

Abstract
Prestin is a membrane protein in the outer hair cell (OHC) that has been shown to be essential for electromotility. OHCs from prestin-null mice do not express prestin, do not have a nonlinear capacitance (the electrical signature of electromotility), and are smaller in size than wild-type OHCs. We sought to determine whether prestin-null OHCs can be transduced to incorporate functional prestin protein in a normal fashion. A recombinant helper-dependent adenovirus expressing prestin and green fluorescent protein (HDAd–prestin–GFP) was created and tested in human embryonic kidney cells (HEK cells). Transduced HEK cells demonstrated membrane expression of prestin and nonlinear capacitance. HDAd–prestin–GFP was then applied to cochlear sensory epithelium explants harvested from wild-type and prestin-null mice at postnatal days 2–3, the age at which native prestin is just beginning to become functional in wild-type mice. At postnatal days 4–5, we investigated transduced OHCs for (1) their prestin expression pattern as revealed by immunofluorescence; (2) their cell surface area as measured by linear capacitance; and (3) their prestin function as indicated by nonlinear capacitance. HDAd–prestin–GFP efficiently transduced OHCs of both genotypes and prestin protein localized to the plasma membrane. Whole-cell voltage clamp studies revealed a nonlinear capacitance in transduced wild-type and prestin-null OHCs, but not in non-transduced cells of either genotype. Prestin transduction did not increase the linear capacitance (cell surface area) for either genotype. In peak nonlinear capacitance, voltage at peak nonlinear capacitance, charge density of the nonlinear capacitance, and shape of the voltage-capacitance curves, the transduced cells of the two genotypes resembled each other and previously reported data from adult wild-type mouse OHCs. Thus, prestin introduced into prestin-deficient OHCs segregates normally to the cell membrane and generates a normal nonlinear capacitance, indicative of normal prestin function.
Keywords: prestin, OHC, nonlinear capacitance, electromotility, adenovirus, gene therapy
Introduction
Prestin is a protein located in the lateral wall plasma membrane of outer hair cells (OHCs) and is a voltage-to-force converter (Adler et al. 2003; Belyantseva et al. 2000a; Dallos and Fakler 2002; Liberman et al. 2002; Ludwig et al. 2001; Zheng et al. 2002; Zheng et al. 2000). Prestin is believed to act in concert with the other structures of the trilaminate OHC lateral wall to generate the relatively large somatic electromotility that is an unusual feature of these cells (Brownell et al. 1985; Evans and Dallos 1993; He et al. 2006; Oghalai et al. 1998b). Electromotility is necessary for the high sensitivity and frequency selectivity of the mammalian cochlea (Brownell et al. 2001; Cheatham et al. 2004; Dallos and Fakler 2002; Oghalai 2004). The electrical signature of prestin function is a nonlinear capacitance, which is thought to reflect anion movement associated with changes in prestin conformation (He et al. 2006; Oliver et al. 2001). Prestin transfection of human embryonic kidney (HEK) cells results in a nonlinear capacitance (Deak et al. 2005; Rajagopalan et al. 2006; Santos-Sacchi 2002; Zheng et al. 2000). In adult OHCs, electromotility and nonlinear capacitance are highly correlated (Ashmore 1990; Dallos and Fakler 2002; Santos-Sacchi 1991a). The time course of prestin expression in postnatal development is consistent with the onset and maturation of electromotility (Belyantseva et al. 2000a; He et al. 1994; Oliver and Fakler 1999). In normal mice, prestin protein begins to be expressed around P2–3 (Winter et al. 2007), and a nonlinear capacitance can first be measured around P5–6 (Abe et al. 2007). Electromotility is not fully functional at an adult level until days 15–18 (Abe et al. 2007).
The lack of prestin in the prestin-null mouse produces OHCs that do not have a nonlinear capacitance, do not have electromotility, and are smaller than wild-type OHCs (Liberman et al. 2002). However, prestin has never been put back into prestin-null OHCs to determine whether any of these properties can be restored. Employing a helper-dependent adenoviral (HDAd) vector, we transduced early postnatal wild-type and prestin-null organ of Corti cultures with prestin and studied them with immunofluorescence and whole-cell voltage clamp. We chose to use HDAd because it could potentially be used in vivo, whereas standard transfection techniques for putting a gene into a cell are typically limited to cultured preparations. HDAd is devoid of viral coding sequences and has been shown to mediate high-efficiency transduction in vivo and to produce a long-term, high-level of transgene expression with negligible chronic toxicity (Ng et al. 2002; Palmer and Ng 2005). In this study, we sought to determine whether transduction of prestin-null OHCs with the gene for prestin would lead to normal prestin protein expression and function.
Material and methods
The Baylor College of Medicine Institutional Animal Care and Use Committee approved the study protocol.
Creation of plasmids
To create the helper-dependent adenovirus expressing prestin and green fluorescent protein (HDAd–prestin–GFP) vector, a Nsi1 linker 5′ATGCAACGCGTGGCGCGCCTGCAT3′, including an Mlu1 restriction site, was inserted into the Nsi1 site in the pCMV-HA–prestin–IRES–hrGFP-1a plasmid (Fig. 1). The prestin cDNA was from the gerbil. The pCMV-HA–prestin–IRES–hrGFP-1a mammalian expression plasmid independently expresses hemagglutinin (HA)-tagged prestin and GFP, which allows for easy identification of transduced cells (Rajagopalan et al. 2006; Sturm et al. 2007). The resultant vector was linearized with Mlu1 and inserted into the compatible Asc I restriction site in the HDAd vector (pΔ25.3E4 vector), which was derived from pΔ28E4 by deletion of the 3 kb Nhe I fragment (Toietta et al. 2002). HDAd–prestin–GFP was produced using the helper virus AdNG163 and 116 cells as described in detail elsewhere (Palmer and Ng 2003; Palmer and Ng 2004). After six to eight serial passages, HDAd–prestin–GFP was isolated in a buffer of 10 mM Tris and 10% glycerol at a concentration of 4.9 × 1012 viral particles (vp) per ml.
FIG. 1.

Creation of HDAd–prestin–GFP vector. A unique Nsi1 linker containing an Mlu1 restriction site was inserted into the pCMV-HA–Prestin–IRES–GFP plasmid. The plasmid was then linearized at the unique Mlu1 site and inserted into a compatible Asc1 restriction site in the pΔ25.3E4 HDAd vector to create HDAd–prestin–GFP vector.
HEK cell culture
HEK 293 cells were maintained in Dulbecco’s modified Eagle medium (DMEM, GIBCO, Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS, Invitrogen), seeded at a density of ∼100,000 cells per well on a 60-mm plate and incubated overnight. The cells were released by applying 2 ml of 0.25% trypsin/ethylenediaminetetraacetic acid (Invitrogen) at 37°C for 5 min, pelleted (800×g), resuspended in 2 ml of DMEM + 10% FBS, and then plated on cover glass slips placed in a 24-well culture dish containing 0.5 ml DMEM + 10% FBS. The concentration was 4,000–10,000 cells/well.
HDAd–prestin–GFP was diluted to a concentration of 1 × 107 vp/μl in 400 μl fresh DMEM + 10% FBS and applied to the wells. Some preparations did not have HDAd–prestin–GFP applied and were defined as controls. The cells were then cultured at 37°C in 5% CO2, and the medium was changed after 24 h. After 48 h, the cells were used for either the immunohistochemical or the physiological studies.
Immature organ of Corti cultures
Prestin wild-type and null mice were derived from a mixed background of 129/SvEv and C57B6/J (obtained from Dr. Jian Zhou in St Jude Children’s Research Hospital, Memphis, TN, USA) and maintained as heterozygote intercrosses (Liberman et al. 2002). Pups at postnatal days 2–3 (P2–P3) were euthanized and the temporal bones removed before the otic capsule was dissected in fresh cold Hank’s balanced salt solution (Invitrogen). The membranous labyrinth was exposed in DMEM containing 10% FBS, 0.01% ampicillin (Invitrogen), and 0.025% FungiZone (Invitrogen). The sensory epithelium from the middle region of the cochlea was isolated in 35 mm Falcon® culture dishes (BD, Franklin Lakes, NJ, USA), and the tectorial membrane was carefully removed. The sensory epithelium was then mounted on a round glass coverslip (Fisher Scientific, IL, USA) with BD Cell-Tak™ Cell (BD Biosciences, San Jose, CA, USA) and placed into each well of a 24-well culture dish containing DMEM.
HDAd–prestin–GFP was applied at a concentration of 1 × 107 vp/μl to the sensory epithelium explant cultures immediately after dissection. Some preparations did not have HDAd–prestin–GFP applied and were defined as controls. The dishes were placed in an incubator (Nuaire, Plymouth, MN, USA) at 37°C in 5% CO2. The DMEM was changed daily. After 48 h, the cells were used for either immunohistochemical or physiological studies.
Immunohistochemical studies
The coverslips containing either HEK 293 cells or organ of Corti preparations were rinsed with PBS two times for 15 min each and then fixed with 4% paraformaldehyde (PFA) for 1 h at room temperature (RT). The preparations were washed twice again and treated for 1 h at RT with 4% normal goat serum (NGS) in PBS + 0.1% Triton X-100 (PBST) to reduce background staining. Mouse anti-HA antibody (Cell Signaling Technology, Beverly, MA, USA) was diluted 1:1,200 in 4% NGS in PBST, applied to the preparations, and incubated at 4°C overnight. The preparations were then washed three times in PBS for 10 min each.
For HEK cell preparations, Alexa Fluor® 594 goat anti-mouse secondary antibody was diluted 1:500 (Invitrogen) and applied for 1 h at RT. For organ of Corti preparations, a mixture of Alexa Fluor® 594 goat anti-mouse secondary antibody diluted 1:500 and Alexa Fluor® 488 phalloidin diluted 1:200 (Invitrogen) was applied for 1 h at RT. As a control, some organ of Corti cultures were stained with the Alexa Fluor® 594 secondary antibody and Alexa Fluor® 488 phalloidin in the absence of primary antibody.
The preparations were washed again with PBS, mounted on microscope slides with Fluoromount G (SouthernBiotech, Hatfield, PA, USA), and sealed with nail polish. Images were captured on a Zeiss LSM 510 confocal microscope (Carl Zeiss Optics Company, Jena, Germany).
Paraffin-embedding and immunolabeling of organ of Corti cultures
Some of the organ of Corti cultures were fixed with 4% PFA, dehydrated in ethanol, clarified in xylenes, and then embedded in paraffin. Serial paraffin sections (7 μm) were cut to obtain cross-sections of the sensory epithelium. After deparaffinization with a series of xylenes and ethanol, the slides were washed with PBS three times and immersed with 4% NGS in PBST for 1 h at RT. The preparations were incubated at 4°C overnight in a mixture of mouse anti-HA (dilution 1:1200) and rabbit anti-myosin VIIa (dilution 1:200, Affinity Bioreagents, Golden, CO, USA).
After washing with PBS the next day, the preparations were immersed in a mixture of Alexa Fluor® 594 goat anti-mouse and Alexa Fluor® 488 goat anti-rabbit secondary antibodies at RT for 1 h. Followed by washing with PBS again, the cultures were mounted on microscope slides with the Vectashield® mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA) and sealed with nail polish. Images were captured on a Zeiss LSM 510 confocal microscope (Carl Zeiss Optics Company).
Electrophysiological measurements
Electrophysiological data were obtained from cells using the whole-cell voltage clamp technique. The extracellular solution contained Ca2+ and K+ channel blockers (122 mM NaCl, 20 mM TEA–Cl, 15 mM CsCl, 10 mM glucose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 1.25 mM CoCl2, and 1.5 mM MgCl2). The pH was 7.3, and the osmolality was 300 mmol/kg. A coverslip containing either HEK 293 cells or the organ of Corti cultures was gently washed with extracellular solution. The coverslip was placed in a chamber and immersed in extracellular solution. The chamber was fixed to the stage of an Axioskop 2 FS upright microscope (Carl Zeiss Optics Company). A ×40 water immersion objective was used to visualize the preparation, and continuous perfusion of fresh extracellular solution was performed throughout the experiment. All recordings were carried out at RT (23 ± 2°C). Borosilicate patch pipettes with resistances ranging from 3 to 6 MΩ were fabricated using a laser-based micropipette puller (P-2000, Sutter Instrument Company, Novato, CA, USA). The intracellular solution also contained channel blockers (145 mM CsCl, 2.0 mM MgCl2, 10 mM glucose, 10 mM HEPES, and 10 mM EGTA). The pH was 7.3, and the osmolality was 315 mmol/kg.
OHCs were patch clamped within the sensory epithelium, using methods previously described (Oghalai et al. 1998a; Rusch and Eatock 1996). Briefly, the OHC to be studied was selected using fluorescence microscopy with an optical filter set designed to visualize GFP (filter set 38, Zeiss). The patch pipette was brought adjacent to the selected cell, and the voltage was zeroed relative to an Ag/AgCl reference electrode in the extracellular solution. Suction was applied to produce an electrical seal (>1 GΩ) between the pipette and cell membrane. Pipette capacitance was then corrected with the compensation circuitry of the patch clamp amplifier (either Axon 200B [Molecular Devices, Union City, CA, USA] or EPC10-2 [HEKA Instruments, Southboro, MA, USA]). A −65 mV holding potential was set, and the whole-cell mode was entered. Voltages have been corrected for a liquid junction potential of 4.7 mV calculated using JPCalc (Barry 1994).
A DC voltage step protocol was used to measure cell capacitance at different voltages (Santos-Sacchi 1991b). With pClamp (version 9, Molecular Devices), the voltage was stepped from hyperpolarizing to depolarizing voltages in either 5 or 10 mV steps and the current recorded at a sample rate of 167 kHz. Each step lasted 12.8 ms. The voltage step protocol was averaged 25 times. As the solutions blocked most ionic currents, voltage errors resulting from series resistance were minimized and thus ignored.
The recorded current was separately integrated for the first and second halves of the step. The second integral was subtracted from the first to determine the charge moved. The charge was divided by the voltage step to give the membrane capacitance, which was defined to be the capacitance at the voltage step midpoint. Arbitrarily, the total capacitance at +70 mV was taken to be the linear capacitance; this value was subtracted from the capacitance measured at all other voltage to calculate the nonlinear capacitance.
The nonlinear capacitance curves (C) were fit to a modified Boltzmann equation as described previously (Oliver and Fakler 1999) to calculate the maximum charge transferred (Qmax), the voltage at peak capacitance (Vpkcm), and the slope factor of the voltage dependence (α):
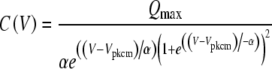 |
The charge density (d) was then calculated to be the number of electrons moved per square micrometer of cell surface area by the equation:
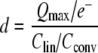 |
where e− is the charge of an electron (1.602 × 10−19 C), Clin is the linear capacitance, and Cconv is the conversion factor for the amount of linear capacitance per unit cell surface area (8 fF/μm2) as previously measured in developing mouse OHCs (Abe et al. 2007).
Curve fitting and statistical analysis
All values and error bars presented are mean±SEM. Curve fitting was performed and plotted using SigmaPlot (9.0, Systat Software) and Origin (Microcal). Statistical significance was performed using Excel (Microsoft Office 2003). The Student’s non-paired two-tailed t-test was used to compare measurements taken from different cells. P values <0.05 were considered statistically significant.
Results
HA–prestin expression and nonlinear capacitance of HEK cells transduced with HDAd–prestin–GFP
We applied HDAd–prestin–GFP to HEK 293 cells to verify that functional prestin protein could be expressed. Confocal images taken 48 h after virus application demonstrated GFP within cell bodies (Fig. 2A). Immunolabeling demonstrated that HA–prestin was expressed within the plasma membrane (Fig. 2B). Non-transduced HEK cells in the same preparation did not demonstrate GFP or HA–prestin expression in the same preparation (Fig. 2C).
FIG. 2.

GFP and HA–prestin expression in HEK 293 cells. A Transduced cells were identified by GFP expression. B HA–prestin immunolabeling was detected in the HEK cell membrane. C A merged image with DAPI nuclear staining demonstrated that only transduced cells expressed GFP and HA–prestin. D Example capacitance versus voltage curves, after subtracting the linear capacitance, in both a GFP-positive cell and a control cell in which HDAd–prestin–GFP was not applied. A nonlinear capacitance was only identified in GFP-positive cells. The solid black line represented Boltzmann fits to the data. Fit parameters were Vpkcm = −74.8 mV, α = 33.8 mV, Qmax = 1040.9 fC. (A), (B), and (C) bar = 10 μm.
To determine if the expressed prestin was functional, we measured the nonlinear capacitance by whole-cell voltage clamp. Representative data from one GFP-positive cell and one control cell in which no HDAd–prestin–GFP was applied are shown (Fig. 2D). Typical bell-shaped curves of capacitance versus voltage were only found in GFP-positive cells (n = 5). Control cells did not have a detectable nonlinear capacitance (n = 5). The average peak nonlinear capacitance in HEK 293 cells was 7.1 ± 1.3 pF, and the average voltage at peak capacitance (Vpkcm) was −71 ± 9 mV. The charge density was 4,432 ± 1,132 e−/μm2. This was similar to charge densities within HEK cells transfected with prestin (range: 3,700–5,500; Rajagopalan et al. 2006; Zheng et al. 2000). These data indicate that the HDAd–prestin–GFP virus was able to transduce cells and lead to the expression of functional prestin protein.
GFP fluorescence in the prestin-null organ of Corti
Immature organ of Corti preparations harvested from prestin-null mice on P2 or P3 were treated with HDAd–prestin–GFP and grown for 2 days in organotypic culture. Confocal images of the whole mount preparations revealed a number of cells that expressed GFP and, therefore, had been transduced. The GFP fluorescence was primarily observed within the hair cell region, the spiral limbus, and the spiral ganglion region (Fig. 3A). High magnification images revealed that GFP was present within OHCs (Fig. 3B). The intensity of the GFP fluorescence varied; some OHCs demonstrated bright fluorescence and some demonstrated weak fluorescence. Transduced inner hair cells (IHCs) were noted rarely. Many supporting cells also expressed GFP.
FIG. 3.

GFP and HA–prestin expression within cochlear sensory epithelium cultures from prestin-null mice. A A confocal image of a whole-mount preparation under low magnification demonstrates that GFP was expressed primarily within the organ of Corti area (white asterisk and rectangles), the spiral limbus (blue asterisk), and the spiral ganglion area (yellow asterisk). B A representative confocal image under high magnification demonstrates transduction of OHCs and supporting cells. Some OHCs had bright GFP fluorescence (white arrow), some OHCs had low GFP fluorescence (blue arrow), and some OHCs had no GFP fluorescence (yellow arrow). Supporting cells were also transduced (arrowheads). C The percentage of GFP-positive and GFP-negative hair cells was determined in 35 images from 7 cochleae [representative areas counted are shown in (A), rectangles]. D A paraffin-embedded cross-section of a cultured preparation demonstrates that HA–prestin was expressed within the organ of Corti (OC, white asterisk), the spiral limbus (SPL, blue asterisk), and the spiral ganglion area (SG, yellow asterisk). These asterisks correspond to those in (A). Myosin VIIa antibody labels the hair cells with green fluorescence. Scale bars: A 200 μm, B 50 μm, D 100 μm.
To determine the efficiency of transduction, we quantified the number of transduced cells in each of five high magnification images from every organ of Corti preparation (rectangles shown in Fig. 3A). In total, seven preparations were studied (total of 35 high magnification images). For all images, the color intensity levels were kept constant to minimize variability between the preparations. All OHCs and IHCs were counted, and every cell with visible GFP (i.e., either bright or dim fluorescence) was considered to be GFP positive. Overall, 45.7 ± 2.64% of 2,179 OHCs and 1.06 ± 1.28% of 659 IHCs expressed GFP (Fig. 3C). As non-transduced supporting cells could not be visualized, the efficiency of supporting cell infection could not be determined. Nevertheless, a total of 235 transduced supporting cells over the same 35 high magnification images were counted. Thus, the relative transduction rate was 235 supporting cells per 2,179 OHCs or roughly 11 transduced supporting cells for every 100 OHCs.
HA–prestin expression in cross-sections of the organ of Corti
Some organ of Corti preparations were sectioned and immunolabeled with antibodies to HA to confirm the sites of gene expression. Double labeling with antibodies to myosin VIIa to localize hair cells was also performed. Under low magnification imaging (Fig. 3D), HA–prestin was found to be expressed within the hair cells region. HA–prestin was also found within the spiral limbus at the area where the tectorial membrane had been attached before it was removed at the time of dissection. The central core region of the modiolus, where spiral ganglion cells exist, also expressed prestin. Several images were selected that demonstrated the various regions of HA–prestin expression and studied under high magnification imaging (Fig. 4). HA–prestin was expressed in several specific cell types, including IHCs, OHCs, Deiter cells, and Hensen cells. The immunolabeling was concentrated within cell membranes. In particular, both the lateral wall plasma membrane and the infranuclear plasma membrane of OHCs were found to contain HA–prestin (Fig. 4A–G). However, we could not determine whether the HA–prestin density was similar or different between these two regions of the OHC because of the 7-μm thickness of the paraffin sections. Nevertheless, these sections clearly demonstrated that stereocilia from inner hair cells OHCs and did not contain HA–prestin expression. No HA–prestin immunolabeling was found in control preparations in which virus was not applied (Fig. 4I).
FIG. 4.

HA–prestin immunolocalization within the organ of Corti of prestin-null cochleae. Selected paraffin-embedded cross-sections of the hair cell region. The orientation is the same for all images, with an IHC on the right and three rows of OHCs on the left. One selected section demonstrates myosin VIIa immunolabeling (green) to identify hair cells (A), HA immunolabeling (red) to identify prestin (B), and a merged image (C). In this example, HA–prestin was expressed in IHCs (white arrowheads), OHCs (yellow arrowheads), and Deiter cells (white arrows). The HA–prestin labeling was primarily within the plasma membrane. D, E, F Another selected section demonstrates HA–prestin expression in OHCs (yellow arrowheads) and Deiter cells (white arrows). Two more selected sections are presented with myosin VIIa and HA–prestin immunolabeling fluorescence merged. A single OHC (G, yellow arrowheads), and a single Hensen cell (H, blue arrow) demonstrated HA–prestin expression. I A control organ of Corti demonstrated no HA–prestin immunolabeling. Scale bar: A–I 10 μm.
Intracellular localization of HA–prestin
To better define the specific areas of the OHC that contained HA–prestin, HA immunolabeling was also performed within prestin-null whole-mount preparations (n = 4 preparations). The preparations were counterstained with phalloidin and Z-stack series of 27 confocal images, each of 0.57-μm thickness, were taken. Every third section from one representative preparation is presented (Fig. 5). Again, no HA–prestin was found at the level of the stereocilia (Fig. 5A). HA–prestin was observed below the cuticular plate region (Fig. 5B). Along the mid-portion of the OHC, there was strong immunolabeling of HA–prestin along the lateral wall and minimal labeling within the axial core (Fig. 5C–F). Weaker immunolabeling was also found within the infranuclear membrane region (Fig. 5G–I). HA–prestin could also be observed within the plasma membrane of several IHCs in this preparation (Fig. 5D–E).
FIG. 5.

HA–prestin immunolocalization within a whole mount preparation of the prestin-null organ of Corti. A–C A Z-stack series of 27 images were taken from a whole-mount preparation, and every third slice is presented. The elongated diagram of an OHC on the right shows the location of each slice. In each slice, an OHC is identified (blue arrows) and enlarged in the inset. In some slices, transduced IHC are identified (yellow arrows). A No HA–prestin was found within the stereociliary membranes. B HA–prestin was found within the plasma membrane at the level of the cuticular plate region. C–F HA–prestin was identified within the lateral wall plasma membrane of OHCs. G–I HA–prestin was also found in the infranuclear membrane, although its immunolabeling intensity was lower than that of the lateral wall. Scale bars: 20 μm.
Whole-cell voltage clamp experiments
Next, we performed whole-cell voltage-clamp experiments to measure the linear and nonlinear capacitance of OHCs in organ of Corti preparations from wild-type and null mice. OHCs were visualized under a ×40 water immersion objective and identified as either being transduced (GFP positive) or non-transduced (GFP negative; Fig. 6A–C). For our transduced sample of voltage-clamped OHCs, we chose those OHCs with the brightest fluorescence. Control OHCs grown in culture in which HDAd–prestin–GFP was not applied (wild-type: n = 6; null: n = 8) did not demonstrate a nonlinear capacitance (Fig. 6D and E). GFP-negative OHCs also did not demonstrate a nonlinear capacitance (wild-type: n = 8; null: n = 6). However, GFP-positive OHCs (wild-type: n = 6; null: n = 6) demonstrated the bell-shaped nonlinear capacitance–voltage relationship that is characteristic of prestin function (Fig. 6D and E).
FIG. 6.

Prestin transduction endows immature OHCs with nonlinear capacitance. Representative examples of the whole-cell voltage clamp technique in transduced immature preparations. A The three rows of OHC V-shaped stereocilia can be noted in the differential interference contrast image. The patch pipette was in contact with an OHC within the third row. B The fluorescence image demonstrates GFP expression within some of the OHCs. C The merged image demonstrates that the patch pipette was in contact with a GFP-positive OHC. After subtracting out the linear membrane capacitance, GFP-positive wild-type (D) and prestin-null (E) OHCs had an obvious voltage-dependent nonlinear capacitance, in contrast to GFP-negative and control OHCs. The solid black lines are Boltzmann fits to the data. Fit parameters: DVpkcm = −53.0 mV, α = 25.7 mV, Qmax = 446.5 fC and EVpkcm = −43.0 mV, α = 27.7 mV, Qmax = 382.0 fC in (E). Scale bar: 40 μm.
Comparison of nonlinear and linear capacitance between the genotypes
The prestin-associated charge movement in transduced OHCs was compared (Fig. 7A). The Qmax was 458 ± 27 fC and 363 ± 81 fC for the wild-type and prestin-null OHCs, respectively (n = 6 for each, p = 0.288). To account for variations in cell surface area, we calculated the transferred charge density (the charge moved per unit cell surface area) as described in “Material and methods.” The charge density was 4,147 ± 460 e−/μm2 for wild-type OHCs and 4,140 ± 1,092 e−/μm2 for null OHCs, and these values were not statistically different (p = 0.995).
FIG. 7.

Comparison of voltage-clamp data between wild-type (WT) and prestin-null (null) OHCs. All values are presented in the text. A The transferred membrane charge density associated with prestin was nearly identical between the genotypes. B After normalizing the nonlinear capacitance curves for each cell to their peak capacitance and aligning their voltage at peak capacitance (Vpkcm), the data were averaged for each genotype. There was no difference in the voltage dependency of prestin between wild-type and null OHCs. C There was no statistically significant difference in Vpkcm between the genotypes. D The linear capacitance was similar between control wild-type OHCs, GFP-negative wild-type OHCs, and GFP-positive wild-type OHCs. Furthermore, the linear capacitance was similar between control null OHCs, GFP-negative null OHCs, and GFP-positive null OHCs. Lastly, there was no statistically difference between the linear capacitance of control wild-type and null OHCs.
Next, the slope factor (α) of the nonlinear capacitance was compared. The nonlinear capacitance curves for each cell were normalized to peak capacitance, and the voltage range was shifted so that Vpkcm was centered. We then averaged the data and found that wild-type and prestin-null OHCs had similar capacitance–voltage relations (Fig. 7B). The average slope factor was 44 ± 3 mV and 43 ± 3 mV in transduced OHCs from wild-type and prestin-null mice, respectively, which were not statistically different (n = 6 for each, p = 0.83).
The average Vpkcm was −51.4 ± 6 mV in transduced OHCs from wild-type mice (n = 6) and −48 ± 8 mV in those from null mice (n = 6; Fig. 7C). This difference was also not statistically significant (p = 0.74). Previous work has shown that, as the wild-type OHC develops and more prestin is incorporated into the membrane, Vpkcm becomes more depolarized (Abe et al. 2007; Oliver and Fakler 1999). We performed a linear regression analysis using all transduced OHCs that we studied (n = 12) to test whether Vpkcm was related to peak nonlinear capacitance. In our sample, there was no significant association between the two values (R2 = 0.119. p = 0.271).
The linear capacitance was then compared to see if prestin transduction was associated with increased cell surface area (Fig. 7D). There was no significant difference between control wild-type (5.6 ± 0.2 pF, n = 6) and null OHCs (5.1 ± 0.3 pF, n = 8, p1 = 0.28). This suggests that immature wild-type and prestin-null OHCs are similar in surface area. In addition, there were no significant differences between the linear capacitance of wild-type GFP-positive (5.8 ± 0.6 pF, n = 6) and control OHCs (p2 = 0.75), and wild-type GFP-positive and GFP-negative OHCs (5.0 ± 0.3 pF, n = 7, p3 = 0.27). Similarly, there were no significant differences between the linear capacitance of null GFP-positive (4.9 ± 0.4 pF, n = 6) and control OHCs (p4 = 0.6) and null GFP-positive and null GFP-negative OHCs (4.1 ± 0.4 pF, n = 6, p5 = 0.23). Thus, prestin transduction was not associated with an increase in cell surface area in either wild-type or prestin-null OHCs.
It should be noted that we did not visually detect electromotility within any of the OHCs that we voltage clamped. However, this does not necessarily mean that electromotility was not present for two reasons. First, in our preparation, the OHCs are viewed from the top and are constrained within the epithelium. Thus, vertical movements generated by a single cell might not be detectable. Second, the size of the electromotile response in adult OHCs varies with cell length. Because immature mouse OHCs are relatively small (15–22 μm long; Abe et al. 2007), even electromotile movements that are a relatively large proportion of the total length (i.e., 5%) might not be visible.
Discussion
Herein, we provide evidence that immature OHCs from wild-type and prestin-null mice can be transduced with prestin using a helper-dependent adenovirus. This was performed in OHCs beginning at P2–3, with testing at P4–5, an age at which native prestin is just beginning to become functional in wild-type mice (Abe et al. 2007) and electromotility is not yet present (Belyantseva et al. 2000a; He et al. 1994; Oliver and Fakler 1999). Our stairstep measurement technique of the nonlinear capacitance was not sensitive enough to detect the low levels of native prestin function in non-transduced OHCs at P4–5. However, this was not an issue in transduced OHCs because of the large amount of functional prestin expressed. While it is likely that any cell type transduced with prestin will have a nonlinear capacitance, these data are the first to demonstrate that prestin transduction rescues normal levels of nonlinear capacitance within prestin-null OHCs.
Our data indicate that both wild-type and prestin-null OHCs that are transduced by HDAd–prestin–GFP express functional prestin protein in a similar manner. The prestin expression pattern in transduced OHCs was found predominantly within the lateral wall like that found in wild-type adult OHCs (Belyantseva et al. 2000a; Cheatham et al. 2005; Zheng et al. 2003). The Z-stack confocal images indicated that prestin was also expressed within the infranuclear plasma membrane of transduced OHCs, although at lower levels than within the lateral wall. This is an expression pattern similar to adult OHCs, which have a lower level of prestin within the infranuclear membrane than the lateral wall (Yu et al. 2006). Alternatively, it is possible that this difference may represent reduced antibody penetration to this tissue depth within the whole-mount preparations, because OHCs from the P2 mouse and the P3 rat demonstrate robust levels (Winter et al. 2007). However, our OHCs were slightly older (P4–5) and contained transduced HA–prestin rather than native prestin. Importantly, neither the paraffin-embedded sections nor the confocal images demonstrate prestin expression within the stereocilia.
As the pCMV promoter provides essentially unregulated transcription of HA–prestin mRNA, it is probable that a near-maximal amount of prestin was present within the plasma membrane of transduced cells during our voltage-clamp experiments. Interestingly, the transferred charge density in transduced OHCs 48 h after HDAd–prestin–GFP application was similar to previously reported charge densities of adult wild-type OHCs (approximately 4,000–10,000 e−/μm2; Abe et al. 2007; Cheatham et al. 2005; Gale and Ashmore 1997; Huang and Santos-Sacchi 1993; Oliver and Fakler 1999; Santos-Sacchi 1991b). In addition, the transferred charge density of transduced HEK cells was similar to that of the transduced OHCs. This suggests that functionally active native prestin protein in adult wild-type OHCs is not normally regulated at the level of transcription.
Indeed, it is possible that the density of functionally active prestin within the lateral wall is controlled by the physical constraints of the plasma membrane surface area and the biophysical interactions between prestin molecules (Wu et al. 2007). The fact that adult wild-type OHCs express prestin in the infranuclear plasma membrane (Yu et al. 2006) similar to our unregulated expression of prestin in transduced OHCs suggests that functionally active prestin is regulated at the posttranscriptional level in OHCs. This theory is also supported by the lack of prestin expression within the stereociliary membranes. Potential mechanisms of prestin regulation may include localized membrane recycling, protein degradation pathways, organization by the lateral wall cytoskeleton (Jensen-Smith and Hallworth 2007), or regulation of mRNA stability. Further comparative studies of prestin protein regulation in transduced IHCs, OHCs, and supporting cells with normal OHCs may help to elucidate these mechanisms.
The Vpkcm we measured in transduced immature OHCs was depolarized relative to that reported in immature and adult wild-type mouse OHCs, which ranged from −90 to −67 mV (Abe et al. 2007). It was, however, similar to that reported for adult rat OHCs (−41 mV; Oliver and Fakler 1999). In addition, the slope factor of the voltage-dependent capacitance (α) was slightly larger in transduced immature OHCs compared to that of adult wild-type OHCs (∼30 mV; Cheatham et al. 2005; Oliver and Fakler 1999), and the slope factor of wild-type mouse OHCs has been shown to be stable from P6 to P18 (Abe et al. 2007). The larger slope factor may in part be because of membrane leakiness at extreme levels of hyperpolarization or depolarization. Electroporation may even be enhanced in the presence of prestin protein (Navarrete and Santos-Sacchi 2006).
However, these variations may also reflect the fact that the immature OHCs of either genotype are missing or have insufficient levels of other components necessary for functionality of prestin in the adult. For example, at this age, the lateral wall cytoskeletal proteins critical for electromotility are not fully integrated, and associated changes in OHC stiffness could modulate the nonlinear capacitance (Iwasa 1993). The role of actin is unclear in that one study demonstrates that its level progressively increases during OHC development (Weaver et al. 1994), while another suggests that its level decreases (Jensen-Smith and Hallworth 2007). Spectrin levels progressively increase in the lateral wall of gerbil OHCs over the first three post-natal weeks (Jenson-Smith and Hallworth 2006). In addition, the cholesterol and/or phospholipid content of the OHC plasma membrane may change during development (Rajagopalan et al. 2007; Sturm et al. 2007). This too may shift the voltage-dependence of prestin-associated charge movement, similar to the effects of alterations of membrane lipids on other transporter proteins (Baukrowitz et al. 1998; Fernandez-Carvajal et al. 2006; Guzman et al. 2007; Hilgemann and Ball 1996).
Adult OHCs from prestin-null mice are smaller than those from wild-type mice and may have less intracellular turgor pressure (Cheatham et al. 2005; Liberman et al. 2002). It is possible that prestin may somehow stimulate the process of OHC lengthening that naturally occurs during development (Abe et al. 2007; Belyantseva et al. 2000b; He et al. 1994; Oliver and Fakler 1999). We have provided the first whole-cell voltage clamp data from immature wild-type and prestin-null OHCs to indirectly assess cell size through the linear capacitance. Immature wild-type and prestin-null OHCs have a similar linear capacitance. In addition, OHCs transduced with prestin did not have a larger linear capacitance than non-transduced OHCs. Whether or not prestin promotes OHC growth during functional maturation is unclear. We only studied OHCs 2 days after incubation with HDAd–prestin–GFP, which may not provide enough time for a “prestin-induced” increase in linear capacitance to occur.
Unanticipated effects of the HDAd vector may also counteract positive effects of prestin on cell size and/or be responsible for the differences in Vpkcm or α. For example, it is possible that HDAd–prestin–GFP alters cell turgor and shifts the voltage dependence of the nonlinear capacitance (Chertoff and Brownell 1994; Kakehata and Santos-Sacchi 1995; Oghalai et al. 2000). Based on the transduction pattern, it is possible that trauma enhances the ability of HDAd to infect a cell. Removing the tectorial membrane may affect the OHC stereocilia and the spiral limbus, increasing HDAd–prestin–GFP uptake in some fashion. There are many different ways to transduce cochlear tissues, some of which might have more transduction efficiency with less toxicity. Previous studies reported viral-mediated gene transfer into the cochlea using herpes simplex type I and vaccinia viruses (Bowers et al. 2002; Chen et al. 2001; Derby et al. 1999; Van de Water et al. 1999), lentiviruses (Han et al. 1999), retroviruses (Zheng et al. 1998), adenoviruses (Jero et al. 2001; Luebke et al. 2001; Mondain et al. 1998; Raphael et al. 1996; Shou et al. 2003; Stover et al. 2000), and adeno-associated viruses (Kho et al. 2000; Lalwani et al. 1998; Stone et al. 2005; Luebke et al. 2001).
Nevertheless, as HDAd can infect the organ of Corti, this virus might potentially be used for gene therapy to treat hearing loss in vivo. On one hand, the presence of human hearing loss because of a prestin mutation (Liu et al. 2003) has been called into question (Tang et al. 2005), and the lack of any other published clinically significant human mutations in prestin to date suggests that prestin-associated hearing loss is rare. On the other hand, prestin transduction in supporting cells may provide some rudimentary form of electromotility that may be valuable in patients with hearing loss of other etiologies.
Acknowledgments
The authors would like to thank Louisa Organ, Jenni Greeson, and Robert Raphael for their gracious help in using their confocal microscope. We would also like to thank William Brownell for helpful comments and suggestions. This study was funded by NIH-NIDCD grant K08 DC006671, The American Hearing Research Foundation, and The Caroline Wiess Law Fund for Research in Molecular Medicine (to JSO), NIDCD DC00354 and DC008134, and NSF BES-0522862 (to FAP), and NIDCD R01DC002290 (to RAE).
References
- Abe T, Kakehata S, Kitani R, Maruya SI, Navaratnam D, Santos-Sacchi J, Shinkawa H. Developmental expression of the outer hair cell motor prestin in the mouse.. J. Membr. Biol. 215(1):49–56, 2007. [DOI] [PMC free article] [PubMed]
- Adler HJ, Belyantseva IA, Merritt RC, Jr, Frolenkov GI, Dougherty GW, Kachar B. Expression of prestin, a membrane motor protein, in the mammalian auditory and vestibular periphery. Hear. Res. 184(1–2):27–40, 2003. [DOI] [PubMed]
- Ashmore JF. Forward and reverse transduction in the mammalian cochlea. Neurosci. Res. Suppl. 12:S39–S50, 1990. [DOI] [PubMed]
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J. Neurosci. Methods. 51(1):107–116, 1994. [DOI] [PubMed]
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP, Fakler B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282(5391):1141–1144, 1998. [DOI] [PubMed]
- Belyantseva IA, Adler HJ, Curi R, Frolenkov GI, Kachar B. Expression and localization of prestin and the sugar transporter GLUT-5 during development of electromotility in cochlear outer hair cells. J. Neurosci. 20(24):RC116, 2000a. [DOI] [PMC free article] [PubMed]
- Belyantseva IA, Frolenkov GI, Wade JB, Mammano F, Kachar B. Water permeability of cochlear outer hair cells: characterization and relationship to electromotility. J. Neurosci. 20(24):8996–9003, 2000b. [DOI] [PMC free article] [PubMed]
- Bowers WJ, Chen X, Guo H, Frisina DR, Federoff HJ, Frisina RD. Neurotrophin-3 transduction attenuates cisplatin spiral ganglion neuron ototoxicity in the cochlea. Mol. Ther. 6(1):12–18, 2002. [DOI] [PubMed]
- Brownell WE, Bader CR, Bertrand D, de Ribaupierre Y. Evoked mechanical responses of isolated cochlear outer hair cells. Science 227(4683):194–196, 1985. [DOI] [PubMed]
- Brownell WE, Spector AA, Raphael RM, Popel AS. Micro- and nanomechanics of the cochlear outer hair cell. Annu. Rev. Biomed. Eng. 3:169–194, 2001. [DOI] [PubMed]
- Cheatham MA, Huynh KH, Gao J, Zuo J, Dallos P. Cochlear function in Prestin knockout mice. J. Physiol. 560(Pt 3):821–830, 2004. [DOI] [PMC free article] [PubMed]
- Cheatham MA, Zheng J, Huynh KH, Du GG, Gao J, Zuo J, Navarrete E, Dallos P. Cochlear function in mice with only one copy of the prestin gene. J. Physiol. 569(Pt 1):229–241, 2005. [DOI] [PMC free article] [PubMed]
- Chen X, Frisina RD, Bowers WJ, Frisina DR, Federoff HJ. HSV amplicon-mediated neurotrophin-3 expression protects murine spiral ganglion neurons from cisplatin-induced damage. Mol. Ther. 3(6):958–963, 2001. [DOI] [PubMed]
- Chertoff ME, Brownell WE. Characterization of cochlear outer hair cell turgor. Am. J. Physiol. 266(2 Pt 1):C467–479, 1994. [DOI] [PubMed]
- Dallos P, Fakler B. Prestin, a new type of motor protein. Nat. Rev. Mol. Cell Biol. 3(2):104–111, 2002. [DOI] [PubMed]
- Deak L, Zheng J, Orem A, Du GG, Aguinaga S, Matsuda K, Dallos P. Effects of cyclic nucleotides on the function of prestin. J. Physiol. 563(Pt 2):483–496, 2005. [DOI] [PMC free article] [PubMed]
- Derby ML, Sena-Esteves M, Breakefield XO, Corey DP. Gene transfer into the mammalian inner ear using HSV-1 and vaccinia virus vectors. Hear. Res 134(1–2):1–8, 1999. [DOI] [PubMed]
- Evans BN, Dallos P. Stereocilia displacement induced somatic motility of cochlear outer hair cells. Proc. Natl. Acad. Sci. U. S. A. 90(18):8347–8351, 1993. [DOI] [PMC free article] [PubMed]
- Fernandez-Carvajal AM, Encinar JA, Poveda JA, de Juan E, Martinez-Pinna J, Ivorra I, Ferragut JA, Morales A, Gonzalez-Ros JM. Structural and functional changes induced in the nicotinic acetylcholine receptor by membrane phospholipids. J. Mol. Neurosci. 30(1–2):121–124, 2006. [DOI] [PubMed]
- Gale JE, Ashmore JF. The outer hair cell motor in membrane patches. Pflugers Arch. 434(3):267–271, 1997. [DOI] [PubMed]
- Guzman GR, Ortiz-Acevedo A, Ricardo A, Rojas LV, Lasalde-Dominicci JA. The polarity of lipid-exposed residues contributes to the functional differences between torpedo and muscle-type nicotinic receptors. J. Membr. Biol. 214(3):131–138, 2006. [DOI] [PubMed]
- Han JJ, Mhatre AN, Wareing M, Pettis R, Gao WQ, Zufferey RN, Trono D, Lalwani AK. Transgene expression in the guinea pig cochlea mediated by a lentivirus-derived gene transfer vector. Hum. Gene. Ther. 10(11):1867–1873, 1999. [DOI] [PubMed]
- He DZ, Evans BN, Dallos P. First appearance and development of electromotility in neonatal gerbil outer hair cells. Hear. Res. 78(1):77–90, 1994. [DOI] [PubMed]
- He DZ, Zheng J, Kalinec F, Kakehata S, Santos-Sacchi J. Tuning in to the amazing outer hair cell: membrane wizardry with a twist and shout. J. Membr. Biol. 209(2–3):119–134, 2006. [DOI] [PubMed]
- Hilgemann DW, Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 273(5277):956–959, 1996. [DOI] [PubMed]
- Huang G, Santos-Sacchi J. Mapping the distribution of the outer hair cell motility voltage sensor by electrical amputation. Biophys. J. 65(5):2228–2236, 1993. [DOI] [PMC free article] [PubMed]
- Iwasa KH. Effect of stress on the membrane capacitance of the auditory outer hair cell. Biophys. J. 65(1):492–498, 1993. [DOI] [PMC free article] [PubMed]
- Jensen-Smith H, Hallworth R. Lateral wall protein content mediates alterations in cochlear outer hair cell mechanics before and after hearing onset. Cell Motil. Cytoskelet. 64(9):705–717, 2007. [DOI] [PMC free article] [PubMed]
- Jenson-Smith HC, Hallworth R. Outer hair cell mechanics are altered by developmental changes in lateral wall protein content. In: Nuttall AL (ed) Auditory Mechanisms Processes and Models. New Jersey, World Scientific, pp187–193, 2006.
- Jero J, Tseng CJ, Mhatre AN, Lalwani AK. A surgical approach appropriate for targeted cochlear gene therapy in the mouse. Hear. Res. 151(1–2):106–114, 2001. [DOI] [PubMed]
- Kakehata S, Santos-Sacchi J. Membrane tension directly shifts voltage dependence of outer hair cell motility and associated gating charge. Biophys. J. 68(5):2190–2197, 1995. [DOI] [PMC free article] [PubMed]
- Kho ST, Pettis RM, Mhatre AN, Lalwani AK. Safety of adeno-associated virus as cochlear gene transfer vector: analysis of distant spread beyond injected cochleae. Mol. Ther. 2(4):368–373, 2000. [DOI] [PubMed]
- Lalwani A, Walsh B, Reilly P, Carvalho G, Zolotukhin S, Muzyczka N, Mhatre A. Long-term in vivo cochlear transgene expression mediated by recombinant adeno-associated virus. Gene. Ther. 5(2):277–281, 1998. [DOI] [PubMed]
- Liberman MC, Gao J, He DZ, Wu X, Jia S, Zuo J. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature 419(6904):300–304, 2002. [DOI] [PubMed]
- Liu XZ, Ouyang XM, Xia XJ, Zheng J, Pandya A, Li F, Du LL, Welch KO, Petit C, Smith RJ, Webb BT, Yan D, Arnos KS, Corey D, Dallos P, Nance WE, Chen ZY. Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss. Hum. Mol. Genet. 12(10):1155–1162, 2003. [DOI] [PubMed]
- Ludwig J, Oliver D, Frank G, Klocker N, Gummer AW, Fakler B. Reciprocal electromechanical properties of rat prestin: the motor molecule from rat outer hair cells. Proc. Natl. Acad. Sci. U. S. A. 98(7):4178–4183, 2001. [DOI] [PMC free article] [PubMed]
- Luebke AE, Foster PK, Muller CD, Peel AL. Cochlear function and transgene expression in the guinea pig cochlea, using adenovirus- and adeno-associated virus-directed gene transfer. Hum. Gene. Ther. 12(7):773–781, 2001. [DOI] [PubMed]
- Mondain M, Restituito S, Vincenti V, Gardiner Q, Uziel A, Delabre A, Mathieu M, Bousquet J, Demoly P. Adenovirus-mediated in vivo gene transfer in guinea pig middle ear mucosa. Hum. Gene. Ther. 9(8):1217–1221, 1998. [DOI] [PubMed]
- Navarrete EG, Santos-Sacchi J. On the effect of prestin on the electrical breakdown of cell membranes. Biophys. J. 90(3):967–974, 2006. [DOI] [PMC free article] [PubMed]
- Ng P, Parks RJ, Graham FL. Preparation of helper-dependent adenoviral vectors. In: Morgan JR (ed) Methods in Molecular Medicine Totowa, NJ, Humana Press, Inc., pp2nd edn.371–388, 2002. [DOI] [PubMed]
- Oghalai JS. The cochlear amplifier: augmentation of the traveling wave within the inner ear. Curr. Opin. Otolaryngol. Head Neck Surg. 12(5):431–438, 2004. [DOI] [PMC free article] [PubMed]
- Oghalai JS, Holt JR, Nakagawa T, Jung TM, Coker NJ, Jenkins HA, Eatock RA, Brownell WE. Ionic currents and electromotility in inner ear hair cells from humans. J. Neurophysiol. 79(4):2235–2239, 1998a. [DOI] [PubMed]
- Oghalai JS, Patel AA, Nakagawa T, Brownell WE. Fluorescence-imaged microdeformation of the outer hair cell lateral wall. J. Neurosci. 18(1):48–58, 1998b. [DOI] [PMC free article] [PubMed]
- Oghalai JS, Zhao HB, Kutz JW, Brownell WE. Voltage- and tension-dependent lipid mobility in the outer hair cell plasma membrane. Science 287(5453):658–661, 2000. [DOI] [PMC free article] [PubMed]
- Oliver D, Fakler B. Expression density and functional characteristics of the outer hair cell motor protein are regulated during postnatal development in rat. J. Physiol. 519(Pt 3):791–800, 1999. [DOI] [PMC free article] [PubMed]
- Oliver D, He DZ, Klocker N, Ludwig J, Schulte U, Waldegger S, Ruppersberg JP, Dallos P, Fakler B. Intracellular anions as the voltage sensor of prestin, the outer hair cell motor protein. Science 292(5525):2340–2343, 2001. [DOI] [PubMed]
- Palmer D, Ng P. Improved system for helper-dependent adenoviral vector production. Mol. Ther. 8(5):846–852, 2003. [DOI] [PubMed]
- Palmer DJ, Ng P. Physical and infectious titers of helper-dependent adenoviral vectors: a method of direct comparison to the adenovirus reference material. Mol. Ther. 10(4):792–798, 2004. [DOI] [PubMed]
- Palmer DJ, Ng P. Helper-dependent adenoviral vectors for gene therapy. Hum. Gene. Ther. 16(1):1–16, 2005. [DOI] [PubMed]
- Rajagopalan L, Patel N, Madabushi S, Goddard JA, Anjan V, Lin F, Shope C, Farrell B, Lichtarge O, Davidson AL, Brownell WE, Pereira FA. Essential helix interactions in the anion transporter domain of prestin revealed by evolutionary trace analysis. J. Neurosci. 26(49):12727–12734, 2006. [DOI] [PMC free article] [PubMed]
- Rajagopalan L, Liu H, Sturm AK, Davidson A, Pereira F, Brownell WE. 2007. Cholesterol Modulates Prestin Function. Abstracts of the Thirtieth Annual Midwinter Research Meeting of the Association for Research in Otolaryngology. Denver, CO.
- Raphael Y, Frisancho JC, Roessler BJ. Adenoviral-mediated gene transfer into guinea pig cochlear cells in vivo. Neurosci. Lett. 207(2):137–141, 1996. [DOI] [PubMed]
- Rusch A, Eatock RA. A delayed rectifier conductance in type I hair cells of the mouse utricle. J. Neurophysiol. 76(2):995–1004, 1996. [DOI] [PubMed]
- Santos-Sacchi J. Reversible inhibition of voltage-dependent outer hair cell motility and capacitance. J. Neurosci. 11(10):3096–3110, 1991a. [DOI] [PMC free article] [PubMed]
- Santos-Sacchi J. Reversible inhibition of voltage-dependent outer hair cell motility and capacitance. J. Neurosci. 11(10):3096–3110, 1991b. [DOI] [PMC free article] [PubMed]
- Santos-Sacchi J. Functional motor microdomains of the outer hair cell lateral membrane. Pflugers Arch. 445(3):331–336, 2002. [DOI] [PubMed]
- Shou J, Zheng JL, Gao WQ. Robust generation of new hair cells in the mature mammalian inner ear by adenoviral expression of Hath1. Mol. Cell Neurosci. 23(2):169–179, 2003. [DOI] [PubMed]
- Stone IM, Lurie DI, Kelley MW, Poulsen DJ. Adeno-associated virus-mediated gene transfer to hair cells and support cells of the murine cochlea. Mol. Ther. 11(6):843–848, 2005. [DOI] [PubMed]
- Stover T, Yagi M, Raphael Y. Transduction of the contralateral ear after adenovirus-mediated cochlear gene transfer. Gene. Ther. 7(5):377–383, 2000. [DOI] [PubMed]
- Sturm AK, Rajagopalan L, Yoo D, Brownell WE, Pereira FA. Functional expression and microdomain localization of prestin in cultured cells. Otolaryngol. Head. Neck. Surg. 136(3):434–439, 2007. [DOI] [PMC free article] [PubMed]
- Tang HY, Xia A, Oghalai JS, Pereira FA, Alford RL. High frequency of the IVS2-2A>G DNA sequence variation in SLC26A5, encoding the cochlear motor protein prestin, precludes its involvement in hereditary hearing loss. BMC Med. Genet. 6:30, 2005. [DOI] [PMC free article] [PubMed]
- Toietta G, Pastore L, Cerullo V, Finegold M, Beaudet AL, Lee B. Generation of helper-dependent adenoviral vectors by homologous recombination. Mol. Ther. 5(2):204–210, 2002. [DOI] [PubMed]
- Van de Water TR, Staecker H, Halterman MW, Federoff HJ. Gene therapy in the inner ear. Mechanisms and clinical implications. Ann. N. Y. Acad. Sci. 884:345–360, 1999. [DOI] [PubMed]
- Weaver SP, Hoffpauir J, Schweitzer L. Distribution of actin in developing outer hair cells in the gerbil. Hear. Res. 72(1–2):181–188, 1994. [DOI] [PubMed]
- Winter H, Braig C, Zimmermann U, Engel J, Rohbock K, Knipper M. Thyroid hormone receptor alpha1 is a critical regulator for the expression of ion channels during final differentiation of outer hair cells. Histochem. Cell Biol. 128(1):65–75, 2007. [DOI] [PubMed]
- Wu X, Currall B, Yamashita T, Parker LL, Hallworth R, Zuo J. Prestin–prestin and prestin–GLUT5 interactions in HEK293T cells. Dev. Neurobiol. 67(4):483–497, 2007. [DOI] [PubMed]
- Yu N, Zhu ML, Zhao HB. Prestin is expressed on the whole outer hair cell basolateral surface. Brain Res. 1095(1):51–58, 2006. [DOI] [PMC free article] [PubMed]
- Zheng JL, Lewis AK, Gao WQ. Establishment of conditionally immortalized rat utricular epithelial cell lines using a retrovirus-mediated gene transfer technique. Hear. Res. 117(1–2):13–23, 1998. [DOI] [PubMed]
- Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature 405(6783):149–155, 2000. [DOI] [PubMed]
- Zheng J, Madison LD, Oliver D, Fakler B, Dallos P. Prestin, the motor protein of outer hair cells. Audiol. Neuro-otol. 7(1):9–12, 2002. [DOI] [PubMed]
- Zheng J, Long KB, Matsuda KB, Madison LD, Ryan AD, Dallos PD. Genomic characterization and expression of mouse prestin, the motor protein of outer hair cells. Mamm. Genome. 14(2):87–96, 2003. [DOI] [PubMed]