

Abstract
We describe a genome-wide characterization of mRNA transcript levels in yeast grown on the fatty acid oleate, determined using Serial Analysis of Gene Expression (SAGE). Comparison of this SAGE library with that reported for glucose grown cells revealed the dramatic adaptive response of yeast to a change in carbon source. A major fraction (>20%) of the 15,000 mRNA molecules in a yeast cell comprised differentially expressed transcripts, which were derived from only 2% of the total number of ∼6300 yeast genes. Most of the mRNAs that were differentially expressed code for enzymes or for other proteins participating in metabolism (e.g., metabolite transporters). In oleate-grown cells, this was exemplified by the huge increase of mRNAs encoding the peroxisomal β-oxidation enzymes required for degradation of fatty acids. The data provide evidence for the existence of redox shuttles across organellar membranes that involve peroxisomal, cytoplasmic, and mitochondrial enzymes. We also analyzed the mRNA profile of a mutant strain with deletions of the PIP2 and OAF1 genes, encoding transcription factors required for induction of genes encoding peroxisomal proteins. Induction of genes under the immediate control of these factors was abolished; other genes were up-regulated, indicating an adaptive response to the changed metabolism imposed by the genetic impairment. We describe a statistical method for analysis of data obtained by SAGE.
INTRODUCTION
The development of innovative techniques to study gene expression combined with the knowledge of the Saccharomyces cerevisiae genome sequence makes it possible to establish an inventory of all yeast transcripts (transcriptome) and to relate these transcripts to the genes from which they originate. Serial Analysis of Gene Expression (SAGE) and hybridization on DNA microarrays are recently described tools for such an analysis (Velculescu et al., 1995, 1997; DeRisi et al., 1997; Wodicka et al., 1997; Zhang et al., 1997; Cho et al., 1998; Holstege et al., 1998). The SAGE technique samples short sequences of 10–14 nucleotides (tags) of individual mRNAs. Determination of the sequence of these tags allows identification of the corresponding genes. The frequency of a tag, representing the steady-state level of the mRNA from which it was derived, gives, with certain limitations, information about the level of gene expression and the amount of protein made. Comparison of transcriptomes yields interesting information about the dynamics of total genome expression attributable to a change in environmental conditions or state of differentiation. In addition, it provides necessary clues to determine the function of those genes whose contribution to cellular life is still unknown.
We were particularly interested in the changes in the mRNA population required for the increase in number and volume of peroxisomes when yeast is faced with fatty acids as a sole carbon source. In S. cerevisiae, the enzymes for degradation of fatty acids are uniquely confined to peroxisomes together with some of the enzymes constituting the glyoxylate cycle (Kunau et al., 1988). Particularly, the genes encoding the β-oxidation enzymes are highly induced, but to what extent the growth on fatty acids also leads to more general alterations in cellular metabolism and organelles other than peroxisomes is not known. It is also unknown to what extent proliferation of the peroxisomal compartment induced by growth on fatty acids requires adjustment in other proteins than those functioning in metabolism, for instance, proteins involved in protein trafficking or proteins involved in peroxisome biogenesis.
To address these questions, we used SAGE to determine the transcriptome of yeast grown on oleate and compared this with the transcriptome of yeast grown on glucose (Velculescu et al., 1997). In the process we developed a statistical method for comparison of SAGE results and the planning of future experiments. We also constructed a SAGE library of mutant yeast cells in which the oleate-induced enlargement of the peroxisomal compartment was abrogated by deletion of the genes encoding transcription factors Pip2p and Oaf1p (Luo et al., 1996; Rottensteiner et al., 1996). This analysis revealed genes that are under the control of Pip2p and Oaf1p but in addition showed genes whose activity increased to survive the genetic impairment imposed to the pip2/oaf1 mutant cells. Here we will relate some of our findings to aspects of peroxisome biogenesis and function.
MATERIALS AND METHODS
Yeast Strains, Culture Conditions, and RNA Isolation
Yeast strains used in this study are BJ1991 (MATα, leu2, trp1, ura3-52, pep4-3, prb1-1122; Jones, 1977) and BJ1991 pip2/oaf1 (MATα, leu2, trp1, ura3-52, pep4-3, prb1-1122, PIP2::KANMX4, OAF1::LEU2; Rottensteiner et al., 1997). Differences in genotype between yeast strains used for glucose (Velculescu et al., 1997) and oleate SAGE libraries were not thought to influence global gene expression patterns, considering the rich carbon sources used. Strains were precultured 24 h on minimal medium containing 0.3% glucose to obtain a derepressed culture. After a shift to medium containing 0.12% oleate, 0.2% Tween 40, 0.3% yeast extract, 0.5% bacto-peptone, and 0.5% potassium phosphate buffer, pH 6.0, the cells were cultured for 18 h at 28°C. Cell growth was stopped by the addition of an equal volume of ethanol (−80°C), and RNA was extracted immediately. mRNA was isolated using the Poly-A-tract kit from Promega (Madison, WI) according to the manufacturer’s protocol.
SAGE Procedure
The SAGE libraries were obtained essentially following the SAGE protocol described (Velculescu et al., 1995). Briefly, mRNA was converted to cDNA using a Life Technologies (Gaithersburg, MD) cDNA synthesis kit and biotin-oligo-(dT)18 (New England Biolabs, Beverly, MA). cDNA was digested with NlaIII, and 3′ cDNAs were isolated using streptavidin magnetic beads (Dynal M280; Dynal, Oslo, Norway). 3′ cDNAs were split into two pools, and SAGE linkers 1 and 2 (synthesized by Eurogentec, Seraing, Belgium) were ligated to pools 1 and 2, respectively. SAGE tags were released with BsmF1 and blunted with T4 polymerase, and the tags from pools 1 and 2 were ligated to each other. A 1:800 dilution of the ligation product was amplified with 28 cycles of PCR and digested with NlaIII. Ditags were isolated from a 12% polyacrylamide gel and self-ligated. Concatemers were isolated from an 8% polyacrylamide gel and cloned into pZERO (Invitrogen, San Diego, CA) digested with SphI. Inserts were amplified by colony PCR using M13 forward and reverse primers. PCR fragments were sequenced via cycle sequencing with the AmpliTaqFS core kit (Applied Biosystems, Foster City, CA) and 2 pmol of Cy5-T7 primer. An MJ Research (Waltham, MA) PT-200 cycler was used to perform 25 cycles (97°C, 15 s; 55°C, 30 s; and 68°C, 30 s). Reactions were loaded on 60-cm-long 4.5% PAGE-PLUS polyacrylamide gels (Amresco, Solon, Ohio) on the European Molecular Biology Laboratory (EMBL) sequencing system with array detectors (Erfle et al., 1997). The 3′ and 5′ sequences were obtained simultaneously by sequencing on the ARAKIS two-dye DNA-sequencing system developed at EMBL (Wiemann et al., 1995). The system allows simultaneous on-line sequencing on both strands (Doublex sequencing), with the two sequencing products obtained in a single sequencing reaction, each labeled with a different fluorescent dye. Up to 2000 bases are thus obtained simultaneously in one sequencing reaction on both strands, which presents an efficient system for identifying large number of sequence tags obtained in one run. Raw sequencing data were evaluated using the GeneSkipper software package (EMBL).
SAGE Data Analysis
Initial data analysis was performed using the SAGE software package version 1.0 (Velculescu et al., 1995; Zhang et al., 1997). The tag list from wild-type cells and pip2/oaf1 cells contained 10,943 and 3847 tags, respectively, of which 577 and 234, respectively, were derived from linker sequences. These tags were excluded from the analysis. The resulting tag lists contained 10,366 total tags from wild-type cells and 3613 tags from pip2/oaf1 cells. We compiled a database of all potential tags of the complete yeast genome (>69,000 10-bp sequences) and linked each tag to the gene annotations in the MIPS database (as of December 9, 1998). Next, we merged this data set with the tags found with SAGE. For estimating the number of different genes of which tags were found, we counted tags located within the ORFs or within the 500-bp 3′ adjacent of the ORFs. When different tags originating from the same gene were found, these tags were pooled to calculate expression levels. We found that tags originated from ∼1700 genes, of which 400 were not identified in the tag list from glucose-grown cells. For most of these 400 genes only one tag was found. Tag numbers were converted to number of mRNA transcripts per cell assuming a total of 15,000 mRNA molecules per cell. mRNA ratios between oleate- and glucose-grown cells were calculated from the tag lists. To avoid division by zero we used a tag value of 1 for tags that were not detected in the tag list from glucose-grown cells (Zhang et al., 1997). Hereby also the genes that are highest expressed are scored as the highest induced. Classification in groups was done according to the yeast protein functional catalogue (Goffeau, 1997; Mewes et al., 1997; also available via the World Wide Web at http://websvr.mips.biochem.mpg.de/proj/yeast). A second database was prepared that permitted searching for genes in specific functional categories. SAGE data sets of oleate grown wild-type and pip2/oaf1 mutant cells are available on request (E-mail: A.Kal@icrf.icnet.uk) and will be made available via the web sites of Munich Information Centre for Protein Sequences (http://websvr.mips.biochem.mpg.de/proj/yeast) and MBC (http://www.molbiolcell.org).
For the nomenclature of genes coding for cytoplasmic ribosomal protein, the guidelines proposed by Mager et al. (1997) were followed.
Statistical Analysis
Two statistical approaches of the number of specific tags found in SAGE can be envisioned. First, the number of tags can be seen as counts and therefore as Poisson distributed values. A test for differences in tag numbers found in two experimental conditions, seemingly based on Poisson distribution statistics, has recently been described (Madden et al., 1997). However, their approach suffers from the disadvantage that it can only be used reliably on tag libraries of similar size.
The second approach is to look at the number of copies of a specific mRNA per cell as a fraction or proportion of the total number of mRNA molecules in that cell. The same proportion (p) of specific tags should be present in the SAGE library of all sequenced tags. Thus:
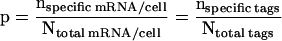 |
In this approach the number of tags found is binomially
distributed because it is the result of the p of each tag to be
identified as being the specific mRNA or not. However, for the high
number of tags sequenced, this binomial distribution can be very well
approximated by a normal distribution with mean p and SD:
SDp = 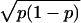 .
The accuracy of the estimation of the proportion depends on the total
number of tags (N) sequenced, its SE being SEp =
SDp/√N. The 95% confidence interval of the observed
number of specific tags can then be calculated as:
.
The accuracy of the estimation of the proportion depends on the total
number of tags (N) sequenced, its SE being SEp =
SDp/√N. The 95% confidence interval of the observed
number of specific tags can then be calculated as:
 |
1 |
In Figure 6A this 95% confidence interval is given for copies per cell up to 100 for our total number of 10,366 sequenced tags. The SE of the difference p1 − p2 of proportions p1 and p2, resulting from samples with sizes N1 and N2, respectively, is given by:
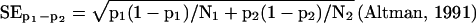 |
2 |
The difference p1 − p2 and the above SE can be used to calculate the test statistic:
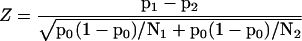 |
3 |
in which p0 is calculated as p0 = (n1 + n2)/(N1 + N2), which is the estimate of the proportions if the null hypothesis is true. Under the null hypothesis this Z statistic is normally distributed and can serve as a statistical test for the difference between the proportions p1 and p2. Comparison of this test and the test based on Poisson-distributed tag counts mentioned above (Madden et al., 1997) shows a difference in sensitivity, the test of Madden et al. (1997) being more conservative. A major advantage of a test that is based on proportions such as the Z test we propose is, however, that it can be used for results based on SAGE libraries of different size.
Figure 6.

Statistical analysis. (A) Ninety-five percent confidence intervals (thin lines) for the number of copies per cell found in the oleate condition (thick line) and critical values for the statistical comparison of these numbers with numbers found in a control condition. Control values that fall outside the critical values can be considered different from the corresponding control value at significance levels of 0.05 (▴), 0.01 (♦) and 0.001 (▪), respectively. Calculations are carried out for a sample size of 10,366 sequenced tags in the oleate condition and 60,633 in the control condition, assuming the total number of mRNA molecules in a yeast cell to be 15,000. (B). Number of tags required in the experimental group to detect a 2- to 20-fold induction in abundance of mRNA compared with the control group from which 60,633 tags already have been sequenced. Calculations were performed with the SAGEstat software, assuming the total number of mRNA molecules in a yeast cell to be 15,000, with α = 0.05 and β = 0.1.
An additional advantage is that this Z test provides a way to calculate the number of tags needed to be sequenced to detect a difference as significant. In statistical testing, the relation between the difference p1 − p2 that can be detected with a two-sided probability of a type I error (incorrect rejection of the null hypothesis) of less then α, as well as a probability of a type II error (failure to detect a true difference p1 − p2) of less then β can generally be expressed as:
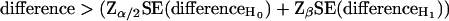 |
4 |
Thus, an equation for the calculation of the difference in proportions that can be detected as significant can be formulated by substitution of Eq. 2 into Eq. 4:
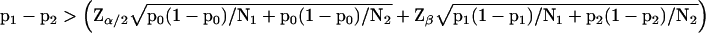 |
5 |
Eq. 5 can be used to calculate critical values for the) comparison of numbers of specific tags found in different experiments (see Figure 6A). Audic and Claverie (1997) derive a formula for a similar calculation of critical values (or what they call first significantly different values). Their critical values and the ones we calculated with Eq. 5 are equal for both 0.01 and 0.05 levels of significance. Assuming N1 and N2 to be equal, Eq. 5 can be rearranged into the following formula for the calculation of the number of tags needed to be sequenced in each of both experimental conditions to detect a difference p1 − p2 with a two-sided significance less then α and a power greater than 1-β:
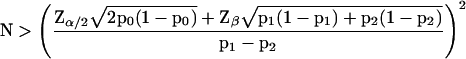 |
6 |
 |
However, when N1 and N2 are not equal, as is the case when one wants to compare an experimental condition with a control condition already published, such a rearrangement of Eq. 5 is not possible. Therefore, the sample size N2, for a given N1, α, β, and a required difference p1 − p2 can only be calculated by an iterative procedure based on Eq. 5. The results of such a calculation for N1 = 60,633, α = 0.05, and β = 0.1 for a range of differences is given in Figure 6B. Similarly, an estimate of the minimal detectable difference p1 − p2 for a given N1, N2, α, and β can only be reached by iteration. A Windows program, SAGEstat, performing these calculations, is available on request (E-mail: j.m.ruijter@amc.uva.nl subject: SAGEstat).
RESULTS
Comparison of SAGE Tag Lists Determined for Glucose-grown and Oleate-grown Yeast
We determined 10,366 SAGE tags (from 10,366 transcripts) originating from ∼1700 different genes obtained from yeast cells grown on medium containing the fatty acid oleate as sole carbon source. Following the specificity criteria set forth by Velculescu et al. (1995, 1997) and assuming a total number of 15,000 mRNA molecules per cell, the number of transcripts from a certain gene was calculated from the number of tags observed in the tag list (Tables 1 and 2). In this way, tag lists of cells cultured under different conditions can be compared with each other to monitor changes in steady-state mRNA levels. Although tag lists cannot account for regulation secondary to transcription and mRNA turnover, such as enzyme feed back control, covalent modification, and protein turnover, they can be used as good indicators for changes in gene expression. Although the mRNA steady-state levels expressed as mRNA copies per cell suggest a certain precision, we like to emphasize that these numbers will vary according to the assumptions made for their calculation (as amply discussed by Velculescu et al., 1997). Here we have used these numbers particularly for comparative purposes to sort our own database in various ways and conformed ourselves to published rules to be able to make simple reference to already existing data. For an in-depth discussion of the applied statistics, see MATERIALS AND METHODS. The calculated mRNA levels from SAGE data obtained from glucose-grown and oleate-grown cells show good correlation with previously published data from Northern blotting experiments (Einerhand et al., 1991; Rottensteiner et al., 1996; Karpichev and Small, 1998).
Table 1.
mRNA levels sorted by copies per cell in oleate-grown cells
| c/c wt ole | c/c wt glu | Ratio ole/glu | c/c pip2/oaf1 ole | Systematic name | Gene name | Description | |
|---|---|---|---|---|---|---|---|
| 1 | 289.4 | 181.6 | 1.6 | 506.5 | YOL086c/YMR303c | ADH1/ADH2 | Alcohol dehydrogenase I |
| 2 | 146.2 | 5.2 | 28.1 | 494.0 | YLR327c | Strong similarity to Stf2p | |
| 3 | 121.6 | 2.0 | 61.4 | 311.4 | YFL014w | HSP12 | Heat shock protein |
| 4 | 117.2 | 0.5 | 236.9 | 91.3 | YOL126c | MDH2 | Malate dehydrogenase, cytoplasmic |
| 5 | 117.2 | 0.0 | 473.8 | 8.3 | YIL160c | POT1 | Acetyl-CoA C-acyltransferase, peroxisomal |
| 6 | 97.0 | 1.2 | 78.4 | 174.4 | YOR374w | ALD7 | Aldehyde dehydrogenase |
| 7 | 92.6 | 0.0 | 374.3 | 220.0 | YCR010c | Strong similarity to Y. ipolytica GPR1 protein and Fun34p | |
| 8 | 86.8 | 248.4 | 0.3 | 49.8 | YPR080w/YBR118w | TEF1/TEF2 | Translation elongation factor eEF1 alpha-A chain, cytosolic |
| 9 | 76.7 | 0.0 | 310.0 | 16.6 | YDR256c | CTA1 | Catalase A, peroxisomal |
| 10 | 73.8 | 3.7 | 19.9 | 4.2 | YJL217w | Hypothetical protein | |
| 11 | 72.4 | 4.2 | 17.2 | 12.5 | YDL078c | MDH3 | Malate dehydrogenase, peroxisomal |
| 12 | 72.4 | 23.0 | 3.1 | 178.5 | YDR077w | SED1 | Abundant cell surface glycoprotein |
| 13 | 68.0 | 21.0 | 3.2 | 207.6 | YPR149w | NCE2 | Involved in nonclassical protein export pathway |
| 14 | 66.6 | 6.7 | 10.0 | 116.2 | YGR086c | Strong similarity to hypothetical protein YPL004c | |
| 15 | 63.7 | 165.5 | 0.4 | 87.2 | YKL060c | FBA1 | Fructose-bisphosphate aldolase |
| 16 | 59.3 | 49.5 | 1.2 | 41.5 | YML028w | TSA1 | Thiol-specific antioxidant |
| 17 | 57.9 | 424.5 | 0.1 | 215.9 | YJR009c/YGR192c | TDH2/TDH3 | Glyceraldehyde-3-phosphate dehydrogenase 2 |
| 18 | 57.9 | 27.2 | 2.1 | 74.7 | YGR209c | TRX2 | Thioredoxin II |
| 19 | 56.4 | 3.0 | 19.0 | 8.3 | YOR383c | Weak similarity to L. mexicana secreted acid phosphatase 2 | |
| 20 | 56.4 | 33.4 | 1.7 | 37.4 | YJR104c | SOD1 | Copper-zinc superoxide dismutase |
| 21 | 56.4 | 43.8 | 1.3 | 58.1 | YDR154c | Questionable ORF | |
| 22 | 55.0 | 6.4 | 8.5 | 224.2 | YBR067c | TIP1 | Temp. shock induced protein of the Srp1/Tip1p family |
| 23 | 50.6 | 56.2 | 0.9 | 70.6 | YDR276c | Strong similarity to Hordeum vulgare blt101 protein | |
| 24 | 47.8 | 0.0 | 193.0 | 0.0 | YGL205w | POX1 | Acyl-CoA oxidase |
| 25 | 46.3 | 72.2 | 0.6 | 4.2 | YGR027c | RPS31A | Ribosomal protein S25.e.c7 |
| 26 | 46.3 | 143.2 | 0.3 | 41.5 | YDL184c | RPL47A | Ribosomal protein |
| 27 | 43.4 | 206.6 | 0.2 | 4.2 | YLR340w/YDL081/ YOL039w/YDL130w | RPLA0/1/2/3 | Acidic ribosomal protein L10.e |
| 28 | 42.0 | 0.0 | 169.6 | 4.2 | YER065c | ICL1 | Isocitrate lyase |
| 29 | 40.5 | 24.0 | 1.7 | 37.4 | YNL055c | POR1 | Mitochondrial outer membrane porin |
| 30 | 75.2 | 0.0 | 304.2 | 199.3 | YIL057c | Strong similarity to YER067w | |
| 31 | 37.6 | 2.2 | 16.9 | 4.2 | YFR033c | QCR6 | Ubiquinol–cytochrome-c reductase 17-kDa protein |
| 32 | 37.6 | 0.5 | 76.0 | 4.2 | YNL202w | SPS19 | 2,4-Dienoyl-CoA reductase, peroxisomal |
| 33 | 37.6 | 0.0 | 152.1 | 37.4 | YOR348c | PUT4 | Proline and gamma-aminobutyrate permease |
| 34 | 36.2 | 33.4 | 1.1 | 0.0 | YGL076c | RPL6A | Ribosomal protein L7.e.A |
| 35 | 36.2 | 0.0 | 146.2 | 0.0 | YOR084w | Hypothetical protein |
Genes encoding peroxisomal proteins are shown in bold type. For calculation of ratios, a tag value of 1 (corresponding to 0.25 c/c) was used to avoid division by zero. wt, wild-type; ole, oleate; glu, glucose.
Table 2.
mRNA levels sorted by level of induction, calculated as ratio (c/c oleate divided by c/c glucose)
| c/c wt ole | c/c wt glu | Ratio ole/glu | c/c pip2/oaf1 ole | Systematic name | Gene name | Description | |
|---|---|---|---|---|---|---|---|
| 1 | 117.2 | 0.0 | 473.8 | 8.3 | YIL160c | POT1 | Acetyl-CoA C-acyltransferase, peroxisomal |
| 2 | 92.6 | 0.0 | 374.3 | 220.0 | YCR010c | Strong similarity to Y. lipolytica GPR1 protein and Fun34p | |
| 3 | 76.7 | 0.0 | 310.0 | 16.6 | YDR256c | CTA1 | Catalase A, peroxisomal |
| 4 | 75.2 | 0.0 | 304.2 | 199.3 | YIL057c | Strong similarity to YER067w | |
| 5 | 117.2 | 0.5 | 236.9 | 91.3 | YOL126c | MDH2 | Malate dehydrogenase, cytoplasmic |
| 6 | 47.8 | 0.0 | 193.0 | 0.0 | YGL205w | POX1 | Acyl-CoA oxidase |
| 7 | 42.0 | 0.0 | 169.6 | 4.2 | YER065c | ICL1 | Isocitrate lyase |
| 8 | 37.6 | 0.0 | 152.1 | 37.4 | YOR348c | PUT4 | Proline and gamma-aminobutyrate permease |
| 9 | 36.2 | 0.0 | 146.2 | 0.0 | YOR084w | Hypothetical protein | |
| 10 | 34.7 | 0.2 | 140.4 | 45.7 | YIL136w | OM45 | Protein of the outer mitochondrial membrane |
| 11 | 31.8 | 0.0 | 128.7 | 4.2 | YFL030w | Similarity to several transaminases | |
| 12 | 28.9 | 0.2 | 117.0 | 124.6 | YOL052c-a | DDR2 | Heat shock protein DDRA2 |
| 13 | 28.9 | 0.2 | 117.0 | 41.5 | YAL054c | ACS1 | Acetyl-CoA synthetase |
| 14 | 28.9 | 0.2 | 117.0 | 20.8 | YIL155c | GUT2 | Glycerol-3-phosphate dehydrogenase, mitochondrial |
| 15 | 28.9 | 0.0 | 117.0 | 8.3 | YLR174w | IDP2 | Isocitrate dehydrogenase, cytosolic |
| 16 | 28.9 | 0.0 | 117.0 | 4.2 | YJR019c | TES1 | Thioesterase, peroxisomal |
| 17 | 27.5 | 0.2 | 111.1 | 166.1 | YKL217w | JEN1 | Carboxylic acid transporter protein |
| 18 | 26.0 | 0.2 | 105.3 | 20.8 | YKL187c | Strong similarity to hypothetical protein YLR413w | |
| 19 | 24.6 | 0.0 | 99.4 | 12.5 | YLR377c | FBP1 | Fructose-1,6-bisphosphatase |
| 20 | 23.2 | 0.0 | 93.6 | 4.2 | YER015w | FAA2 | Long-chain fatty acid–CoA ligase |
| 21 | 21.7 | 0.2 | 87.7 | 54.0 | YMR107w | Hypothetical protein | |
| 22 | 20.3 | 0.2 | 81.9 | 0.0 | YJL153c | INO1 | Myo-inositol-1-phosphate synthase |
| 23 | 20.3 | 0.0 | 81.9 | 0.0 | YNL009w | IDP3 | Isocitrate dehydrogenase, NADP-dependent |
| 24 | 20.3 | 0.0 | 81.9 | 16.6 | YNL117w | MLS1 | Malate synthase 1 |
| 25 | 97.0 | 1.2 | 78.4 | 174.4 | YOR374w | ALD7 | Aldehyde dehydrogenase |
| 26 | 37.6 | 0.5 | 76.0 | 4.2 | YNL202w | SPS19 | 2,4-Dienoyl-CoA reductase, peroxisomal |
| 27 | 18.8 | 0.0 | 76.0 | 4.2 | YIL125w | KGD1 | 2-Oxoglutarate dehydrogenase complex E1 component |
| 28 | 18.8 | 0.0 | 76.0 | 8.3 | YKR097w | PCK1 | Phosphoenolpyruvate carboxykinase |
| 29 | 17.4 | 0.0 | 70.2 | 0.0 | YOL155c | Similarity to glucan 1,4-alpha-glucosidase Sta1p and YAR066w | |
| 30 | 15.9 | 0.0 | 64.3 | 12.5 | YLR155c/157c/ 158c/160c | ASP3A/B/C/D | l-Asparaginase II |
| 31 | 15.9 | 0.0 | 64.3 | 4.2 | YGR244c | Strong similarity to rumen fungus beta-succinyl CoA synthetase | |
| 32 | 121.6 | 2.0 | 61.4 | 311.4 | YFL014w | HSP12 | Heat shock protein |
| 33 | 14.5 | 0.2 | 58.5 | 37.4 | YDR070c | Hypothetical protein | |
| 34 | 14.5 | 0.0 | 58.5 | 12.5 | YBR046c | ZTA1 | Similarity to ζ-crystallin |
| 35 | 13.0 | 0.2 | 52.6 | 0.0 | YCR062w | Similarity to Ytp1p protein |
Genes encoding peroxisomal proteins are shown in bold type. For calculation of ratios, a tag value of 1 (corresponding to 0.25 c/c) was used to avoid division by zero. wt, wild-type; ole, oleate; glu, glucose.
When genes were graphically displayed in order of decreasing tag frequency, a surprisingly small number of mRNAs were found to be present at high steady-state levels. This was already observed for glucose-grown cells (Velculescu et al., 1997) and is shown in Figure 1A. The same was true for oleate-grown cells (Figure 1C). Only 0.1% of the genes was represented at ∼100 copies per cell (c/c) or more; 0.5% at 50 c/c or more; 5% at 2 c/c or more, whereas most mRNAs (>90%) were present at <2 c/c or were not expressed at all.
Figure 1.

Display of steady-state mRNA levels in yeast grown on different carbon sources. Each bar represents one mRNA species. (A) Display of mRNA levels in glucose-grown wild-type yeast. The 500 highest expressed genes are sorted by levels of mRNA copies per cell. (B) mRNA levels in oleate-grown wild-type cells. The genes are sorted in the same order as in A. (C) mRNA levels of the 500 highest expressed genes in oleate-grown wild-type cells. The genes are sorted by levels of mRNA copies per cell. (D) mRNA levels in oleate-grown pip2/oaf1 cells. The genes are sorted in the same order as in C.
The nature of the tags representing the abundant mRNAs was totally different between the two conditions of growth. This was illustrated by displaying the mRNA levels observed in oleate-grown cells arranged in the same order as mRNA levels of glucose-grown cells (Figure 1B). Here we observed a complete change in the mRNA landscape. The abundant mRNAs of oleate-grown cells now appeared as peaks at positions where the same mRNAs were present at low levels in the graph of the glucose-grown cells. These were the mRNAs that were sorted as the high abundance group in the graph of Figure 1C. The ratios between copies per cell on oleate and glucose represent the fold induction of expression. When c/c on oleate is divided by c/c on glucose, a high ratio indicates a high induction on oleate. Calculation of these ratios showed that many high abundant mRNAs on oleate were also highly induced (Tables 1 and 2). The error in the values of these ratios is subject to variation dependent on the tag numbers used for their calculation. Again, like the c/c, we have used these ratios not as absolute values but only as general indicators to help us in the interpretation of the information present in the database.
The peaks in Figure 1B alerted us to the genes that were highly expressed when cells were grown on oleate. A number of such genes were already known from previous work using Northern blotting and reporter gene studies (Einerhand et al., 1991; Rottensteiner et al., 1996; Karpichev and Small, 1998). Examples are genes encoding enzymes of the β-oxidation pathway such as acyl-coenzyme A (CoA) oxidase (FOX1/POX1) and thiolase (FOX3/POT1) and other genes encoding peroxisomal matrix proteins such as catalase A (CTA1) and malate dehydrogenase isoform 3 (MDH3). Indeed, the number of mRNAs observed was also correspondingly high: acyl-CoA oxidase, 55 c/c; thiolase, 120 c/c; catalase, 84 c/c; and malate dehydrogenase 3, 72 c/c. These numbers were substantially lower in glucose-grown cells (0, 0, 0, and 4 c/c, respectively). Several genes with unknown functions that were highly expressed proved to encode novel peroxisomal proteins: YNL009W encodes Idp3p (23 c/c), a peroxisomal NADP-dependent isocitrate dehydrogenase (van Roermund et al., 1998), and YJR019C encodes Tes1p (29 c/c), a peroxisomal acyl-CoA thioesterase (Kal, 1997).
In the Yeast Genome Directory 1997 (Goffeau, 1997), genes with known function or with homology to genes with known function have been grouped in functional categories. This allowed graphical display of only those genes that fall into such a category and permitted a more in-depth analysis of a particular biological process. A few examples will be discussed below; the complete data set is available on the Internet.
Biogenesis and Function of Peroxisomes
For growth on oleate peroxisomes are required, because in yeast these organelles exclusively house the β-oxidation enzymes necessary for fatty acid degradation. As a consequence, in oleate-grown cells the number and volume of peroxisomes are greatly increased compared with cells grown on other carbon sources, particularly glucose. This is illustrated in Figure 2A. The highest transcript frequencies corresponded with mRNAs encoding the β-oxidation enzymes and other peroxisomal enzymes directly or indirectly involved in fatty acid metabolism. Control of gene expression in these cases was very tight, because for most of these genes not a single tag was found in the glucose tag list despite the overwhelming number of 60,000 tags determined (Velculescu et al., 1997). Induction extended also over a large range considering the high number of mRNA copies per cell. This situation was totally different for PEX genes coding for peroxins, proteins involved in the biogenesis of peroxisomes and involved in import of proteins into peroxisomes (Figure 2A). For most PEX genes it has been reported that they are induced when cells are grown on oleate. However, the abundance of mRNAs encoding peroxins with known functions was still <6 c/c, e.g., components of the protein import pathway such as the peroxisomal-targeting signal receptor Pex5p and the Pex5p-docking proteins Pex13p and Pex14p (Elgersma et al., 1996; Erdmann and Blobel, 1996; Gould et al., 1996; Albertini et al., 1997). This seemed to be a general feature; also, mRNAs coding for proteins of the nuclear pore complex, the mitochondrial Tim and Tom proteins, and the components of the protein import machinery of the endoplasmic reticulum were low abundant or absent in both the glucose and oleate tag lists. Because much about peroxisome metabolism and biogenesis is still unknown, we like to caution that the “peroxisome” functional category is plainly incomplete. Clues for additional candidates of this category can be found in the data set. For instance, YJR019c (TES1) is an unknown reading frame that by definition will not show up in the peroxisome functional category. It is, however, a good example to illustrate the potential use of the data set. TES1 is expressed at 29 c/c in oleate-grown cells and hardly expressed in glucose-grown cells (<0.3 c/c; Table 2). The encoded reading frame shows homology to acyl-CoA thioesterases of Escherichia coli, Haemophilus influenzae, and human. Further biochemical analysis showed that the encoded protein had acyl-CoA thioesterase activity and is located in the peroxisomal matrix. Like the majority of genes encoding peroxisomal matrix proteins, the TES1 gene is regulated by the Pip2p and Oaf1p transcription factors. Upon deletion of the TES1 gene we have not yet observed an overt phenotype (Kal, 1997). This example shows how candidates for the peroxisome functional category can be traced. In addition, it is an example of a gene that we missed for lack of a phenotype in our genetic screens applied thus far.
Figure 2.

mRNA levels of genes encoding peroxins and peroxisomal matrix enzymes. (A) Bars in the front row represent mRNA levels in glucose-grown wild-type cells (wt glu); bars in the back row represent mRNA levels in oleate-grown wild-type cells (wt ole). Note that all peroxins localize to the left, whereas all matrix enzymes localize to the right, stressing the differences in expression levels between the two classes of genes. (B). Bars in the back row represent mRNA levels in oleate-grown wild-type cells (wt ole); bars in the front row represent mRNA levels in oleate-grown pip2/oaf1 cells (p/o ole). *, Statistically significant difference in expression levels (p < 0.05).
Mitochondria
For growth on oleate and other nonfermentable carbon sources, a functional mitochondrial compartment is essential. We therefore wondered whether genes encoding mitochondrial proteins would be up-regulated or down-regulated as a group depending on the carbon source. Figure 3 illustrates that on average the frequency of mRNAs coding for mitochondrial proteins was higher in oleate-grown cells compared with glucose-grown cells. It indicated that cells more heavily rely on mitochondrial metabolism when they grow on oleate (see DISCUSSION). However, in a number of cases, tag frequencies are substantially increased above the average enrichment, suggesting that mitochondria require fine tuning to adjust to growth on oleate. Some adjustments, such as elevated expression of the mitochondrial glycerol-3-phosphate dehydrogenase (Gut2p), can be explained in terms of changing metabolism (see below); other changes ask for further research, such as the mitochondrial outer membrane protein OM45 (35 c/c in oleate grown cells vs. <0.3 c/c in glucose grown cells).
Figure 3.

mRNA levels of genes encoding mitochondrial proteins. The 100 highest expressed genes in wild-type cells grown in oleate medium are displayed in the back row (wt ole) and are sorted by numbers of mRNA copies per cell. The front row represents the mRNA levels for the same genes of wild-type cells grown on glucose (wt glu).
Communication among Peroxisomes, Cytosol, and Mitochondria
The end products of β-oxidation, NADH and acetyl-CoA, generated inside peroxisomes, must be exported across the impermeable membrane to cytoplasm and mitochondria for ATP generation and biosynthetic processes (Elgersma and Tabak, 1996). Reduction equivalents are proposed to shuttle from the peroxisomal matrix to the cytoplasm in the form of malate in a process requiring peroxisomal malate dehydrogenase (Figure 4). Indeed, not only the gene encoding peroxisomal malate dehydrogenase (MDH3, 72 c/c) but also the gene encoding cytosolic malate dehydrogenase (MDH2, 120 c/c) was highly expressed considering the number of mRNAs per cell, and both genes were specifically induced considering the corresponding numbers in glucose-grown cells (4 and 0 c/c, respectively).
Figure 4.

Transport of reducing equivalents among peroxisomes, cytoplasm, and mitochondria requires membrane-localized metabolite transporters. Indicated are proposed shuttles for malate/oxaloacetate, isocitrate/2-oxoglutarate, and glycerol-3-phosphate/dihydroxyacetonephosphate. The question mark indicates a cytosolic glycerol-3-phosphate dehydrogenase, of which the activity was measured in the cytosol (van Roermund, Hettema, and Tabak, unpublished observations).
After export from peroxisomes, the now cytoplasmic NADH can in part be oxidised by mitochondrial oxidases facing the cytoplasm (de Vries and Marres, 1987; Larsson et al., 1998) or shuttled into the respiratory chain by the mitochondrial glycerol-3-phosphate dehydrogenase isoenzyme Gut2p (Figure 4). This was indicated by the appreciable induction of the GUT2 gene (Ronnow and Kielland-Brandt, 1993) (30 c/c).
Although the net flow of NADH is directed outward to the cytosol, a net inward flow of NADPH goes toward peroxisomes. The NADP-dependent isocitrate dehydrogenase Idp3p is involved in the production of NADPH in the peroxisomal matrix, to enable the action of the peroxisomal 2,4 dienoyl-CoA reductase Sps19p, which is required for degradation of unsaturated fatty acids with double bonds at even positions (Gurvitz et al., 1997; Henke et al., 1998; van Roermund et al., 1998). The existence of an isocitrate/2-oxoglutarate redox shuttle across the peroxisomal membrane (Figure 4) is indicated by the tandem induction of both the IDP3 gene (20 c/c on oleate, <0.3 c/c on glucose) and the IDP2 gene (29 c/c on oleate, <0.3 c/c on glucose) encoding peroxisomal isocitrate dehydrogenase Idp3p and cytosolic isocitrate dehydrogenase Idp2p, respectively.
There are two mechanisms for acetyl-CoA to leave the peroxisomes. The first is in the form of acetyl-carnitine; the second is in the form of succinate, generated in the glyoxylate cycle (Tabak et al., 1995; van Roermund et al., 1995). Part of the acetyl-CoA is transferred to the mitochondria for ATP production, whereas another part is assimilated to C4 and C6 compounds (Elgersma and Tabak, 1996). For this latter process the glyoxylate cycle and gluconeogenesis pathway are essential. Two enzymes are unique components of the glyoxylate cycle: malate synthase I and isocitrate lyase I. The remaining enzymes of the cycle are used for other purposes as well. This was underscored by the number of mRNAs observed for the (peroxisomal) enzymes that are essential for operation of this cycle (Figure 5A). Indeed, all the glyoxylate cycle enzymes were induced in oleate compared with glucose. We have left out the result for peroxisomal malate dehydrogenase 3, because it was shown that this enzyme does not participate in the glyoxylate cycle. Instead, Mdh3p is involved in the shuttling of reduction equivalents from peroxisomes to the cytosol (van Roermund et al., 1995). Further assimilation to C6 sugars is mediated by the gluconeogenesis pathway. The genes encoding the enzymes isocitrate lyase (ICL1), fructose 1,6-bisphosphatase (FBP1), and phosphoenolpyruvate carboxykinase (PCK1) are essential for gluconeogenesis. Their mRNAs are present at 25–42 c/c in oleate-grown cells (Figure 5A). Remarkably, in the tag list of glucose-grown cells, no hits for these mRNAs were scored. Thus, the gluconeogenesis enzymes are indeed very specific for oleate-grown cells. In addition, most of the glycolytic household enzymes are of course required in this process.
Figure 5.

mRNA levels of genes encoding proteins for gluconeogenesis, glyoxylate cycle, and heat shock and stress response. (A) Bars in the front row represent mRNA levels in glucose-grown wild-type cells (wt glu); bars in the back row represent mRNA levels in oleate-grown wild-type cells (wt ole). (B). Bars in the front row represent mRNA levels in oleate grown pip2/oaf1 cells (p/o ole); bars in the back row represent mRNA levels in oleate grown wild-type cells (wt ole). *, Statistically significant difference in expression levels (p < 0.05).
Heat Shock Proteins and Stress Response
The comparison also indicated that mRNAs coding for certain members of the heat shock and stress family of proteins occur at significantly higher steady-state levels in oleate-grown compared with glucose-grown cells (Figure 5A): HSP12, TIP1, SSA1, PIR3, DDR2, YRO2, SIR4, and HSP26 mRNAs. This was not simply due to a general stress response, because the levels of other stress indicators, e.g., catalase T mRNAs (CTT1), remained <0.3 c/c in the two growth conditions, and expression of genes encoding the 30- and 60-kDa heat shock proteins (HSP30 and HSP60) remained practically unchanged.
The Transcriptome of pip2/oaf1 Mutant Cells
Transcription of the genes coding for the major enzymes of peroxisomal metabolism is controlled by the transcription factors Pip2p and Oaf1p. The constitutive Oaf1p and oleate-inducible Pip2p bind as a heterodimer to an upstream activation sequence called “oleate response element” (ORE) and activate transcription of genes containing an ORE in their promoter (Karpichev et al., 1997; Rottensteiner et al., 1997). To investigate the consequences of the loss of these transcription factors and to identify additional, unknown genes subjected to their control, we cultured a pip2/oaf1 double mutant strain on oleate-containing medium. A SAGE library of 3613 tags was prepared from the mRNA derived from this culture. When this library was compared with the one obtained from wild-type cells grown on oleate, two different effects could be discerned: 1) a number of highly expressed genes under the control of Pip2p/Oaf1p were down-regulated to much lower levels, comparable with the levels encountered in yeast cells growing on nonfermentable carbon sources such as glycerol or ethanol, for which no major contribution of peroxisomes is required (derepression level) (Figures 1D and 2B and Tables 1 and 2); and 2) in response to the inefficient β-oxidation, cells made more efficient use of alternative carbon sources present in the growth medium (enforced with yeast extract). For instance, genes required for proline import and catabolism were induced in wild-type cells grown on oleate (PUT1 and PUT4) and even further induced in the mutant (PUT1). The induction of dicarboxylic acid transporters and permeases JEN1 and DIP5 also suggested that the cells switched to alternative carbon sources.
In addition to these metabolic adaptations, several genes that are known to be induced by stress were also induced in the pip2/oaf1 strain (e.g., HSP12, TIP1, DDR2, HSP26, and HSP30) (Figure 5B), whereas certain genes encoding cytosolic ribosomal proteins were down-regulated (Table 1). Possibly, the loss of Pip2p and Oaf1p transcription factors and the resulting inability to deal with oleate as a carbon source was a stressful condition, which the cells attempted to compensate.
The absence of high-level expression of certain oleate-induced genes in the pip2/oaf1 strain compared with the wild-type strain made it possible to identify genes that were thus far unknown to be under the control of Pip2p and Oaf1p. Examples are the TES1 gene encoding a peroxisomal acyl-CoA thioesterase (Kal, 1997) and the IDP3 gene encoding a peroxisomal NADP-dependent isocitrate dehydrogenase (van Roermund et al., 1998). For other genes encoding proteins with unknown functions that were expressed highly in wild-type cells on oleate but not in pip2/oaf1 mutant cells, e.g., YOR084W, further investigation of their suspected role in peroxisome function is required.
DISCUSSION
Here we report a transcriptome analysis using SAGE of S. cerevisiae growing on the fatty acid oleate as the sole carbon source. The availability of a transcriptome of glucose-grown yeast cells (Velculescu et al., 1997) made it possible to investigate the changes in mRNA levels that result from the presence of one or the other carbon source in the growth medium.
Growth of yeast on oleate requires the presence of β-oxidation enzymes to metabolize this fatty acid. In S. cerevisiae the β-oxidation enzymes are exclusively located in peroxisomes, and the peroxisomal compartment is increased in number and volume in oleate-grown cells (Veenhuis et al., 1987). We hoped that SAGE analysis would alert us to new genes involved in peroxisome biogenesis and function. In particular, we were interested in those genes that went unnoticed in the genetic screens applied thus far, for instance, genes that upon mutation would confer lethality to the cell or genes coding for proteins the loss of which did not result in a clear phenotype. SAGE is perfectly suited for application to S. cerevisiae because the total genome has been sequenced, and ∼6300 reading frames longer than 100 amino acids were identified (Goffeau, 1997). This allows assignment of almost all tags to genes with great precision. Ironically, the most abundant tag provided an exception. Here, the high DNA sequence conservation between the ADH1 and ADH2 genes encoding the isoenzymes alcohol dehydrogenase 1 and 2 caused ambiguity. From the separation of the Adh isoenzymes by PAGE under native conditions followed by determination of enzyme activity (Williamson et al., 1980), we concluded that in oleate-grown cells the tags were derived from the ADH2 gene; the tags from glucose-grown cells originated from the ADH1 gene (our unpublished data). Another imperfection of SAGE is that it cannot detect genes that lack a recognition site for the anchoring enzyme (NlaIII). An example is the TPI1 gene, which functions in glycolysis and is highly expressed in glucose-grown cells (Alber and Kawasaki, 1982).
When we compared both tag lists and limited ourselves to statistically significant changes (see MATERIALS AND METHODS and Figure 6 for a discussion of our statistical analysis), we observed that the number of mRNAs that was clearly different under both conditions was rather small: ∼100. This comprised only 2% of the total number of genes in the genome; however, it contributed a substantial portion of the total number of 15,000 mRNAs per cell: >20%. Most of these mRNAs that significantly increase in oleate-grown cells encoded enzymes required for adaptation of metabolism to the new carbon source. This confirms the dictum of Christian de Duve (1984) in his book A Guided Tour of the Living Cell: “The internal affairs of a living cell are mainly concerned with biogenesis and energy production.” The genes encoding these enzymes were under very strict control. Many of the abundant mRNAs in oleate-grown cells were almost or totally absent in the glucose-grown cells.
We were rather surprised to find that mRNAs coding for proteins functioning in the maintenance of cellular structures, for instance proteins that support trafficking of proteins in the cell, were present at very low numbers of copies per cell in both growth conditions. This holds even for cases in which a specific alteration takes place when cells are faced with another carbon source. In glucose-grown cells only a few small peroxisomes are present; in oleate-grown cells, however, their number and volume are strongly increased. But also in the latter case the peroxins (proteins involved in the biogenesis of peroxisomes) were represented with exceptionally low numbers of copies per cell. Considering this strict rule, it is remarkable that Pex11p forms an exception. Induction and transcriptional control of the PEX11 gene resembles that of genes coding for enzymes involved in fatty acid metabolism. Indeed, preliminary experiments suggest that Pex11p function is related to transport of metabolites across the peroxisomal membrane (van Roermund, Hettema, and Tabak, unpublished observations) rather than to proliferation of peroxisomes (Erdmann and Blobel, 1995; Passreiter et al., 1998). Another case illustrating the low copy number of certain mRNAs concerns the transcription factors involved in peroxisome proliferation. We previously reported that Oaf1p and Pip2p form a heterodimeric complex that binds to OREs, present in many promoters of genes coding for peroxisomal enzymes (Rottensteiner et al., 1997). The OAF1 gene is constitutively expressed, but the PIP2 gene is induced by autoregulation in oleate-containing growth medium. Both mRNAs were present at only very low levels, and the number of tags collected in this study was too low to detect a statistically significant induction of PIP2 mRNA. However, although we were not able to monitor alterations in the level of transcription factors or, for that matter, in components of signal transduction routes, the changes of expression levels of the target genes evoked by the action of these components were clearly demonstrated. Thus, 10,000 tags were sufficient to visualize the changes in mRNAs coding for components of metabolism.
The analysis of the SAGE library from pip2/oaf1 mutant cells provided a valuable tool to identify new genes involved in peroxisomal functioning. In addition, the attempts of the mutant cells to adapt to the absence of efficient metabolism of fatty acids were illustrated by elevated levels of mRNAs for alternative pathways, such as the uptake and metabolism of proline (PUT1 and PUT4) and the uptake of dicarboxylic acids (JEN1 and DIP5). The stressful condition was reflected in the induction of genes encoding heat shock and stress proteins.
The ability to visualize the behavior of each gene with respect to its contribution to cellular life is of particular interest to study the interactions and ways of communication between the major compartments of the cell. The β-oxidation of fatty acids takes place in peroxisomes, but further metabolism of its end products, NADH and acetyl-CoA, requires the participation of other compartments of the cell, particularly cytosol and mitochondria. The impermeability of the peroxisomal membrane to small molecules requires dedicated transporters in the membrane, comparable with the situation in mitochondria. Candidate genes coding for such proteins can be traced by screening the tag list for genes that are induced on oleate and code for proteins with multiple membrane spans or otherwise hydrophobic character (e.g., Pex11p; see above). We are currently determining the cellular localization of several of such proteins and studying the ones confined to peroxisomes in more detail.
Acetyl-CoA is in part converted to succinate via the glyoxylate cycle or to glucose via the gluconeogenesis pathway. Our SAGE analysis confirmed these predictions convincingly. We have argued that peroxisomal malate dehydrogenase (Mdh3p) is used to regenerate NAD+ for continuation of β-oxidation, rather than being an intrinsic part of the glyoxylate cycle (van Roermund et al., 1995). The simultaneous induction of both MDH3 and MDH2 (the latter encoding cytosolic malate dehydrogenase) reinforces the proposal of a malate/oxaloacetate shuttle to transfer reduction equivalents across the peroxisomal membrane. The strong induction on oleate of the GUT2 gene, encoding glycerol-3-phosphate dehydrogenase located at the mitochondrial inner membrane, suggests how mitochondria can tap part of the cytosolically delivered NADH for production of ATP.
To degrade polyunsaturated fatty acids, double bonds at even positions must be relocated to uneven positions in a reductive process requiring NADPH (Gurvitz et al., 1997). The induction on oleate of the genes encoding peroxisomal and cytosolic isocitrate dehydrogenase (IDP3 and IDP2, respectively) suggests that an isocitrate/2-oxoglutarate shuttle exists to expedite the transfer of reduction equivalents from the cytosol into the peroxisomes to maintain the level of NADPH (Henke et al., 1998; van Roermund et al., 1998).
Large amounts of information can already be abstracted from a comparison of steady-state conditions: glucose-grown, oleate-grown, and transcription factor-deficient cells. For more detailed insight in the dynamic transition of one state into the other, the SAGE technique is less suitable because of its low throughput capacity. In that respect, the application of the DNA microarray technique is more promising. However, SAGE data are very robust and quantitative. Dynamic ranges >200-fold in gene expression can be determined. Furthermore, by obeying some basic rules for data management, tag lists obtained by different research groups can be integrated into larger databases, and the increasing tag numbers can further improve the statistical confidence in the analysis.
Evaluating our comparison of transcriptomes of yeast cells grown on different carbon sources, we expect that detailed knowledge of metabolism and its change to altering conditions can be gained from such studies. In addition, these studies can provide clues to discover functions of novel genes identified in sequencing projects. Considering the strong conservation of biological principles during evolution, these genome-wide model studies in yeast may help to understand certain pathological conditions in human.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to Victor Velculescu and Kenneth Kinzler for generously providing SAGE software, protocols, and data from glucose-grown cells and for advice during the course of this project. We thank Werner Mewes for support in data analysis, Ted Young for advice on discriminating between alcohol dehydrogenases I and II, and Piet Borst, Ewald Hettema, Fred Meijer, Ton Muijsers, Ronald Plasterk, Carlo van Roermund, and Ron Wanders for stimulating discussions. This project was financially supported by the Netherlands Foundation for Chemical Research (Stichting Scheikundig Onderzoek Nederland)–Netherlands Foundation for Scientific Research (Nederlandse Organisatie voor Wetenschappelÿk Onderzoek) and by the European Functional Analysis Network.
Abbreviations used:
- c/c
copies per cell
- CoA
coenzyme A
- EMBL
European Molecular Biology Laboratory
- ORE
oleate response element
- SAGE
Serial Analysis of Gene Expression
Footnotes
Online version of this article contains a complete data set. Online version available at www.molbiolcell.org.
REFERENCES
- Alber T, Kawasaki G. Nucleotide sequence of the triose phosphate isomerase gene of Saccharomyces cerevisiae. J Mol Appl Genet. 1982;1:419–434. [PubMed] [Google Scholar]
- Albertini M, Rehling P, Erdmann R, Girzalsky W, Kiel JAKW, Veenhuis M, Kunau WH. Pex14p, a peroxisomal membrane protein binding both receptors of the two PTS-dependent import pathways. Cell. 1997;89:83–92. doi: 10.1016/s0092-8674(00)80185-3. [DOI] [PubMed] [Google Scholar]
- Altman DG. Practical Statistics for Medical Research. London: Chapman-Hall; 1991. pp. 162–163. [Google Scholar]
- Armitage P, Berry G. Statistical Methods in Medical Research. Oxford: Blackwell Scientific Publications; 1987. pp. 179–185. [Google Scholar]
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Cho RJ, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- De Duve C. A Guided Tour of the Living Cell. New York: Scientific American Books, W.H. Freeman; 1984. [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- de Vries S, Marres CA. The mitochondrial respiratory chain of yeast. Structure and biosynthesis and the role in cellular metabolism. Biochim Biophys Acta. 1987;895:205–239. doi: 10.1016/s0304-4173(87)80003-4. [DOI] [PubMed] [Google Scholar]
- Einerhand AW, Voorn-Brouwer TM, Erdmann R, Kunau WH, Tabak HF. Regulation of transcription of the gene coding for peroxisomal 3-oxoacyl-CoA thiolase of Saccharomyces cerevisiae. Eur J Biochem. 1991;200:113–122. doi: 10.1111/j.1432-1033.1991.tb21056.x. [DOI] [PubMed] [Google Scholar]
- Elgersma Y, Kwast L, Klein A, Voorn-Brouwer T, van den Berg M, Metzig B, America T, Tabak HF, Distel B. The SH3 domain of the Saccharomyces cerevisiae peroxisomal membrane protein Pex13p functions as a docking site for Pex5p, a mobile receptor for the import PTS1-containing proteins. J Cell Biol. 1996;135:97–109. doi: 10.1083/jcb.135.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma Y, Tabak HF. Proteins involved in peroxisome biogenesis and functioning. Biochim Biophys Acta. 1996;1286:269–283. doi: 10.1016/s0304-4157(96)00012-3. [DOI] [PubMed] [Google Scholar]
- Erdmann R, Blobel G. Giant peroxisomes in oleic acid-induced Saccharomyces cerevisiae lacking the peroxisomal membrane protein Pmp27p. J Cell Biol. 1995;128:509–523. doi: 10.1083/jcb.128.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann R, Blobel G. Identification of Pex13p a peroxisomal membrane receptor for the PTS1 recognition factor. J Cell Biol. 1996;135:111–121. doi: 10.1083/jcb.135.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erfle H, Ventzki R, Voss H, Rechmann S, Benes V, Stegemann J, Ansorge W. Simultaneous loading of 200 sample lanes for DNA sequencing on vertical and horizontal, standard and ultrathin gels. Nucleic Acids Res. 1997;25:2229–2230. doi: 10.1093/nar/25.11.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau AEA. The yeast genome directory. Nature. 1997;387S:5–105. [PubMed] [Google Scholar]
- Gould SJ, Kalish JE, Morrell JC, Bjorkman J, Urquhart AJ, Crane DI. Pex13p is an SH3 protein of the peroxisome membrane and a docking factor for the predominantly cytoplasmic PTS1 receptor. J Cell Biol. 1996;135:85–95. doi: 10.1083/jcb.135.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvitz A, Rottensteiner H, Kilpelainen SH, Hartig A, Hiltunen JK, Binder M, Dawes IW, Hamilton B. The Saccharomyces cerevisiae peroxisomal 2,4-dienoyl-CoA reductase is encoded by the oleate-inducible gene SPS19. J Biol Chem. 1997;272:22140–22147. doi: 10.1074/jbc.272.35.22140. [DOI] [PubMed] [Google Scholar]
- Henke B, Girzalsky W, Berteaux-Lecellier V, Erdmann R. IDP3 encodes a peroxisomal NADP-dependent isocitrate dehydrogenase required for the beta-oxidation of unsaturated fatty acids. J Biol Chem. 1998;273:3702–3711. doi: 10.1074/jbc.273.6.3702. [DOI] [PubMed] [Google Scholar]
- Holstege FCP, Jennings EG, Wyrick JJ, Lee TI, Hengarter CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- Jones EW. Proteinase mutants of Saccharomyces cerevisiae. Genetics. 1977;85:23–33. doi: 10.1093/genetics/85.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kal AJ. Saccharomyces cerevisiae, Ph.D. thesis. University of Amsterdam, Academic Medical Center; 1997. Transcriptional Regulation of Genes Encoding Peroxisomal Proteins. [Google Scholar]
- Karpichev IV, Luo Y, Marians RC, Small GM. A complex containing two transcription factors regulates peroxisome proliferation and the coordinate induction of beta-oxidation enzymes in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:69–80. doi: 10.1128/mcb.17.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpichev IV, Small GM. Global regulatory functions of Oaf1p and Pip2p (Oaf2p), transcription factors that regulate genes encoding peroxisomal proteins in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:6560–6570. doi: 10.1128/mcb.18.11.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunau WH, Buhne S, de la Garza M, Kionka C, Mateblowski M, Schultz-Borchard U, Thieringer R. Comparative enzymology of beta-oxidation. Biochem Soc Trans. 1988;16:418–420. doi: 10.1042/bst0160418. [DOI] [PubMed] [Google Scholar]
- Larsson C, Påhlman I-L, Ansell R, Rigoulet M, Adler L, Gustafsson L. The importance of the glycerol 3-phosphate shuttle during aerobic growth of Saccharomyces cerevisiae. Yeast. 1998;14:347–357. doi: 10.1002/(SICI)1097-0061(19980315)14:4<347::AID-YEA226>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Luo Y, Karpichev IV, Kohanski R, Small GM. Purification, identification, and properties of a Saccharomyces cerevisiae oleate-activated upstream activating sequence-binding protein that is involved in the activation of. POX1. J Biol Chem. 1996;271:12068–12075. doi: 10.1074/jbc.271.20.12068. [DOI] [PubMed] [Google Scholar]
- Madden SL, Galella EA, Zhu JS, Bertelsen AH, Beaudry GA. SAGE transcript profiles for p53-dependent growth regulation. Oncogene. 1997;15:1079–1085. doi: 10.1038/sj.onc.1201091. [DOI] [PubMed] [Google Scholar]
- Mager WH, Planta RJ, Ballesta J-PG, Lee JC, Mizuta K, Suzuki K, Warner JR, Woolford J. A new nomenclature for the cytoplasmic ribosomal proteins of Saccharomyces cerevisiae. Nucleic Acids Res. 1997;25:4872–4875. doi: 10.1093/nar/25.24.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes HW, Albermann K, Heumann K, Liebl S, Pfeiffer F. MIPS—a database for protein sequences, homology data and yeast genome information. Nucleic Acids Res. 1997;25:28–30. doi: 10.1093/nar/25.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passreiter M, Anton M, Lay D, Frank R, Harter C, Wieland FT, Gorgas K, Just WW. Peroxisome biogenesis—involvement of Arf and coatomer. J Cell Biol. 1998;141:373–383. doi: 10.1083/jcb.141.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnow B, Kielland-Brandt MC. GUT2, a gene for mitochondrial glycerol 3-phosphate dehydrogenase of Saccharomyces cerevisiae. Yeast. 1993;9:1121–1130. doi: 10.1002/yea.320091013. [DOI] [PubMed] [Google Scholar]
- Rottensteiner H, Kal AJ, Filipits M, Binder M, Hamilton B, Tabak HF, Ruis H. Pip2p: a transcriptional regulator of peroxisome proliferation in the yeast Saccharomyces cerevisiae. EMBO J. 1996;15:2924–2934. [PMC free article] [PubMed] [Google Scholar]
- Rottensteiner H, Kal AJ, Hamilton B, Ruis H, Tabak HF. A heterodimer of the Zn(2)Cys(6) transcription factors Pip2p and Oaf1p controls induction of genes encoding peroxisomal proteins in Saccharomyces cerevisiae. Eur J Biochem. 1997;247:776–783. doi: 10.1111/j.1432-1033.1997.00776.x. [DOI] [PubMed] [Google Scholar]
- Tabak HF, Elgersma Y, Hettema E, Franse MM, Voorn-Brouwer T, Distel B. Transport of proteins and metabolites across the impermeable membrane of peroxisomes. Cold Spring Harb Symp Quant Biol. 1995;60:649–655. doi: 10.1101/sqb.1995.060.01.069. [DOI] [PubMed] [Google Scholar]
- van Roermund CW, Elgersma Y, Singh N, Wanders RJ, Tabak HF. The membrane of peroxisomes in Saccharomyces cerevisiae is impermeable to NAD(H) and acetyl-CoA under in vivo conditions. EMBO J. 1995;14:3480–3486. doi: 10.1002/j.1460-2075.1995.tb07354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Roermund CWT, Hettema EH, Kal AJ, van den Berg M, Tabak HF, Wanders RJA. Peroxisomal beta-oxidation of polyunsaturated fatty acids in Saccharomyces cerevisiae: isocitrate dehydrogenase provides NADPH for reduction of double bonds at even positions. EMBO J. 1998;17:677–687. doi: 10.1093/emboj/17.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenhuis M, Mateblowski M, Kunau WH, Harder W. Proliferation of microbodies in Saccharomyces cerevisiae. Yeast. 1987;3:77–84. doi: 10.1002/yea.320030204. [DOI] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Zhou W, Vogelstein J, Basrai MA, Bassett DE, Jr, Hieter P, Vogelstein B, Kinzler KW. Characterization of the yeast transcriptome. Cell. 1997;88:243–251. doi: 10.1016/s0092-8674(00)81845-0. [DOI] [PubMed] [Google Scholar]
- Wiemann S, Stegemann J, Grothues D, Bosch A, Estivill X, Schwager C, Zimmermann J, Voss H, Ansorge W. Simultaneous on-line DNA sequencing on both strands with two fluorescent dyes. Anal Biochem. 1995;224:117–121. doi: 10.1006/abio.1995.1015. [DOI] [PubMed] [Google Scholar]
- Williamson VM, Bennetzen J, Young ET, Nasmyth K, Hall BD. Isolation of the structural gene for alcohol dehydrogenase by genetic complementation in yeast. Nature. 1980;283:214–216. doi: 10.1038/283214a0. [DOI] [PubMed] [Google Scholar]
- Wodicka L, Dong HL, Mittmann M, Ho MH, Lockhart DJ. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotech. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhou W, Velculescu VE, Kern SE, Hruban RH, Hamilton SR, Vogelstein B, Kinzler KW. Gene expression profiles in normal and cancer cells. Science. 1997;276:1268–1272. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.