

Abstract
SA-IGIV is a human polyclonal immunoglobulin containing elevated levels of antibodies specific for the fibrinogen-binding MSCRAMM protein clumping factor A (ClfA). In vitro, SA-IGIV specifically recognized ClfA that was expressed on the surface of Staphylococcus aureus and inhibited bacterial adherence to immobilized human fibrinogen by >95%. Moreover, SA-IGIV efficiently opsonized ClfA-coated fluorescent beads and facilitated phagocytosis by human polymorphonuclear leukocytes. To determine its potential therapeutic efficacy, SA-IGIV was evaluated in combination with vancomycin in a rabbit model of catheter-induced aortic valve infective endocarditis (IE) caused by methicillin-resistant S. aureus (MRSA). The combination therapy was more effective than vancomycin alone in sterilizing all valvular vegetations when used therapeutically during early (12-h) IE. The combination therapy resulted in clearance of bacteremia that was significantly faster than that of vancomycin alone in animals with well-established (24-h) IE. Therefore, in both early and well-established MRSA IE, the addition of SA-IGIV to a standard antibiotic regimen (vancomycin) increased bacterial clearance from the bloodstream and/or vegetations.
The incidence of methicillin-resistant Staphylococcus aureus (MRSA) infections continues to rise. According to the Centers for Disease Control and Prevention, the incidence of nosocomial infections in intensive care unit patients due to MRSA increased by 40% in 1999 compared to that of the previous 4 years in the United States (4). Vancomycin, the drug of choice for such infections, is often suboptimal, and indeed, the first documented case of an infection caused by vancomycin-resistant S. aureus in the United States has recently been reported (5). The increased incidence of antibiotic resistance observed in S. aureus clinical isolates has underscored the need for alternatives to current antibiotic strategies (6). An emerging option in this regard is antibody-based immunotherapy approaches, via targeting of epitopes on critical virulence proteins expressed in vivo. Mounting evidence suggests that microbial adherence is central to the initiation and metastatic spread of S. aureus infections. Therefore, the MSCRAMM (microbial surface components recognizing adhesive matrix molecules) family of proteins, which play a central role in adherence to host tissues, represents a cadre of potential antigenic candidates for the development of novel immunotherapies (8, 24).
One well-characterized MSCRAMM protein that is a candidate target for immunotherapy is clumping factor A (ClfA), a fibrinogen-binding adhesin expressed on the surface of nearly all strains of S. aureus (3, 17, 18). It is well established that ClfA recognizes the C terminus of the γ chain of human fibrinogen (10, 19, 26) and that the interaction between ClfA and fibrinogen can be inhibited by antibodies raised against the A domain of ClfA (18). In vivo data suggest that ClfA plays a critical role in the induction and persistence of experimental endovascular infections. For example, mutant strains of S. aureus which lack ClfA expression were less virulent in a rat endocarditis model when the rats were challenged with low levels of bacteria (21). Furthermore, it has recently been shown that when commensal organisms, such as Streptococcus gordonii or Lactococcus lactis, are genetically engineered to express ClfA, the bacteria become more virulent in a rat infective endocarditis (IE) model (20, 25).
The present study was designed to further characterize potential antibody therapies against S. aureus, with a particular focus on ClfA as an antigenic target. As a first step towards the development of an anti-ClfA immunotherapy, we have developed a hyperimmunoglobulin (SA-IGIV) derived from plasma donors with naturally occurring high titers of anti-ClfA immunoglobulin G (IgG). It has been shown that human antibody preparations containing high titers of anti-ClfA-specific IgG can be successfully used as a prophylactic agent to decrease mortality in a murine model of S. aureus sepsis (13). Similarly, we have found that human anti-ClfA antibodies have potent prophylactic efficacy when they are tested in a rabbit model of MRSA-induced IE (data not shown). In this report, we have demonstrated that the anti-ClfA antibodies in the SA-IGIV preparation are able to recognize the staphylococcal cell surface and specifically inhibit S. aureus adherence to immobilized fibrinogen. Further, we have shown that the anti-ClfA antibodies present in SA-IGIV can function as an efficient opsonin in an in vitro assay of human polymorphonuclear leukocyte (PMN) opsonophagocytosis. Finally, we have shown the therapeutic efficacy of SA-IGIV, when used in combination with the glycopeptide antibiotic vancomycin, in a rabbit model of catheter-induced aortic valve IE caused by MRSA.
MATERIALS AND METHODS
Bacteria.
S. aureus strain 67-0 is an oxacillin (methicillin)-resistant wound isolate (provided courtesy of Henry Chambers, University of California San Francisco and San Francisco General Hospital, San Francisco), previously determined to be virulent in an animal model of IE (2). Newman spa::kan, a protein A knockout mutant of S. aureus strain Newman, L. lactis(pKS80), and L. lactis ClfA+ transfected strains were provided by Timothy Foster (Trinity College, Dublin, Ireland).
Clf40 recombinant protein.
Clf40 is a recombinant protein corresponding to the A domain (amino acids 40 to 559) of the ClfA molecule. The fibrinogen-binding domain of ClfA is completely encompassed by the Clf40 construct (23). The recombinant protein contains an N-terminal 6-His tag and was purified from Escherichia coli lysates by metal affinity chromatography on a chelating Sepharose Fast Flow resin (Amersham Biosciences, Piscataway, N.J.) followed by Q Sepharose (Amersham Biosciences) chromatography.
SA-IGIV antibody.
SA-IGIV is a sterile, solvent- and detergent-treated liquid preparation of highly purified IgG. Plasma donors with elevated titers of anti-ClfA antibody were selected from the general donor population for the manufacture of SA-IGIV. SA-IGIV was manufactured under good manufacturing practices by Massachusetts Public Health Biological Laboratories (Jamaica Plain, Mass.) using cold ethanol fractionation (7). The resulting product had an anti-ClfA titer that was approximately five times greater than that measured in random commercial lots of immunoglobulins for intravenous use (IGIV) prepared from unselected donors (data not shown).
Flow cytometry.
The recognition of ClfA expressed on the bacterial cell surface by SA-IGIV was detected by flow cytometry. Bacteria were washed in phosphate-buffered saline (PBS) and resuspended in 10 ml of a 100-μg/ml concentration of rabbit IgG (Sigma, St. Louis, Mo.) in PBS and incubated for 30 min on ice. Twenty microliters of blocked bacteria was added to 0.5 ml of a 2-mg/ml concentration of SA-IGIV. All tubes were vortexed and incubated on ice for 30 min. Following the incubation, the bacteria were pelleted by centrifugation, and the supernatant was discarded. The bacteria were washed twice in cold PBS-2.5% bovine serum albumin (BSA). One-half milliliter of a 1:200 dilution of phycoerythrin (PE)-conjugated F(ab′)2 fragment of affinity-purified goat anti-human IgG (heavy and light chains) (Rockland, Inc., Gilbertsville, Pa.) was added to each tube, and the tubes were incubated on ice for 30 min. The bacteria were washed twice and finally resuspended in PBS-2.5% BSA. FL2 fluorescence (transmittance at 585/42 nm) was measured with a FACSCalibur (Becton-Dickinson, Mountain View, Calif.) flow cytometer equipped with an argon-ion laser (excitation at 488 nm).
Inhibition of fibrinogen binding.
To eliminate any effect of protein A binding to inhibitory antibodies, the protein A mutant strain Newman spa::kan was used in the inhibition assay. An overnight stationary culture of Newman spa::kan was washed in cold Hanks' balanced salt solution, and the bacterial suspension was adjusted to an optical density at 600 nm of 1.0. The cells were then labeled with 5 μM Syto13 fluorescent dye (Molecular Probes, Eugene, Oreg.). The bacteria were blocked by incubating them with 0.1 mg of rabbit IgG/ml (Sigma) on ice for 30 min, pelleted by centrifugation, and resuspended in 0.5 volume of Hanks' balanced salt solution with 1% BSA. A 0.25-ml sample of each test antibody (SA-IGIV or a control preparation of SA-IGIV preabsorbed with S. aureus) was diluted in Hanks' balanced salt solution with 1% BSA, mixed with 0.25 ml of bacterial suspension in titer tubes (Bio-Rad Laboratories, Hercules, Calif.), and allowed to incubate for 30 min at 2 to 8°C. Immulon II HB microtiter plates (Dynex Technologies Inc., Chantilly, Va.) were coated with 1.25 μg of human fibrinogen/ml (Enzyme Research Labs, South Bend, Ind.) and stored overnight at 4°C. The fibrinogen-coated microtiter plates were washed four times with 350 μl of 1× PBS-0.05% Tween 20 per well by using a Skanwasher (Skatron, Sterling, Va.). One hundred microliters of each bacterium-antibody reaction mixture was added to duplicate wells of the plate. A sample containing bacteria alone was used as a 100% binding control, and buffer alone was used as the 0% binding control. Samples were incubated on the plate for 1 h at 2 to 8°C. Nonadherent bacteria were removed by washing the plates four times with 350 μl of 1× PBS-0.05%Tween 20 per well. One hundred microliters of Hanks' balanced salt solution was added to each well, and the intensity of the fluorescence was measured with an HTS7000 fluorescent plate reader (Perkin-Elmer, Wellesley, Mass.) (excitation wavelength, 485 nm; emission wavelength, 535 nm). To demonstrate the specificity of the inhibition activity, serial dilutions of Clf40 protein were incubated with the bacteria in addition to 2 mg of SA-IGIV/ml.
OP assay.
The opsonophagocytic (OP) method was based on previously reported techniques for evaluating OP activity with antigen-coated fluorescent beads (1, 9, 14, 15, 16). ClfA-coated beads were used as a surrogate for the S. aureus organism to evaluate the OP activity associated specifically with the anti-ClfA antibodies in SA-IGIV and to eliminate any activity contributed by other anti-staphylococcal antibodies that might have been present. Fluoresbrite yellow-green carboxylate microspheres (diameter, 1 μm) were coated with target antigens by using the manufacturer's carbodiimide kit (Polysciences Inc., Warrington, Pa.). Protein adsorption to the beads was verified by measuring the concentration of free protein remaining in the supernatant after antigen coupling by use of a bicinchoninic acid total protein assay (Pierce, Rockford, Ill.). Primary human PMNs from healthy volunteers were purified from freshly harvested, heparinized peripheral blood by using Lympholyte-poly (Cedarlane Labs, Hornby, Ontario, Canada). The cell concentration was adjusted to 8 × 106 cells/ml with Hank's balanced salt solution-1% BSA (OP buffer). For each OP test, 5 × 106 antigen-coated microspheres were opsonized with 2.5 mg of SA-IGIV antibody/ml for 30 min at 37°C in a total volume of 450 μl of OP buffer. Fifty microliters of a 1:5 dilution of guinea pig complement (ICN Biomedical, Aurora, Ohio) was then added, and the mixture was incubated for an additional 30 min at 37°C. The opsonized microspheres were washed, resuspended in 0.5 ml of OP buffer, and mixed with 0.25 ml of the PMN preparation (2 × 106 cells). The microspheres and cells were incubated together for 30 min at 37°C with end-over-end rotation. Ice-cold OP buffer (0.25 ml) was added, and the samples were kept on ice until analysis on a FACSCalibur flow cytometer. Data were collected using the FL1 photomultiplier (transmittance at 530/30 nm) after excitation with a 488-nm argon-ion laser. Electronic gating was used to analyze bead uptake by PMNs. The phagocytic product (i.e., mean number of beads per cell multiplied by the percentage of fluorescent PMNs) was calculated for each reaction as described in detail previously (14, 15, 16).
Animal model of IE.
All animal experimentation was performed in accordance with the guidelines for animal health and welfare required by the Research and Education Institute at Harbor-UCLA Medical Center. Female outbred New Zealand White rabbits (Irish Farms, Corona, Calif.) underwent carotid artery-to-left-ventricle catheterization as previously described (28). At 24 h postcatheterization, the rabbits received an inoculum of 5 × 106 CFU of S. aureus 67-0 intravenously in the marginal ear vein (representing a 95% infective dose for this strain for inducing IE). At either 12 h (early IE) or 24 h (well-established IE) postinfection, the animals were randomized into the following groups: no therapy, vancomycin alone, or vancomycin plus SA-IGIV. Animals in the vancomycin-alone group received vancomycin (Abbott Laboratories, Chicago, Ill.) at a dose of 7.5 mg/kg of body weight intravenously twice a day for four consecutive days after baseline blood cultures were obtained. This dose regimen of vancomycin has shown efficacy in IE caused by this MRSA strain in prior studies (L. I. Kupferwasser, S. M. Shapiro, and A. S. Bayer, Abstr. 103rd Gen. Meet. Am. Soc. Microbiol. 2003, abstr. A-027, p. 6, 2003). Animals in the group receiving vancomycin plus SA-IGIV were given vancomycin therapy plus a single dose of 200 mg of SA-IGIV/kg intravenously via the marginal ear vein. At 24, 48, 72, and 96 h post-SA-IGIV administration, blood cultures were obtained from all treated animals. Following the 96-h time point, the animals were sacrificed and the hearts and kidneys were aseptically removed. Vegetations and renal abscesses from these organs were then quantitatively cultured as previously described (28). Quantitative culture data were expressed as either log10 CFU per milliliter of blood (cultures) or log10 CFU per gram of tissue (vegetations and renal abscesses). Animals with culture-positive vegetations were considered to have active IE irrespective of the bacterial densities.
Statistical analysis.
In the rabbit IE model, proportional data between groups (e.g., proportion of vegetations rendered culture negative) were analyzed by Fisher's exact test. Continuous data (e.g., bacterial densities in vegetations) were analyzed by the Kruskal-Wallis analysis of variance with post hoc correction for multiple comparisons between groups analyzed by Tukey's post hoc test. A P value of <0.05 was considered statistically significant.
RESULTS
Recognition of S. aureus by SA-IGIV.
It was necessary to demonstrate that the antibodies present in the SA-IGIV preparation were capable of recognizing native epitopes expressed on the surface of S. aureus. To this end, bacteria were incubated with SA-IGIV, and bound antibodies were detected with a fluorescently labeled anti-human IgG F(ab′)2 antibody. To eliminate protein A-mediated antibody binding, a protein A knockout mutant of S. aureus strain Newman (Newman spa::kan) was used. Eighty-seven percent of Newman spa::kan cells exhibited antibody surface binding after exposure to SA-IGIV (Fig. 1A). To demonstrate that some of this reactivity is due to the presence of ClfA-specific antibodies in SA-IGIV, we performed a similar experiment with an L. lactis strain transfected with a ClfA expression vector. Antibodies in SA-IGIV were able to recognize the surface of the L. lactis ClfA+ strain but did not recognize a control L. lactis strain which lacks ClfA expression (Fig. 1B). These data demonstrated that SA-IGIV was capable of recognizing native epitopes expressed on the surface of S. aureus and that some of this recognition is attributable to ClfA-specific antibodies present in SA-IGIV.
FIG. 1.
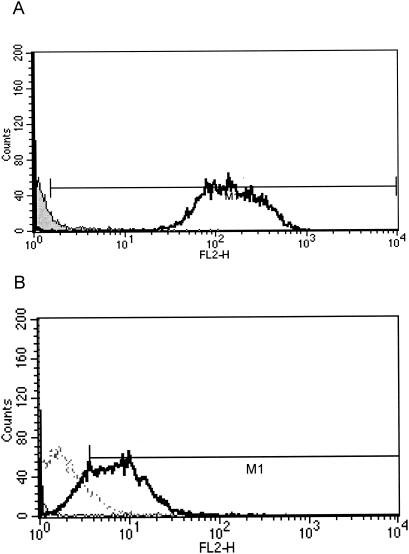
Flow cytometric analysis of SA-IGIV binding to S. aureus. (A) S. aureus strain Newman spa::kan was stained with SA-IGIV and a PE-conjugated F(ab′)2 anti-human IgG secondary antibody as described in Materials and Methods (open histogram). Bacteria incubated with secondary antibody alone (shaded histogram) served as a negative control. (B) SA-IGIV recognition of L. lactis ClfA+ (solid line) or L. lactis(pKS80) (broken line) was measured as described for panel A. All samples were analyzed for PE fluorescence with a Becton-Dickinson FACSCalibur flow cytometer. Results were gated for single bacteria by using forward versus side scatter. The data represent 10,000 collected events for each sample.
Inhibition of S. aureus binding to fibrinogen.
A binding inhibition test was performed to determine if ClfA-specific antibodies in SA-IGIV were capable of mitigating the interaction between ClfA and fibrinogen. Fluorescently labeled Newman spa::kan bacteria were incubated with increasing concentrations of SA-IGIV and then incubated on fibrinogen-coated microtiter plates. Nonadherent bacteria were removed, and the remaining fluorescence was measured. Concentration-dependent inhibition of S. aureus binding to fibrinogen was observed with SA-IGIV (Fig. 2A). Forty percent inhibition was observed with as little as 0.03 mg of SA-IGIV/ml, and >95% inhibition was achieved at 2 mg/ml. As a specificity control, SA-IGIV was preabsorbed with S. aureus cells, and then this absorbed antibody was used in place of SA-IGIV in the inhibition assay (Fig. 2A, negative control). There was little or no inhibition observed with this control antibody preparation, indicating that the inhibition of fibrinogen binding was due to antibodies specific for the surface of S. aureus. To determine if anti-ClfA antibodies in SA-IGIV were responsible for the inhibition of fibrinogen binding, increasing concentrations of soluble Clf40 protein were added to mixtures of 2 mg of SA-IGIV/ml and S. aureus prior to incubation on fibrinogen-coated plates. The presence of soluble Clf40 protein completely blocked the ability of SA-IGIV to inhibit S. aureus binding to fibrinogen (Fig. 2B). These data support the conclusion that ClfA-specific antibodies present in SA-IGIV recognize the fibrinogen-binding domain of ClfA and inhibit the ability of S. aureus to adhere to human fibrinogen-coated surfaces.
FIG. 2.
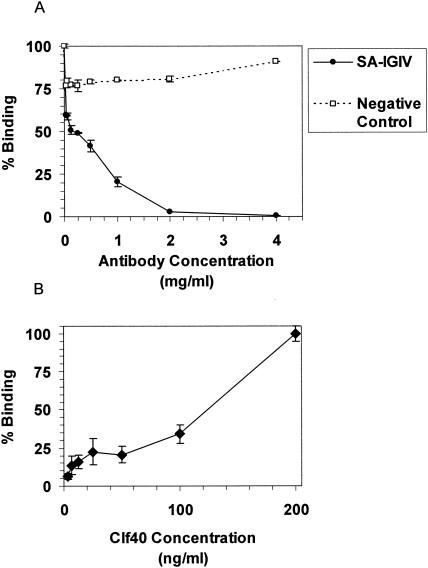
Inhibition of S. aureus binding to fibrinogen. Fluorescently tagged S. aureus strain Newman spa::kan was incubated with different concentrations of SA-IGIV (solid line) or with SA-IGIV which had previously been absorbed with S. aureus to remove specific antibody (control, dashed line) (A) and an inhibiting concentration of SA-IGIV (2 mg/ml) and increasing concentrations of soluble Clf40 protein (B). Each reaction mixture was then incubated on fibrinogen-coated microtiter plates, and nonadherent bacteria were washed away. The amounts of adherent fluorescent bacteria were then measured. Background fluorescence (sample without added bacteria) was subtracted from all measurements. Samples with bacteria alone were used as 100% binding controls. Data are presented as the means ± standard deviations of results of duplicate samples.
Opsonizing activity of ClfA-specific antibodies in SA-IGIV.
Although antibodies in SA-IGIV can recognize the surfaces of S. aureus organisms and can inhibit the bacteria from adhering to fibrinogen-coated surfaces, the ability of this antibody to act as an opsonin to facilitate OP clearance of bacteria may play a critical role in its overall therapeutic effectiveness in vivo. To specifically delineate the opsonic capacity of anti-ClfA antibodies present in SA-IGIV, fluorescent microspheres were coated with Clf40 and incubated with a saturating concentration (2.5 mg/ml) of SA-IGIV and complement. Antigen-coated beads were used in place of S. aureus organisms to eliminate the opsonizing effects of other anti-staphylococcal antibodies that may be present in SA-IGIV. The opsonized beads were incubated with freshly isolated human PMNs, and the association of PMNs with fluorescent beads was quantitated by flow cytometry. Controls included the addition of all reactants with the exception of SA-IGIV and the use of control beads lacking the Clf40 antigen. The results are shown in Fig. 3. When SA-IGIV was used, the resulting phagocytic product was 4.5-fold greater than that in control reactions. To determine if the cell-associated fluorescence was due to simple surface adherence or phagocytosis, reaction samples were analyzed by transmission electron microscopy. The results of transmission electron microscopy showed that beads were internalized by the PMNs in membrane-lined vesicles, and very few, if any, beads were surface associated (data not shown). The results clearly demonstrate that ClfA-specific antibodies present in SA-IGIV can act as opsonins to mediate the recognition and uptake of ClfA-coated particles by human PMNs.
FIG. 3.
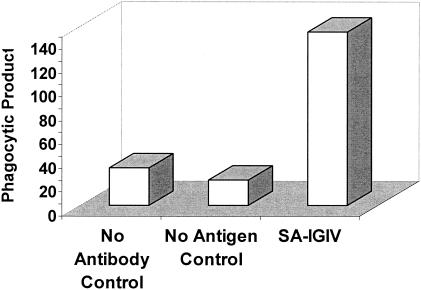
Opsonic capacity of Clf40-specific antibodies present in SA-IGIV. Clf40-coupled fluorescent beads were incubated with 2.5 mg of SA-IGIV/ml and complement. The opsonized beads were then incubated with human PMNs for 30 min at 37°C with end-over-end rotation. Fluorescent bead association with PMNs was quantitated by flow cytometry. The data are presented as the phagocytic product of each reaction. No Antibody Control, all reactants with the exception of SA-IGIV; No Antigen Control, Clf40 conjugated beads replaced with unconjugated beads.
In vivo impact of antibody on MRSA IE.
To determine whether the in vitro properties of SA-IGIV would translate into in vivo efficacy, SA-IGIV was tested in two models of experimental IE. To establish parameters for the model, we performed a pilot analysis of the potential efficacy of SA-IGIV alone in established IE. Two animals received SA-IGIV (200 mg/kg) intravenously 12 h after induction of IE, while two animals were untreated controls. In the untreated controls, blood cultures obtained at 24 h postinfection yielded high-level bacteremia (mean, 3.77 log10 CFU/ml); both animals died by 48 h postinfection. For animals receiving SA-IGIV, serial blood cultures showed an initial decrease in the level of bacteremia compared to that of untreated controls at 24 h (mean, 2.0 log10 CFU/ml). However, over the ensuing 48 h, bacterial densities in these blood cultures reached levels virtually identical to those of untreated controls. Therefore, we did not include an antibody-alone group in the larger studies but rather investigated the effect of SA-IGIV in combination with vancomycin. To determine if there were differences in treating animals with early or well-established IE, SA-IGIV was administered at either 12 or 24 h after MRSA challenge. In animals with early IE (12 h postinfection), vancomycin and vancomycin plus SA-IGIV were equally efficacious at reducing the extent of bacteremia, with all blood cultures sterilized (data not shown). Both treatment regimens also provided similar levels of protection from metastatic renal seeding (Fig. 4A). However, 100% of valvular vegetations were sterilized by the antibiotic-antibody combination in comparison to only 43% of those receiving antibiotic alone (P < 0.025) (Fig. 4B). In animals with well-established MRSA IE, the clearance of bacteremia was substantially faster and more complete in the antibiotic-antibody-treated group than in the antibiotic-alone group (Fig. 5). Among animals treated with vancomycin alone, 67% remained culture positive at 72 h (Fig. 5A), whereas all blood samples were rendered culture negative by the combination therapy by this time point (P = 0.0110) (Fig. 5B). The effect of SA-IGIV was less dramatic in the target tissue samples. In vegetations, both vancomycin alone and vancomycin plus SA-IGIV produced similar reductions in bacterial densities compared to that in the untreated control group (P < 0.05) (data not shown). Likewise, bacterial densities in kidney tissues were similar for the vancomycin-treated and vancomycin-plus-SA-IGIV-treated groups, and neither treatment provided significant reductions relative to that of the control group (data not shown).
FIG. 4.
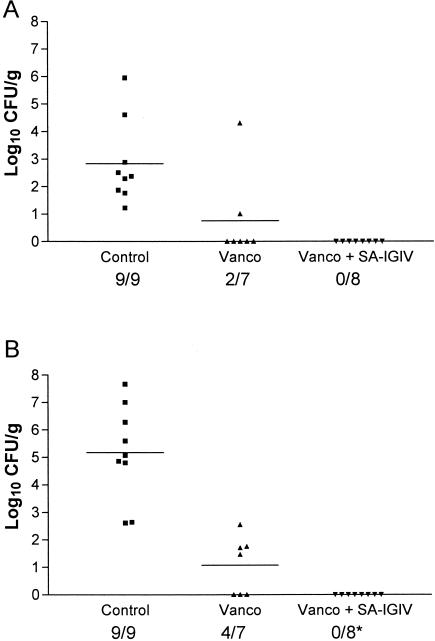
Clearance of MRSA from target organs (12 h). At 12 h after induction of MRSA IE, rabbits received one of the following: no therapy (Control), vancomycin alone (Vanco), or vancomycin plus SA-IGIV (Vanco + SA-IGIV). The data are presented as log10 CFU per gram measured in kidney lesions (A) and cardiac valve vegetations (B). Group means are indicated by horizontal lines. Numbers beneath the x axis indicate the number of infected samples per total number of samples evaluated. The asterisk indicates a significant difference between the number of infected tissues and that of the control (P < 0.025).
FIG. 5.
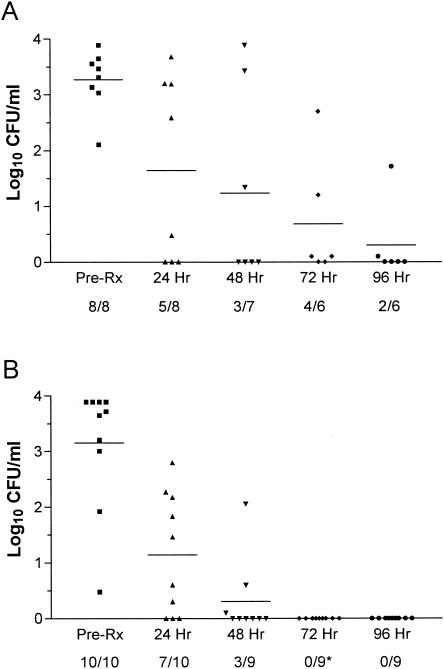
Bloodstream clearance of MRSA in rabbits with IE (24 h). At 24 h after induction of MRSA IE, rabbits received either vancomycin alone (A) or vancomycin plus SA-IGIV (B). The data are presented as log10 CFU per milliliter of blood. Group means are indicated by horizontal lines. Numbers beneath the x axis indicate the number of positive blood cultures per total number of blood samples evaluated. The asterisk indicates a significant difference between the number of positive blood cultures and that of the control (P = 0.0110).
DISCUSSION
IGIV are approved for a wide variety of clinical indications, including replacement therapy in primary and secondary immunodeficiency, pediatric AIDS, allogeneic bone marrow transplantation, Kawasaki's disease, and Guillain-Barré syndrome (22). Many clinical studies have also been conducted to assess the efficacy of IGIV treatment in the prevention of sepsis (27). In fact, at least 20 separate studies have reported the use of IGIV for the prophylaxis of neonatal infections (12). Despite some indications of efficacy, it is clear that the current IGIV preparations have quite different potencies against specific pathogens and that a customized approach designed to target specific pathogenic organisms would likely achieve more consistent and salutary results.
In an effort to create a more consistent and potent IGIV for the prevention and treatment of S. aureus infections, we have formulated a hyperimmunoglobulin with an increased antibody titer against a major, pathogenically important MSCRAMM protein, ClfA. The product was prepared from human plasma donors who were screened for the presence of high-titer, anti-Clf40 (ClfA A domain) IgG. A rabbit model of MRSA IE was used to evaluate the potential therapeutic efficacy of this product in mitigating the course of a multisystem staphylococcal infection. Products similar to SA-IGIV have been demonstrated to have prophylactic efficacy against S. aureus infection (at doses of 200 mg/kg) in both a rodent model of sepsis (13) and in the rabbit IE model (data not shown). In both models, prophylaxis with nonselected, normal human IGIV preparations had no effect on the course of disease. In the present study, we assessed the therapeutic potential of the same dose of SA-IGIV (200 mg/kg) in the rabbit IE model. Of note, the SA-IGIV dose chosen was also well below typical clinical doses of IGIV, which can range from 500 mg/kg to 1 g/kg (12, 27). A pilot study performed with a small number of animals indicated that SA-IGIV alone only transiently impacted the course of early MRSA IE (i.e., a modest, early reduction in the extent of bacteremia). We therefore chose to evaluate the benefit of SA-IGIV therapy in combination with a clinically relevant standard antibiotic therapy (vancomycin). Our choice of vancomycin dosage stemmed from long experience in using this antibiotic in the rabbit IE model against the MRSA 67-0 strain. Vancomycin has been successfully used to treat strain 67-0-mediated IE in the rabbit model at doses of 7.5 to 20 mg/kg when administered intravenously twice daily (2, 11; Kupferwasser et al., Abstr. 103rd Gen. Meet. Am. Soc. Microbiol. 2003). To increase the likelihood of divulging potential salutary effects of SA-IGIV-antibiotic combination treatment, the lowest effective vancomycin dose (7.5 mg/kg) was used. In both early and well-established infections, the addition of SA-IGIV to vancomycin increased the rate of bacterial clearance from blood and/or selected target tissues. When administered 12 h after induction of IE, the combination of vancomycin and SA-IGIV resulted in the complete elimination of bacteria from the heart valve vegetations and kidneys of treated rabbits. This result clearly demonstrated that the administration of SA-IGIV plus vancomycin early in the infection process may be potentially curative under conditions in which vancomycin alone is only modestly effective. Even when the infection was allowed to progress for an additional 12 h, at which time blood and target tissues contained large concentrations of MRSA, the beneficial effects of SA-IGIV treatment over that of vancomycin alone were clearly demonstrable in terms of clearance of bacteremia. The results reported here establish that passive immunotherapy against an MSCRAMM protein can be effectively used in conjunction with a standard antimicrobial regimen to amplify treatment outcomes of an established S. aureus infection.
The mechanism(s) of the salutary effect of SA-IGIV may reflect both antiadhesion and opsonic properties of this antibody. Flow cytometry showed that antibodies present in the SA-IGIV preparation were capable of recognizing antigens on the surface of S. aureus and, based on the intensity of the fluorescence, the bacterial surface appeared to be highly decorated with IgG. Some of this signal was certainly due to the anti-ClfA antibodies present in SA-IGIV, although it is likely that antibodies with specificities for other staphylococcal antigens also contributed to this signal. It is reasonable to speculate that many of these antibodies may be opsonizing and therefore may also contribute to the efficacy of SA-IGIV in vivo. Using an in vitro test for opsonization, SA-IGIV was shown to be capable of triggering recognition and uptake of Clf40-coated microspheres by human PMNs. Therefore, the ClfA-specific antibodies in SA-IGIV may act as a potent opsonin which would enable recognition and clearance of bacteria by phagocytic cells. Immune clearance of bacteria by this mechanism is likely to play an important role in the therapeutic efficacy of this antibody in the rabbit IE model. This enhanced OP property of SA-IGIV may have been reflected in the facilitated blood culture clearance in antibody-treated animals in this study.
In addition to the potential therapeutic effects of opsonizing S. aureus, SA-IGIV has the capacity to inhibit S. aureus binding to fibrinogen. Fibrinogen binding mediated by ClfA has been shown to be a key virulence factor for S. aureus, particularly in endovascular infections such as IE (20, 21, 25). Inhibition of fibrinogen binding would likely interfere with the capacity of S. aureus to spread hematogenously, bind to sites of vascular damage in IE (e.g., to kidneys), and reseed the damaged heart valve. Thus, this antibody may limit the capacity of S. aureus to colonize and replicate at these sites and may reduce the bacterial burden and the overall severity of disease.
Studies to adjudicate the relative impact of SA-IGIV on S. aureus adhesion and S. aureus opsonophagocytosis in vivo are in progress. The implications for the therapeutic use of SA-IGIV in the clinical setting warrant further studies to evaluate the biological role of this antibody in invasive S. aureus infections.
Acknowledgments
We thank Y.-Q. Xiong for assistance with the animal IE model. We also thank Robert Apkarian of the Integrated Microscopy & Microanalytical Facility of Emory University for performing transmission electron microscopy of the opsonophagocytosis assay samples.
REFERENCES
- 1.Bassoe, C.-F. 2002. Assessment of phagocyte functions by flow cytometry, p. 9.19.1-9.19.22. In J. P. Robinson, Z. Darzynkiewicz, P. N. Dean, A. R. Hibbs, A. Orfao, P. S. Rabinovitch, and L. L. Wheeless (ed.), Current protocols in cytometry. John Wiley & Sons, Inc., New York, N.Y. [DOI] [PubMed]
- 2.Bayer, A. S., C. Li, and M. Ing. 1998. Efficacy of trovafloxacin, a new quinolone antibiotic, in experimental staphylococcal endocarditis due to oxacillin-resistant strains. Antimicrob. Agents Chemother. 42:1837-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Booth, M. C., L. M. Pence, P. Mahasreshti, M. C. Callegan, and M. S. Gilmore. 2001. Clonal associations among Staphylococcus aureus isolates from various sites of infection. Infect. Immun. 69:345-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention. 2000. Semiannual report. Aggregated data from the National Nosocomial Infections Surveillance (NNIS) system, June 2000. Centers for Disease Control and Prevention, Atlanta, Ga.
- 5.Centers for Disease Control and Prevention. 2002. Staphylococcus aureus resistant to vancomycin—United States, 2002. Morb. Mortal. Wkly. Rep. 51(26):565-567. [PubMed] [Google Scholar]
- 6.Chambers, H. F. 2001. The changing epidemiology of Staphylococcus aureus? Emerg. Infect. Dis. 7:178-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohn, E. J., J. L. Oncley, L. E. Strong, W. L. Hughes, Jr., and S. H. Armstrong, Jr. 1944. Chemical, clinical and immunological studies on the products of human plasma fractionation. I. The characterization of the protein fractions of human plasma. J. Clin. Investig. 23:417-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Foster, T. J., and M. Hook. 1998. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 6:484-488. [DOI] [PubMed] [Google Scholar]
- 9.Guy, B., C. Testart, S. Gimenez, V. Sanchez, P. Lheritier, D. Rossin, M. Mignon, B. Danve, and E. Trannoy. 2000. Comparison of polymorphonuclear cells from healthy donors and differentiated HL-60 cells as phagocytes in an opsonophagocytic assay using antigen-coated fluorescent beads. Clin. Diagn. Lab. Immunol. 7:314-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hawiger, J., S. Timmons, D. D. Strong, B. A. Cottrell, M. Riley, and R. F. Doolittle. 1982. Identification of a region of human fibrinogen interacting with staphylococcal clumping factor. Biochemistry 21:1407-1413. [DOI] [PubMed] [Google Scholar]
- 11.Hirano, L., and A. S. Bayer. 1991. β-Lactam-β-lactamase-inhibitor combinations are active in experimental endocarditis caused by β-lactamase-producing oxacillin-resistant staphylococci. Antimicrob. Agents Chemother. 35:685-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenson, H. B., and B. H. Pollock. 1997. Meta-analyses of the effectiveness of intravenous immune globulin for prevention and treatment of neonatal sepsis. Pediatrics 99(2):e2. [Online.] http://pediatrics.aappublications.org. [DOI] [PubMed]
- 13.Josefsson, E., O. Hartford, L. O'Brien, J. M. Patti, and T. Foster. 2001. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J. Infect. Dis. 184:1572-1580. [DOI] [PubMed] [Google Scholar]
- 14.Lehmann, A. K., A. Halstensen, J. Holst, and C. F. Bassoe. 1997. Functional assays for evaluation of serogroup B meningococcal structures as mediators of human opsonophagocytosis. J. Immunol. Methods. 200:55-68. [DOI] [PubMed] [Google Scholar]
- 15.Lehmann, A. K., A. Halstensen, and C. F. Bassoe. 1998. Flow cytometric quantitation of human opsonin-dependent phagocytosis and oxidative burst responses to meningococcal antigens. Cytometry 33:406-413. [DOI] [PubMed] [Google Scholar]
- 16.Lehmann, A. K., A. Halstensen, I. S. Aaberge, J. Holst, T. E. Michaelsen, S. Sørnes, L. M. Wetzler, and H.-K. Guttormsen. 1999. Human opsonins induced during meningococcal disease recognize outer membrane proteins PorA and PorB. Infect. Immun. 67:2552-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDevitt, D., P. Francois, P. Vaudaux, and T. J. Foster. 1994. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol. Microbiol. 11:237-248. [DOI] [PubMed] [Google Scholar]
- 18.McDevitt, D., P. Francois, P. Vaudaux, and T. J. Foster. 1995. Identification of the ligand-binding domain of the surface located fibrinogen receptor (clumping factor) of Staphylococcus aureus. Mol. Microbiol. 16:895-907. [DOI] [PubMed] [Google Scholar]
- 19.McDevitt, D., T. Nanavaty, K. House-Pompeo, E. Bell, N. Turner, L. McIntire, T. Foster, and M. Hook. 1997. Characterization of the interaction between the Staphylococcus aureus clumping factor (ClfA) and fibrinogen. Eur. J. Biochem. 247:416-424. [DOI] [PubMed] [Google Scholar]
- 20.Meier, P. S., J. M. Entenza, P. Vaudaux, P. Francioli, M. P. Glauser, and P. Moreillon. 2001. Study of Staphylococcus aureus pathogenic genes by transfer and expression in the less virulent organism Streptococcus gordonii. Infect. Immun. 69:657-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moreillon, P., J. M. Entenza, P. Francioli, D. McDevitt, T. J. Foster, P. François, and P. Vaudaux. 1995. Role of Staphylococcus aureus coagulase and clumping factor in the pathogenesis of experimental endocarditis. Infect. Immun. 63:4738-4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nydegger, U. E., P. J. Mohasci, R. Escher, and A. Morell. 2000. Clinical use of intravenous immunoglobulins. Vox Sang. 78(Suppl. 2):191-195. [PubMed] [Google Scholar]
- 23.O'Connell, D. P., T. Nanavaty, D. McDevitt, S. Gurusiddappa, M. Hook, and T. J. Foster. 1998. The fibrinogen-binding MSCRAMM (clumping factor) of Staphylococcus aureus has a Ca2+-dependent inhibitory site. J. Biol. Chem. 273:6821-6829. [DOI] [PubMed] [Google Scholar]
- 24.Patti, J. M., B. L. Allen, M. J. McGavin, and M. Hook. 1994. MSCRAMM mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 48:585-617. [DOI] [PubMed] [Google Scholar]
- 25.Que, Y.-A., P. François, J.-A. Haefliger, J.-M. Entenza, P. Vaudaux, and P. Moreillon. 2001. Reassessing the role of Staphylococcus aureus clumping factor and fibronectin-binding protein by expression in Lactococcus lactis. Infect. Immun. 69:6296-6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strong, D. D., A. P. Laudano, J. Hawiger, and R. F. Doolittle. 1982. Isolation, characterization, and synthesis of peptides from human fibrinogen that block the staphylococcal clumping reaction and construction of a synthetic clumping particle. Biochemistry 21:1414-1420. [DOI] [PubMed] [Google Scholar]
- 27.Werdan, K. 1999. Supplemental immune globulins in sepsis. Clin. Chem. Lab. Med. 37:341-349. [DOI] [PubMed] [Google Scholar]
- 28.Yeaman, M. R., J. Lee, and A. S. Bayer. 1999. Experimental Candida endocarditis, p. 1709-1720. In O. Zak and M. A. Sande (ed.), Handbook of animal model infections. Academic Press, New York, N.Y.