration profiles with the subsequent therapeutic response in uncomplicated malaria. The pharmacokinetic properties of quinine in malaria are altered; in the acute phase of falciparum malaria there is a contraction in the apparent volume of distribution of quinine and a reduction in systemic clearance, which results in elevated concentrations in blood (13). Increased levels of plasma protein binding reduce the free fraction in blood. This probably explains why the concentrations in plasma in the first days of treatment in the present study were higher in those with a slower resolution of fever and a slower clearance of parasites. In children, in whom the therapeutic response may be worse than that in adults because of a lack of background immunity, this decline in the concentrations in blood in the second half of the treatment course may lead to treatment failure (2). This led to a suggestion 20 years ago that the dose of quinine in children should be increased by 50% in the second half of the treatment course (2).
In this study the relationship between plasma quinine concentrations and the subsequent therapeutic response was examined by using data obtained prospectively from chemotherapeutic studies. In one of those studies, rifampin was combined with quinine in a therapeutic trial (6). Rifampin, which alone has very weak antimalarial activity, resulted in a marked increase in systemic clearance, presumably by inducing hepatic cytochrome P450 and increasing biotransformation. This led to a reduction in quinine concentrations in blood and a high treatment failure rate. These data confirm earlier suggestions that it is necessary to maintain concentrations of quinine, or indeed, any rapidly eliminated antimalarial drug, above the MPCs for the entire 7-day treatment course (14). This is because parasite killing by quinine does not usually exceed 1,000-fold per asexual cycle (in this trial the mean value was estimated to be 250-fold), and therefore, it is necessary for the concentrations in plasma to exceed the MPC during four asexual cycles (8 days) in order to ensure eradication of any infection with ≥109 malaria parasites. In this small series, the profiles of the concentrations of quinine and its main metabolite, 3OH-Q (which has approximately 1/10 of the antimalarial activity of quinine), in plasma in the first 48 h of treatment were no different in patients who were cured and those whose infections recrudesced. Thus, maximum parasiticidal effects, and, therefore, maximum parasite killing, can be assumed in this first cycle of drug exposure. Thereafter, the profiles of the concentrations in plasma diverged and there were lower levels of quinine in the plasma of patients whose infections recrudesced. This suggested submaximal killing after the first asexual parasite cycle. The relationship between concentration and effect cannot be defined precisely in this small series because parasites were not taken systematically for in vitro culture. Therefore, in vitro data from a large series conducted in Thailand were used to provide an average slope for the linear portion of the sigmoid concentration-effect relationships. If it is assumed that the slopes of the in vivo and in vitro concentration-effect relationships were similar, these preliminary data allow fitting of the concentration-effect or dose-response curve to the observed profiles. For an unrestrained parasite multiplication rate of 10, then, an approximate MPC of 7.25 μg/ml (the concentration giving 99.99% of the maximum effect) and an MIC of 0.7 μg/ml can be deduced. As the concentration-effect relationship is flat at the top of the sigmoid curve, concentrations much lower than the MPC still result in almost maximal parasite killing. For example, a concentration of 3.4 μg/ml would be expected to give 99.9% of the maximum killing rate. Much higher concentrations would be required for these effects in severe malaria because of increased plasma protein binding and consequent lower concentrations of free drug. There is considerable variability in in vitro susceptibility between different parasite isolates (approximate IC50 range in Thailand, 50 to 700 ng/ml). Furthermore, the values of the slopes for the concentration-effect relationship ranged from 1.9 to 5, with an average of 3 (1), and so these estimates based on a small number of treatment failures can be regarded only as rough approximations. The estimates were relatively sensitive to changes in slope and unrestrained parasite multiplication rate but were robust to changes in the maximum parasite killing rate (Emax). Taking an average value for plasma protein binding in uncomplicated malaria of 15% (5, 10), this would suggest an in vivo average IC90 (approximating the MIC) of 105 ng/ml for free quinine. If the true parasite multiplication rate was 6 per cycle and not 10 per cycle and the concentration-effect slope increased from 3 to 3.5, the estimated MIC rises to 1.17 μg/ml and the corresponding IC90 is 176 ng/ml.
Overall these data confirm the importance of maintaining adequate drug concentrations in blood throughout a treatment course and ensuring that rapidly eliminated drugs are present at levels above the MPC in a nonimmune patient for four asexual parasite cycles (14). In the case of quinine treatment in Thailand, this means a 7-day treatment course that provides concentrations in plasma of ≥6 μg/ml throughout the treatment course to ensure a cure. This emphasizes the importance of providing an adequate treatment course and also complete adherence to the prescribed drug regimen in order to optimize cure rates.
Table 2a.
| 3OH-Q
|
AUC for quinine/AUC for 3OH-Q
|
|||||||
|---|---|---|---|---|---|---|---|---|
| AU0-2 (μg · day/ml) | AUC3-7 (μg · day/ml) | AUC0-7 (μg · day/ml) | AUC0-2/ AUC3-7 | Tmax (days) | Cmax (μg/ml) | AUC0-2 | AUC3-7 | AUC0-7 |
| 1.37 (0.31-2.68) | 4.35 (1.92-8.90) | 5.67 (2.84-10.73) | 0.29 (0.07-0.67) | 3.00 (1.00-7.00) | 1.26 (0.60-2.07) | 13.29 (4.94-64.19) | 8.45 (2.77-17.27) | 9.48 (4.01-17.63) |
| 1.61 (0.87-3.05) | 3.25 (2.45-6.58) | 4.90 (3.98-7.92) | 0.50 (0.20-0.98) | 2.00 (2.00-5.00) | 1.37 (0.84-2.19) | 8.43 (4.89-22.14) | 3.81 (3.08-22.67) | 5.34 (4.10-22.56) |
| 1.39 (0.31-3.05) | 4.24 (1.92-8.90) | 5.55 (2.84-10.73) | 0.30 (0.07-0.98) | 3.00 (1.00-7.00) | 1.26 (0.60-2.19) | 13.29 (4.89-64.19) | 8.65 (2.77-22.67) | 9.48 (4.01-22.56) |
Acknowledgments
This study was supported by the Wellcome Trust-Mahidol University-Oxford Tropical Medicine Research Programme, funded by the Wellcome Trust of the Great Britain.
REFERENCES
- 1.Brockman, A., R. N. Price, M. van Vugt, D. G. Heppner, D. Walsh, P. Sookto, T. Wimonwattrawatee, S. Looareesuwan, N. J. White, and F. Nosten. 2000. Plasmodium falciparum antimalarial drug susceptibility on the northwestern border of Thailand during five years of extensive artesunate-mefloquine use. Trans. R. Soc. Trop. Med. Hyg. 94:537-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chongsuphajaisiddhi, T., A. Sabcharoen, and P. Attanath. 1981. In vivo and in vitro sensitivity of falciparum malaria to quinine in Thai children. Ann. Trop. Paediatr. i:21-26. [DOI] [PubMed]
- 3.Earle, D. P., R. W. Berliner, J. V. Taggart, W. J. Welch, C. G. Zubrod, N. Bowman-Wise, T. C. Chalmers, R. L. Grief, and J. A. Shannon. 1948. Studies on the chemotherapy of the human malarias. II. Method for the quantitative assay of suppressive antimalarial action in falciparum malaria. J. Clin. Investig. 27:75-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Looareesuwan, S., P. Wilairatana, S. Vanijanonta, D. Kyle, and K. Webster. 1992. Efficacy of quinine-tetracycline for acute uncomplicated falciparum malaria in Thailand. Lancet i:367-370. [DOI] [PubMed]
- 5.Nontprasert, A., S. Pukrittayakamee, D. E. Kyle, S. Vanijanonta, and N. J. White. 1996. Antimalarial activity and interactions between quinine, dihydroquinine, and 3-hydroxyquinine against P. falciparum in vitro. Trans. R. Soc. Trop. Med. Hyg. 90:553-555. [DOI] [PubMed] [Google Scholar]
- 6.Pukrittayakamee, S., S. Prakongpan, S. Wanwimolruk, R. Clemens, S. Looareesuwan, and N. J. White. 2003. Adverse effect of rifampin on quinine efficacy in uncomplicated falciparum malaria. Antimicrob. Agents Chemother. 47:1509-1513. [DOI] [PMC free article] [PubMed]
- 7.Pukrittayakamee, S., P. Pitisuttithum, H. Zhang, A. Jantra, S. Wanwimolruk, and N. J. White. 2002. Effects of cigarette smoking on quinine pharmacokinetics in malaria. Eur. J. Clin. Pharmacol. 58:315-319. [DOI] [PubMed] [Google Scholar]
- 8.Pukrittayakamee, S., S. Looareesuwan, D. Keeratithakul, T. M. E. Davis, P. Teja-Isavadharm, B. Nagachinta, A. Weber, A. L. Smith, D. Kyle, and N. J. White. 1997. A study of the factors affecting the metabolic clearance of quinine in malaria. Eur. J. Clin. Pharmacol. 52:487-493. [DOI] [PubMed] [Google Scholar]
- 9.Pukrittayakamee, S., W. Supanaranond, S. Looareesuwan, S. Vanijanonta, and N. J. White. 1994. Quinine in severe falciparum malaria: evidence of declining efficacy in Thailand. Trans. R. Soc. Trop. Med. Hyg. 88:324-327. [DOI] [PubMed] [Google Scholar]
- 10.Silamut, K., R. Hough, T. Eggelte, S. Pukrittayakamee, and N. J. White. 1955. Simple methods for assessing quinine pre-treatment in acute malaria. Trans. R. Soc. Trop. Med. Hyg. 89:665-667. [DOI] [PubMed] [Google Scholar]
- 11.Simpson, J. A., L. Aarons, W. E. Collins, G. Jeffery, and N. J. White. 2002. Population dynamics of untreated Plasmodium falciparum malaria within the adult human host during the expansion phase of the infection. Parasitology 124:247-263. [DOI] [PubMed] [Google Scholar]
- 12.Wanwimolruk, S., S. M. Wong, H. Zhang, P. F. Coville, and R. J. Walker. 1995. Metabolism of quinine in man: identification of a major metabolite, and effects of smoking and rifampicin pretreatment. J. Pharm. Pharmacol. 47:957-963. [DOI] [PubMed] [Google Scholar]
- 13.White, N. J., S. Looareesuwan, D. A. Warrell, M. J. Warrell, D. Bunnag, and T. Harinasuta. 1982. Quinine pharmacokinetics and toxicity in cerebral and uncomplicated falciparum malaria. Am. J. Med. 73:564-572. [DOI] [PubMed] [Google Scholar]
- 14.White, N. J. 1997. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob. Agents Chemother. 41:1413-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Health Organization, Division of Control of Tropical Diseases. 1990. Severe and complicated malaria. Trans. R. Soc. Trop. Med. Hyg. 84(Suppl. 2):1-65. [PubMed] [Google Scholar]