

Abstract
Long-term antibiotic treatment is required to cure tuberculosis. Targeted antibiotics should improve the efficacy of treatment by concentrating the drugs close to the bacteria. The aim of the present study was to synthesize targeted conjugates. For this purpose, we used mannose as a homing device to direct norfloxacin into macrophages. Dextran was used as the polymer bearing both mannose and norfloxacin. Using different peptide spacer arms to link norfloxacin to dextran, we demonstrated that norfloxacin acts as an antibiotic only when it is released in its native form. Also, targeting by using mannose as a homing device is required to achieve antimycobacterial activity in vivo. Thus, norfloxacin, which is inactive against mycobacteria in its native form in vivo, can be transformed into an active drug by targeting.
A variety of bacterial and protozoan pathogens reside within host macrophages. These pathogens are largely protected against many of the host's defense mechanisms, i.e., antibodies and/or complement. This intracellular mode of life also means that the pathogens can potentially become quiescent or latent. Examples of facultative or obligate intracellular pathogens include the agents responsible for chronic bacterial diseases such as brucellosis, trachoma, and tuberculosis and those responsible for protozoan diseases such as toxoplasmosis and leishmaniasis.
In this report, we focus on bacterial infections and, more precisely, on infections caused by mycobacteria, which are facultative intracellular pathogens. They escape killing during phagocytosis by blocking phagosome-lysosome fusion. Intracellular mycobacteria are also largely protected against drugs. Consequently, it is difficult to destroy the pathogen while leaving the host cells intact. The main aim of this work was to design a conjugate able to target and to deliver a drug into the phagosomal vacuole, such that it is brought into close contact with the engulfed bacteria.
The targeting of drugs into cells, more specifically, into phagocytic cells, was proposed by De Duve et al. (5), who developed the notion of lysosomotropic drugs or carriers. Different vehicle molecules have been prepared and tested, but the results were disappointing. Some of these molecules were active in vitro, but they were frequently unable to find their targets in vivo or to deliver the drug into the infected cells in an active form.
The temporary conjugation of a drug to a homing device through a macromolecular carrier could induce the endocytosis of the drug by the target cell via a specific receptor and, following this first step, subcellular distribution of the drug to sites where the bacilli are localized. The use of mannosyl ligands to target macrophages has been proposed (9).
Water-soluble polyfunctional polymers can accommodate several homing device residues, thus increasing the affinity for the target (10). Similarly, the linking of several drug molecules may increase the rate at which the drug is delivered to the target. A conjugate that is large enough to impair renal filtration may optimize drug delivery (1).
We report on the development of a macromolecular vehicle based on dextran linked to mannose, to target the macrophage, and norfloxacin, to be delivered into the phagosomal vacuole in its active form. We describe the synthesis of conjugates in which norfloxacin was linked to the macromolecular carrier through two different peptide arms. The relative in vitro antibiotic efficacies of the different norfloxacin macromolecular prodrugs were determined, and we tested the in vivo antibiotic activities of the macromolecular prodrugs against intracellular Mycobacterium bovis (BCG strain) in mice.
MATERIALS AND METHODS
Materials.
Norfloxacin was obtained from Sigma-Aldrich (Bornem, Belgium). Gly-Phe, Z-Gly-paranitrophenylester, methoxy (OMe) Ser (Ser-OMe), and Ala-Leu were purchased from Bachem (Bubendorf, Switzerland). para-Nitrophenyl-chloroformate was obtained from Merck (Darmstadt, Germany), and bovine cathepsin B was obtained from Fluka (Buchs, Switzerland). Dextran (Pharmacia, Uppsala, Sweden) was repurified to get fractions with weight-average molar masses of 64,000, 90,000, or 95,000 g mol−1 and with a weight-average molar mass/number-average molar mass ratio of 1.2. All other chemicals were purchased from Acros (Beerse, Belgium).
Synthesis of Leu-norfloxacin. (i) Synthesis of N-(tert-butoxycarbonyl)-l-Leu (Boc-Leu).
Leu (0.58 g, 4.5 mmol) was dissolved in a mixture of dioxane (10 ml) and 0.5 M NaOH (10 ml). After the mixture was cooled to 0°C, di-tert-butylpyrocarbonate (1.08 g, 4.95 mmol) was added. The reaction mixture was stirred for 1 h at 0°C and then for 4 h at room temperature. The dioxane was evaporated. The resulting aqueous solution was acidified to pH 3 with a KHSO4 solution (1 M) and extracted with ethyl acetate (two times with 100 ml each time). The ethyl acetate solution was dried on Na2SO4 and evaporated until it was dry.
(ii) Synthesis of Boc-Leu-pentafluorophenyl ester (Boc-Leu-PFP).
Boc-Leu (3.82 mmol) and pentafluorophenol (0.82 g, 4.44 mmol) were dissolved in dry tetrahydrofuran (20 ml). After the mixture was cooled to 0°C, dicyclohexylcarbodiimide (0.86 g, 4.16 mmol) was added. The reaction mixture was stirred for 1 h at 0°C and overnight at room temperature. The precipitate that formed during the reaction was filtered, and the filtrate was evaporated under vacuum. The residue was dissolved in ethyl acetate (30 ml), and the solution was filtered. The filtrate was evaporated. Rf (dichloromethane-methanol; 9/1) = 0.65. Infrared (film), 1,790 cm−1: PFP ester.
(iii) Synthesis of Boc-Leu-norfloxacin.
Norfloxacin was silylated in dichloromethane by adding two equivalents of N-methyl-N-(trimethylsilyl)trifluoroacetamide. Silylated norfloxacin and Boc-Leu-PFP were dissolved in dichloromethane in equivalent quantities. The solution was stirred overnight. The residue was dissolved in dichloromethane-methanol (9/1), extracted twice with water, and then purified by chromatography on a silica (normal phase) column.
1H nuclear magnetic resonance (NMR) (CDCl3) δ 8.70 (1H, s, C-2 norfloxacin), 8.15 (1H, d, C-5 norfloxacin), 6.85 (1H, d, C-8 norfloxacin), 4.70 (1H, m, CH, Leu), 4.35 (2H, q, CH2 norfloxacin), 4.1 to 3.5 (8H, m, 4-CH2, piperazine), 1.75 (1H, m, CH, Leu), 1.61 (3H, t, CH3, norfloxacin), 1.5 (2H, m, CH2, Leu), 1.40 (9H, m, Boc), 0.95 to 0.85 (2-CH3, Leu) ppm.
To obtain Leu-norfloxacin, the Boc group was removed by using trifluoroacetic acid.
Synthesis of Gly-Phe-Gly-Gly-(α-norfloxacin)OMe (Fig. 1). (i) Synthesis of benzyloxycarbonyl-Gly-Gly(α-norfloxacin)-OMe [Z-Gly-Gly(α-norfloxacin)-OMe]. (a) Z-Gly-Ser-OMe.
FIG. 1.
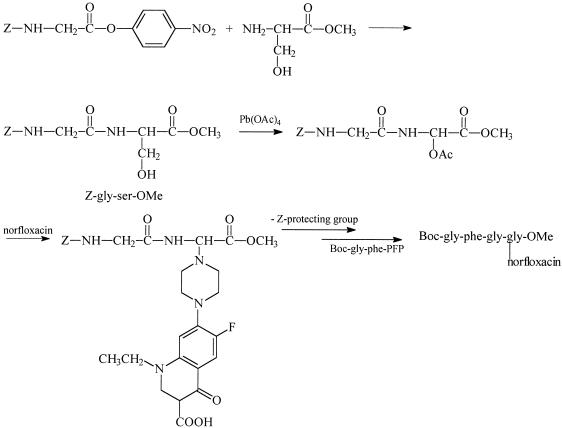
Schematic representation of Gly-Phe-Gly-Gly-(α-norfloxacin)-OMe synthesis.
Benzyloxycarbonyl-protected Gly-para-nitrophenylester (Z-Gly-para-nitrophenylester; 1 g, 3 mmol), Ser-OMe (0.46 g, 3 mmol), and N-methylmorpholine (3 ml) were dissolved in dimethylformamide (15 ml). The solvent was evaporated after 48 h, and the residue was purified by chromatography (dichloromethane-isopropanol; gradient from 98/2 to 88/12). Pure Z-Gly-Ser-OMe was obtained with a yield of 93%. Rf (dichloromethane-isopropanol; 9/1) = 0.4.
1H NMR (MeOD) δ 3.95 to 3.70 (7H, m, CH2 Gly + CH2 Ser + CH3 methyl ester), 4.55 (1H, m, CH Ser), 5.15 (2H, s, CH2 benzyl), 7.35 (5H, m, Phe) ppm.
(b) Z-Gly-Gly(α-OAc)-OMe.
Z-Gly-Ser-OMe (200 mg, 0.65 mmol), lead acetate [Pb(OAc)4; 340 mg, 0.97 mmol], and molecular sieves (pore size, 4 Å; 550 mg), which were used to trap water, were refluxed in dry ethyl acetate (20 ml) for 3 h. After the mixture was cooled, the mixture was filtered on Celite, and the solvent was evaporated. Pure Z-Gly-Gly(α-OAc)-OMe was obtained with a yield of 95%. Rf (dichloromethane-methanol; 9/1) = 0.5.
1H NMR (500 MHz, CDCl3) δ 7.35 (5H, m, phenyl), 6.40 (1H, d, CH of acetoxy [OAc]-substituted Gly), 5.15 (2H, s, CH2 benzyl), 4.00 (2H, m, CH2 Gly), 3.80 (3H, s, methylester), 2.10 (3H, s, CH3 of OAc) ppm.
(c) Z-Gly-Gly(α-norfloxacin)-OMe.
Z-Gly-Gly(α-OAc)-OMe (200 mg, 0.59 mmol) was dissolved in dimethylformamide (10 ml). Norfloxacin (189 mg, 0.59 mmol) dissolved in dimethylformamide and triethylamine (82 μl, 0.59 mmol) were added, and the mixture was stirred for 36 h. The solvent was evaporated, and Z-Gly-Gly(α-norfloxacin)-OMe was extracted with dichloromethane, then 50 mM HCl solution (three times), and finally, water (three times). Pure Z-Gly-Gly(α-norfloxacin)-OMe was obtained with a yield of 89%. Rf (dichloromethane-methanol-acetic acid; 95/5/0.1) = 0.14.
1H NMR (CDCl3) δ 8.65 (1H, s, H on C-2 norfloxacin), 7.93 (1H, d, H on C-5 norfloxacin), 7.40 (5H, m, phenyl), 7.05 (1H, d, NH-substituted Gly), 6.77 (1H, d, H on C-8 norfloxacin), 5.60 (1H, t, NH Gly), 5.45 (1H, d, CH), 5.15 (2H, s, CH2 benzyl), 4.31 (2H, q, CH2 norfloxacin), 4.00 (2H, d, CH2 Gly), 3.83 (3H, s, CH3 methyl ester), 3.30, 2.85, and 2.74 (8H, m, CH2 groups of piperazine), 1.60 (3H, t, CH3 norfloxacin) ppm.
(ii) Synthesis of Gly-Gly-(α-norfloxacin)-OMe.
Z-Gly-Gly(α-norfloxacin)-OMe (320 mg, 0.54 mmol) was dissolved in dry methanol (MeOH; 10 ml) and HBr in acetic acid (0.2 ml). To this solution, 10% Pd/C (320 mg) was added. After 24 h of stirring under H2 pressure, the catalyst was filtered and the filtrate was concentrated. Pure Gly-Gly-(α-norfloxacin)-OMe was obtained with a yield of 64%. Rf (dichloromethane-methanol-acetic acid; 95/5/0.1) = 0.0.
1H NMR (dimethyl sulfoxide [DMSO]-d6) δ 8.97 (1H, s, H on C-2 norfloxacin), 7.93 (1H, d, H on C-5 norfloxacin), 7.15 (1H, d, H on C-8 norfloxacin), 5.23 (1H, s, CH), 4.60 (2H, q, CH2 norfloxacin), 3.71 (3H, s, CH3 methyl ester), 3.55 to 2.7 (10H, m, CH2 Gly and 4-CH2 piperazine), 1.60 (3H, t, CH3 norfloxacin) ppm.
(iii) Synthesis of Boc-Gly-Phe-Gly-Gly(α-norfloxacin)-OMe.
Boc-Gly-Phe-O-PFP (137 mg, 0.28 mmol) and Gly-Gly(α-norfloxacin)-OMe (130 mg, 0.28 mmol) were dissolved in dimethylformamide (10 ml), and N-methylmorpholine (500 μl) was added. The dimethylformamide was evaporated after 48 h. Extraction with dichloromethane was followed by evaporation and purification by chromatography with a gradient (from dichloromethane-methanol [9/1] to dichloromethane-methanol-acetic acid [9/1/0.1]). Pure Boc-Gly-Phe-Gly-Gly(α-norfloxacin)-OMe was obtained with a yield of 70%. Rf (dichloromethane-methanol-acetic acid; 9/1/0.1) = 0.29.
1H NMR (DMSO-d6) δ 8.90 (1H, s, H C-2 norfloxacin), 8.61 (1H, m, NH), 8.48 (1H, m, NH), 8.15 (1H, m, NH), 7.85 (1H, m, H C-5 norfloxacin), 7.20 (6H, m, phenyl + H on C-8 norfloxacin), 6.88 (1H, m, NH), 5.23 (1H, m, CH), 4.50 (3H, m, CH Phe and CH2 norfloxacin), 3.85 (2H, m, CH2 Gly), 3.72 (3H, s, CH3), 3.60 to 3.40 (2H, m, CH2 Gly), 3.28 (4H, m, 2-CH2 piperazine), 3.05 (1H, m, HB Phe), 2.75 (5H, m, HA Phe + 2-CH2 piperazine), 1.35 (12H, m, Boc and CH3 norfloxacin) ppm.
(iv) Coupling of Gly-Phe-Gly-Gly(α-norfloxacin)-OMe to activated dextran (Fig. 2).
FIG. 2.
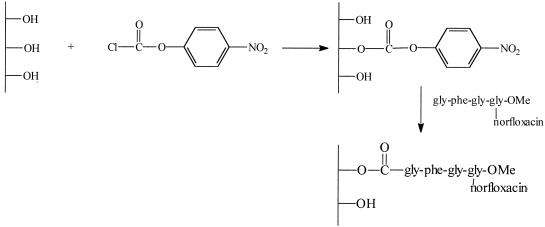
Schematic representation of dextran-Gly-Phe-Gly-Gly-(α-norfloxacin)-OMe synthesis.
Chloroformate-activated dextran (13), with 15 mol% linear carbonates, as measured by spectrophotometry (402 nm, ɛ = 18,400 l mol−1 cm−1), was dissolved in DMSO-pyridine (1/1). Gly-Phe-Gly-Gly(α-norfloxacin)-OMe was added to the solution. The Boc-protecting group was first removed by dissolving Boc-Gly-Phe-Gly-Gly(α-norfloxacin)-OMe (50 mg, 65 μmol) in trifluoroacetic acid (2 ml). The mixture was stirred for 30 min. After evaporation of the solvent, the residue was dried and added to the activated dextran. After 48 h, the conjugate was precipitated in ethanol-ether (1/1) and then dissolved in NaOH (0.1 M, 8 ml) and purified by preparative gel permeation chromatography (Sephadex G25; Pharmacia). The substitution degree of norfloxacin was measured by UV spectroscopy at 278 nm and 1H NMR.
The degree of substitution was determined by comparison of integration of the signals by 1H NMR (D2O) δ 4.95 ppm (H-1 dextran) and δ 7.20 ppm (6H, phenyl and H on C-8 norfloxacin).
(v) Synthesis of the conjugate dextran-Gly-Phe-Ala-Leu-norfloxacin.
The dextran-Gly-Phe-Ala-Leu-norfloxacin conjugate was synthesized as described previously (4).
(vi) Mannosylation.
Mannosylation (Fig. 3) was carried out as described previously (6, 13).
FIG. 3.
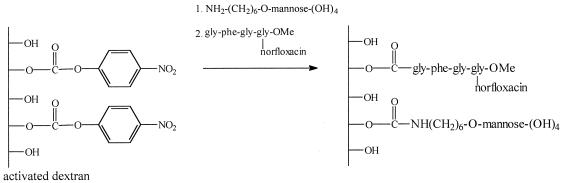
Schematic representation of mannosylated dextran-Gly-Phe-Gly-Gly-(α-norfloxacin)-OMe synthesis.
Release of norfloxacin from the conjugates. (i) Chemical release.
One milligram of the polymeric norfloxacin conjugate was dissolved in 1 ml of either 0.1 M phosphate buffer (pH 7.4) or 0.28 M citrate-phosphate buffer (pH 5.5) at 37°C. At predetermined time intervals, samples were taken and analyzed by high-pressure liquid chromatography (PLRP-S column, 100 Å, 5-μm Achrom; Zulte) with a mixture of acetonitrile-methanol-tetrahydrofuran-acetic acid-water (228/ 39/16/138/1,582) as the eluent. The flow rate was 0.75 ml/min. The products were detected spectophotometrically at 278 nm.
(ii) Proteolysis.
The conjugate (1 mg) was dissolved in 2 ml of a citrate-phosphate buffer (0.28 M pH 5.5) containing reduced glutathione (5 mM), EDTA (1 mM), and bovine cathepsin B (250 μg). Aliquots were taken at regular intervals and analyzed by high-pressure liquid chromatography.
Bacteriology. (i) Bacteria.
The bacteria used were Escherichia coli K12 (ATCC 25290), Brucella melitensis (ATCC 739), Staphylococcus aureus (ATCC 6538), and M. bovis (BCG strain 1173P2; Institut Pasteur).
(ii) MICs and MBCs.
The antimicrobial activities, i.e., the MICs and minimal bactericidal concentrations (MBCs), of native norfloxacin and Leu-norfloxacin were determined. The comparison of native norfloxacin and Leu-norfloxacin was based on the use of equimolar amounts of norfloxacin. Norfloxacin was dissolved in HCl (1 M), and Leu-norfloxacin was dissolved in water. The MIC was determined by adding 105 CFU of bacteria to tubes containing 1 ml of serial twofold dilutions of norfloxacin and Leu-norfloxacin in broth (Mueller-Hinton broth; Difco). After 18 h at 37°C, the MIC was defined as the lowest concentration of the tested molecule that prevented bacterial growth. When no bacterial growth was observed, samples of medium (100 μl) were plated on a suitable agar medium. After incubation at 37°C, colonies were detected. The MBC was defined as the lowest concentration of the tested molecule at which no viable bacteria were present in the sample.
(iii) In vitro antimycobacterial activities of norfloxacin and of its macromolecular conjugates.
One hundred microliters of Middlebrook 7H9 medium (Difco) containing 104 CFU of BCG (105 per ml) was placed in each well of a microtiter plate (Nunc) containing 50 μl of appropriate dilutions of the different preparations to be tested. After 4 days at 37°C, 50 μl of 7H9 medium containing 10 μCi of [3H]uracil (Amersham) per ml was added for 18 h. The labeled bacteria were harvested onto glass fiber filters and subjected to liquid scintillation counting. The results are expressed as the mean ± standard deviation counts per minute for triplicate culture wells.
In vivo assays. (i) Animals.
Specific-pathogen-free C57BL/6 mice (age, 6 weeks) were obtained from Iffa-Credo (Saint-Germain sur l'Arbesle, France).
(ii) Microorganisms and infection.
M. bovis BCG 1173P2 (Institut Pasteur) was grown on Sauton medium for 14 days. The bacilli were then homogenized with stainless steel balls in the same medium to a concentration of 50 mg ml−1 (107 viable units per mg) (8). Vials containing the BCG suspension were stored at −70°C until use. Mice were infected by intravenous injection of 0.5 ml containing 106 viable units.
(iii) Treatment of mice.
Starting from the 6th day after infection, the mice (five per group) were injected intraperitoneally twice a day for 5 days with the different test molecules. Phosphate-buffered saline (PBS) and isoniazid (0.5 mg per mouse per day) were used as negative and positive control treatments, respectively. The macromolecular conjugates containing norfloxacin were diluted in PBS, and each mouse was injected with the equivalent of 0.25 mg of norfloxacin per day. Native norfloxacin was injected as a control (0.5 mg per day).
(iv) BCG growth in lungs.
BCG growth was monitored by counting the number of viable units in the lungs of the mice 2 days after the end of treatment. This 2-day washout period allowed the elimination of persisting native antibiotics. The lung tissues were homogenized, and suitable dilutions were plated on Middlebrook 7H11 medium. The number of CFU was counted after 21 days at 37°C.
Statistics.
The means for each group of five mice were tested by one-way analysis of variance by the Tukey-Kramer multiple-comparison test (Instat; GraphPad Software, San Diego, Calif.).
RESULTS
Synthesis.
The conjugates designed to target an antibiotic into macrophages included dextran as the carrier, norfloxacin as the antibiotic, and, sometimes, mannose as a homing device. To try to produce a carrier that was able to release native norfloxacin, the antibiotic was linked to the carrier by means of two different chemical bonds, an amide bond and an α bond, borne by two different tetrapeptide linkers.
When norfloxacin was linked to Gly-Phe-Ala-Leu through an amide bond, Leu-norfloxacin was released in the presence of cathepsin B. Thus, Leu-norfloxacin was synthesized to study its in vitro antimicrobial efficacy.
In Gly-Phe-Gly-Gly(α-norfloxacin)-OMe, norfloxacin was linked to the carrier through an α bond. The structures of the four norfloxacin conjugates, consisting of two different linkers with or without mannose, are shown in Fig. 4. The chemical characteristics of the four conjugates are summarized in Table 1.
FIG. 4.
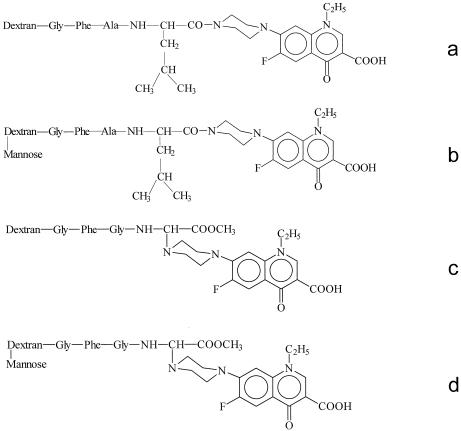
Schematic representation of the structures of the different conjugates studied. Norfloxacin was linked to the carrier either through an amide bond (a and b) or through an α bond (c and d), and the conjugates were mannosylated (b and d) or not (a and c).
TABLE 1.
Chemical characteristics of the four norfloxacin conjugates
| Product | Mw of dextran | Mannose concn (mol%) | Norfloxacin concn (% [wt/wt]) | Norfloxacin concn (mol%) |
|---|---|---|---|---|
| Dextran-GFGG-α-norfloxacin | 95,000 | 4.9 | 3.3 | |
| Dextran-GFAL-norfloxacin | 95,000 | 7.1 | 4.3 | |
| Mannosylated dextran-GFGG-α-norfloxacin | 64,000 | 2.3 | 8.2 | 5.5 |
| Mannosylated dextran-GFAL-norfloxacin | 90,000 | 1.7 | 8.3 | 5.2 |
Norfloxacin release.
We first studied the macromolecular conjugates bearing two different bonds to link norfloxacin, but no mannose, in vitro to determine their capacity to release norfloxacin. This was done in the presence and absence of cathepsin B, because the peptide arms linking norfloxacin to its macromolecular carrier were susceptible to this lysosomal enzyme. Less than 0.2% of the norfloxacin was spontaneously released from the Gly-Phe-Ala-Leu tetrapeptide arm, i.e., the arm bearing an amide bond, in 24 h at both plasma pH (pH 7.4) and lysosome pH (pH 5.5) (Fig. 5). Moreover, cathepsin B, which degrades the peptide arm, did not hydrolyze the amide bond between norfloxacin and the last amino acid (Leu), meaning that Leu-norfloxacin was released (4). The antimicrobial efficacy of Leu-norfloxacin was found to be negligible (see below).
FIG. 5.
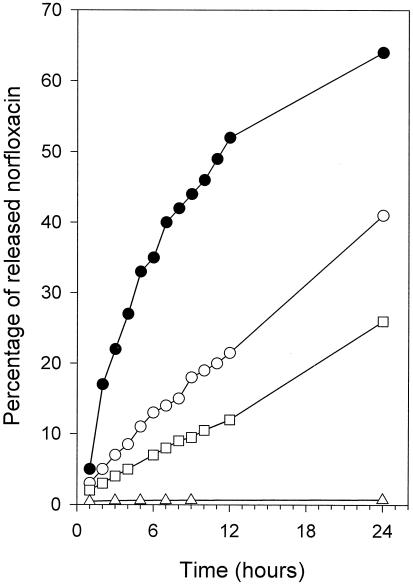
In vitro release of norfloxacin from macromolecular conjugates as a function of time. Open triangles, amide bond; open squares, α bond, pH 7.4; open circles, α bond, pH 5.5; closed circles, α bond, pH 5.5, in the presence of cathepsin B.
In contrast to this behavior, about 25% of the norfloxacin was spontaneously released from Gly-Phe-Gly-Gly(α-norfloxacin)-OMe, which bears an α bond, after 24 h at pH 7.4, whereas about 40% of the norfloxacin was spontaneously released at pH 5.5 (Fig. 5). In the presence of cathepsin B, norfloxacin was released (only the release at pH 5.5 was studied because the optimum pH of cathepsin B is between 4.5 and 6) from the peptide bearing an α bond about four times more rapidly, as shown by the initial release rates (Fig. 5). Regardless of whether the release mechanism was chemical or enzymatic, the Gly-Phe-Gly-Gly(α-norfloxacin)-OMe peptide always released native norfloxacin.
Microbiology.
As the conjugate consisting of norfloxacin linked to the Gly-Phe-Ala-Leu tetrapeptide arm through an amide bond released Leu-norfloxacin, we wondered whether this compound had any antibacterial activity. Consequently, we determined the MICs and MBCs of Leu-norfloxacin for E. coli, S. aureus, and B. melitensis. Whereas norfloxacin was active against these bacteria in vitro, Leu-norfloxacin was inactive under the same conditions (Table 2).
TABLE 2.
MICs and MBCs of norfloxacin and Leu-norfloxacin for E. coli, S. aureus, and B. melitensis
| Bacterium | Norfloxacin
|
Leu-norfloxacin
|
||
|---|---|---|---|---|
| MICa | MBCa | MICa | MBCa | |
| E. coli K-12 | 0.078 | 0.156 | 10 | >10 |
| S. aureus | 0.625 | 1.25 | 10 | >10 |
| B. melitensis | 0.625 | 2.5 | 2.5 | 10 |
The results are expressed in milligrams of equivalent norfloxacin liter−1.
As the ultimate goal of this work was to design a macromolecular prodrug active against intracellular bacteria, particularly members of the genus Mycobacterium, we tested the activities of the conjugates prepared during this work against M. bovis BCG in vitro. The antibacterial activities of native norfloxacin and the dextran-norfloxacin conjugates containing either the amide bond or the α bond were compared at equal norfloxacin concentrations (Fig. 6). The antibacterial activity of the conjugate bearing an α bond was similar to that of native norfloxacin. The conjugate in which an amide bond links leucine to norfloxacin was at least 20 times less active than the other conjugate.
FIG. 6.
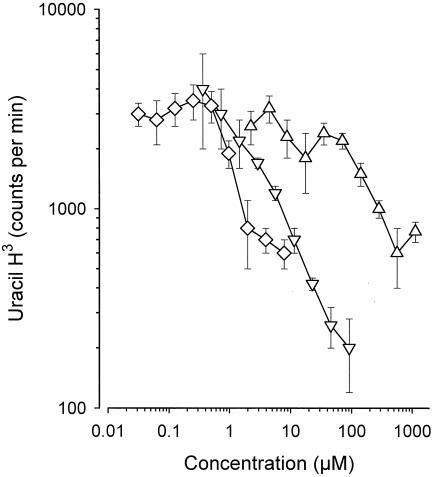
In vitro anti-M. bovis BCG activities of norfloxacin (diamonds) and its nonmannosylated macromolecular conjugates containing an α bond (inverted triangles) or an amide bond (triangles) as a function of the molar concentration of native or conjugated norfloxacin present in the medium. Bacterial multiplication was assessed by measuring the incorporation of [3H]uracil by M. bovis.
In a second series of in vitro experiments with M. bovis (Fig. 7), we compared the bactericidal activities of native norfloxacin and the conjugate of norfloxacin containing an α bond to the activity of the reference antibiotic, isoniazid. Isoniazid was about 10 times more active than norfloxacin at an equal molar concentration. Likewise, the dextran-norfloxacin conjugate was slightly less active than norfloxacin alone. As expected, the presence of mannose on the dextran carrier had no effect on the antibiotic activity in vitro, as no macrophages were present in this test (results not shown).
FIG. 7.
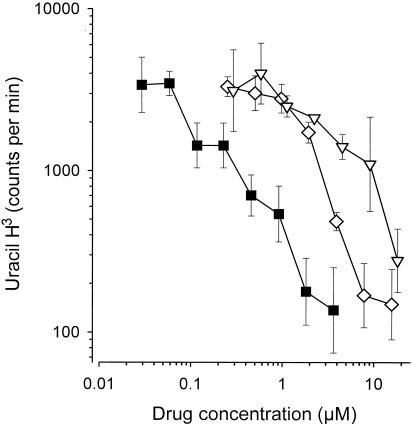
In vitro anti-M. bovis BCG activities of isoniazid (closed squares), norfloxacin (diamonds), and the nonmannosylated macromolecular conjugate of norfloxacin containing an α bond (inverted triangles) as a function of the molar concentration of active drug (i.e., isoniazid or norfloxacin) in the medium. Bacterial multiplication was assessed by measuring the incorporation of [3H]uracil by M. bovis.
In vivo assays.
To test these molecules under conditions resembling the clinical situation as closely as possible, we compared the antibacterial activities of native norfloxacin and the different macromolecular conjugates of norfloxacin in mice infected with M. bovis BCG. PBS was also tested as a negative control, and isoniazid was tested as a positive control. The results, expressed as the number of CFU per lung, are presented in Fig. 8. As expected, isoniazid was active against M. bovis BCG, whereas PBS was not. Despite its in vitro activity, native norfloxacin was not active in vivo, probably because it is rapidly filtered by the kidneys (2). Only the conjugate containing both an α bond and mannose was as active as isoniazid against M. bovis BCG. The other conjugates, which contained either an amide bond with or without mannose or an α bond without mannose, were all inactive.
FIG. 8.
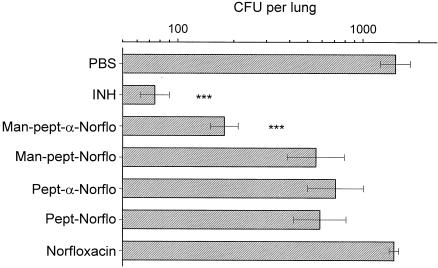
In vivo anti-M. bovis BCG activities of isoniazid (INH), PBS, norfloxacin, and the four macromolecular conjugates of norfloxacin. These conjugates were mannosylated (Man) or not and contained either an α bond or an amide bond. The antimicrobial activities, expressed as the number of CFU per organ, were measured in the lungs of mice (n = 5). Error bars indicate standard deviations. ***, a highly significant difference (P < 0.001) compared to the results for the control (PBS-treated) group. No significant differences were observed for the other groups.
DISCUSSION
The medical importance of killing intracellular bacteria has led to the development of different macromolecular prodrugs that can be targeted to macrophages. Members of our group have previously developed several macromolecular prodrugs that include norfloxacin as the antibiotic (4). To enable it to be released preferentially in the lysosomes, the antibiotic was linked to the carrier through one of two tetrapeptides, Gly-Phe-Ala-Leu or Gly-Phe-Leu-Gly, both of which may be cleaved by lysosomal proteases such as cathepsin B (11). In these compounds, an amide bond linked the piperazine ring of norfloxacin to the C-terminal amino acid (i.e., leucine or glycine). As this amide bond is not destroyed by cathepsin B in vitro, Leu-norfloxacin and Gly-norfloxacin, respectively, are released (4). These derivatives, as well as Lys-norfloxacin, had no activity against E. coli, S. aureus, and B. melitensis (this study and unpublished results). Another norfloxacin derivative, 4-bromoethyloxy-carbonyl-norfloxacin, displayed only low levels of activity against E. coli. This residual activity is due to chemical hydrolysis at pH 7.4, which leads to the subsequent release of native norfloxacin at proportions of 7% after 1 day and 18% after 3 days (7). All these observations demonstrate that the release of native norfloxacin is essential for antibacterial activity.
As it is critical that the antibiotic be released in its active form, we linked norfloxacin to the carrier through an N—C bond to the C-terminal glycine (called the α bond) of Gly-Phe-Gly-Gly: the piperazine ring of norfloxacin substitutes for an α hydrogen atom of the methylene group of the glycine residue to form an α-substituted glycine, as suggested by Nichifor and Schacht (12). The drug forms the lateral chain of the α-substituted glycine. When the atom (of the drug forming the lateral chain) bound to glycine is N, O, or S, the α-substituted glycine is unstable unless its amino group is engaged in an amide bond. Under the conditions of our study, the amino group of the glycine derivative was linked to another glycine residue, its COOH was blocked by a methanol residue, and it bore norfloxacin linked through the secondary amine of its piperazine ring. In the presence of cathepsin B, the C-terminal amide bond of Gly-Phe-Gly-Gly(α-norfloxacin)-OMe was hydrolyzed, releasing the unstable α-substituted glycine, which spontaneously decomposed to release native norfloxacin.
In vitro, about 1% norfloxacin was released from the tetrapeptide Gly-Phe-Gly-Gly(α-norfloxacin)-OMe per hour at the blood pH. This slow norfloxacin release explains its in vitro activity against M. bovis. Gly-Phe-Gly-Gly(α-norfloxacin)-OMe is also susceptible to proteolysis by cathepsin B, which can occur in the lysosome. In the lysosomal environment, the proteolytic release of norfloxacin from the linking arm may be more rapid than that in vitro, as several different proteases with different specificities are present. This peptide can be used for the lysosome-controlled release of norfloxacin because it is separated from its temporary carrier after uptake of the macromolecular prodrug by the macrophages. A release occurring mainly in the phagolysosome, i.e., in close contact with the pathogenic bacteria, should avoid the renal elimination of the antibiotic before it has time to act on the bacteria.
Among the four conjugates tested, only the conjugate containing both mannose as a homing device and an α bond was active in vivo against M. bovis BCG. Several conclusions can be drawn from these results.
First, the fact that native norfloxacin did not exert an antibacterial effect in vivo demonstrates the importance of and the interest in drug targeting. This antibiotic, which was active in vitro and inactive in vivo, became active in vivo when it was carried by a targeted macromolecule.
Second, only the breaking of the α bond was able to release native and, thus, active norfloxacin. This observation was true in vitro as well as in vivo.
Third, targeting of the macrophage is necessary: only the mannosylated conjugate was active in vivo. Moreover, our results indicate that antibiotic was released close to the bacteria, thus supporting the observations of Clemens and Horwitz (3).
Finally, as the conjugate containing an amide bond had no antimicrobial activity, it can be concluded that the carrier had no antimicrobial effect by itself.
REFERENCES
- 1.Aubrée-Lecat, A., M.-C. Duban, S. Demignot, M. Domurado, P. Fournié, and D. Domurado. 1993. Influence of barrier-crossing limitations on the amount of macromolecular drug taken up by its target. J. Pharmacokinet. Biopharm. 21:75-98. [DOI] [PubMed] [Google Scholar]
- 2.Carbon, C., B. Régnier, G. Saimot, J.-L. Vildé, and P. Yéni. 1994. Médicaments anti-infectieux, p. 123-146. Médecine Sciences Flammarion, Paris, France.
- 3.Clemens, D. L., and M. A. Horwitz. 1996. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J. Exp. Med. 184:1349-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coessens, V., E. H. Schacht, and D. Domurado. 1997. Synthesis and in vitro stability of macromolecular prodrugs of norfloxacin. J. Control. Release 47:283-291. [Google Scholar]
- 5.De Duve, C., T. de Barsy, B. Poole, A. Trouet, and P. Tulkens. 1974. Lysosomotropic agents. Biochem. Pharmacol. 23:2495-2531. [DOI] [PubMed] [Google Scholar]
- 6.Domurado, M., D. Domurado, S. Vansteenkiste, A. De Marre, and E. Schacht. 1995. Glucose oxidase as a tool to study in vivo the interaction of glycosylated polymers with the mannose receptor of macrophages. J. Control. Release 33:115-123. [Google Scholar]
- 7.Gac, S. 1998. Ph.D. thesis. Université de Montpellier, Montpellier, France.
- 8.Gheorghiu, M., and P. H. Lagrange. 1983. Viability, heat stability and immunogenicity of four BCG-vaccines prepared from four different BCG strains. Ann. Immunol. (Institut Pasteur) 134C:125-147. [DOI] [PubMed]
- 9.Gordon, S., and S. Rabinowitz. 1989. Macrophages as targets for drug delivery. Adv. Drug Deliv. Rev. 4:27-47. [Google Scholar]
- 10.Kéry, V., J. J. F. Krepinsky, C. D. Warren, P. Capek, and P. D. Stahl. 1992. Ligand recognition by purified human mannose receptor. Arch. Biochem. Biophys. 298:49-55. [DOI] [PubMed] [Google Scholar]
- 11.Kopecek, J., P. Rejmonova, J. Dohl, M. Baudys, and V. Kostka. 1983. Polymers containing enzymatically degradable bonds. Degradation of oligopeptide sequence in HMPA by cathepsin B. Makromol. Chem. 174:2009-2020. [Google Scholar]
- 12.Nichifor, M., and E. Schacht. 1994. Synthesis of peptide derivatives of 5-fluorouracil. Tetrahedron 50:3747-3760. [Google Scholar]
- 13.Vansteenkiste, S., A. De Marre, and E. Schacht. 1992. Synthesis of glycosylated dextrans. J. Bioact. Compat. Polym. 7:4-14. [Google Scholar]