

Abstract
Although drug-induced and age-related hearing losses are frequent otologic problems affecting millions of people, their underlying mechanisms remain uncertain. The inner ear is exclusively endowed with a positive endocochlear potential (EP) that serves as the main driving force for the generation of receptor potential in hair cells to confer hearing. Deterioration of EP leads to hearing loss or deafness. The generation of EP relies on the activity of many ion transporters to establish active potassium (K+) cycling within the inner ear, including K+ channels, the Na–K–2Cl co-transporter (NKCC1), and the α1 and α2 isoforms of Na,K–ATPase. We show that heterozygous deletion of either NKCC1, α1-Na,K–ATPase, or α2–Na,K–ATPase independently results in progressive, age-dependent hearing loss with minimal alteration in cochlear morphology. Double heterozygote deletion of NKCC1 with α1–Na,K–ATPase also shows a progressive, though delayed, age-dependent hearing loss. Remarkably, double heterozygote deletion of NKCC1 with α2–Na,K–ATPase demonstrates a striking preservation of hearing threshold both initially and with age. Measurements of the EP confirm the anticipated drop in potential for genotypes that demonstrate age-dependent hearing loss. The EP generated by the NKCC1 + α2-Na,K–ATPase double heterozygote, however, is maintained at a level comparable to that of the control condition, suggesting a potential advantage in this combination of ion transporter modification. These observations provide insight into the detailed mechanisms of EP generation, and results of combination-knockout experiments may have important implications in the future treatment of drug-induced and age-related hearing losses.
Keywords: age-related hearing loss, endocochlear potential, gene targeting, potassium transport, stria vascularis
INTRODUCTION
The exquisite sensitivity of cochlear hair cells is derived from the unique, organ-specific endocochlear potential (EP; Davis 1957; von Bekesy 1950). The EP provides the driving force for hair cell transduction, and its elimination results in profound deafness (Davis 1957; Flagella et al. 1999). It is believed that the EP is generated by the high throughput of K+ across the intermediate cells (IC) of the stria vascularis (StV) into the intrastrial (IS) space of the inner ear (Kuijpers and Bonting 1970; Salt et al. 1987).
At the lateral wall, perilymphatic K+ is taken up by type II and IV fibrocytes within the spiral ligament by means of α2–Na,K–ATPase and NKCC1 (Crouch et al. 1997; Ichimiya et al. 1994; McGuirt and Schulte 1994; Mizuta et al. 1997; Schulte and Steel 1994). These fibrocytes are coupled to strial basal cells and intermediate cells by gap junctions through which K+ passes (Kikuchi et al. 1995; Spicer and Schulte 1996; Xia et al. 1999). The extrusion of K+ from the ICs into the IS space is facilitated by Kir4.1 (a.k.a. KCNJ10), and pharmacologic blockade or genetic knockout of this channel has been shown to diminish EP and cause deafness (Marcus et al. 2002; Nie et al. 2005; Wangemann et al. 2004). Potassium is transported out of the IS space and into the marginal cells (MC) of the StV by α1–Na,K–ATPase in conjunction with Na–K–2Cl co-transporter (NKCC1), both located on the MC basolateral membrane (Offner et al. 1987; Salt and DeMott 1999). K+ secretion into the endolymph is completed via KCNQ1 in MC apical membranes (Casimiro et al. 2004; Lee et al. 2000; Takeuchi and Ando 1998; Takeuchi et al. 2000).
Inhibitors of NKCC1 and Na,K–ATPase (such as furosemide and ouabain) produce a sharp but reversible decline in EP, suggesting dynamic regulation (Rybak and Morizono 1982; Shugyo et al. 1990). Besides α1–Na,K–ATPase, which is expressed in the mammalian body ubiquitously, α2–Na,K–ATPase and NKCC1 are expressed in a predominantly cell-specific manner, suggesting distinct cellular function (Kaplan et al. 1996; Lingrel et al. 2003). The decline in activity of the Na,K–ATPase isoforms within the inner ear has been shown to correlate well with the decline in EP with aging (Gratton et al. 1996; Gratton et al. 1997; Gratton et al. 1995; Sakaguchi et al. 1998). In addition, a reduction in the level of NKCC1 expression, as seen in heterozygote mutant NKCC1+/− mice, results in elevated hearing threshold relative to wild-type littermates (Flagella et al. 1999). These findings, coupled to the evidence of age-related dependence of NKCC1 expression in the StV, adds to the mounting evidence that age-induced hearing loss and the activity of NKCC1 and Na,K–ATPase are strongly linked (Hequembourg and Liberman 2001; Schulte and Schmiedt 1992; Spicer et al. 1997).
Based on the two-cell model of EP generation, we propose that the rate-limiting steps in K+ throughput within the StV lie in the transport dynamics of both absorption at the spiral ligament fibrocytes by α2–Na,K–ATPase and NKCC1 and secretion at the stria via α1–Na,K–ATPase and NKCC1 in the MC. Disruption of either transport mechanism would lead to an imbalance in K+ flux through the StV, a reduction in EP, and hearing loss. However, we propose that downregulation of both absorption and secretion simultaneously may decrease the overall K+ throughput yet still preserve the overall balance in K+ absorption and secretion, thus, maintaining EP generation more successfully than single site disruption alone.
To test these hypotheses, we performed targeted gene mutation for NKCC1, α1–Na,K–ATPase, and α2–Na,K–ATPase individually. We measured the effects that single gene mutations had on hearing function and cochlear morphology. We then performed double targeted deletion of NKCC1 in combination with either α1–Na,K–ATPase or α2–Na,K–ATPase and documented the effects on hearing function and cochlear morphology in each of these double mutation states. Finally, we measured the EP in each of the single and double knockout models to assess the effects of each set of genetic manipulations on EP generation. Results of these experiments confirm the vital importance of each of these ion transporters in the generation and maintenance of EP, as heterozygote mutation of each of these ion transporters produces a model for age-related hearing loss. In addition, double heterozygote experiments indicate a cooperative crosstalk between NKCC1 and Na,K–ATPase, which may confer a greater degree of hearing conservation with age. This benefit was greatest in the α2–Na,K–ATPase and NKCC1 interaction, suggesting the relative importance of the role that α2–Na,K–ATPase plays in lateral wall K+ flux.
METHODS
All procedures described were approved by the University of California, Davis Institutional Animal Care and Use Committee, and adhered to the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Generation of mutant mice by embryonal stem cell/gene targeting technology
Conventional gene targeting technology was used to generate NKCC1−/−, α1–Na,K–ATPase−/−, and α2–Na,K–ATPase−/− null mutants, and NKCC1+/−, α1–Na,K–ATPase+/−, and α2–Na,K–ATPase+/− heterozygote mutants in Black Swiss–129/SvJ mice, as described in previous studies (Flagella et al. 1999). Homozygous NKCC1−/− mice have been studied in previous experiments and show a 28% incidence of death around the time of weaning, whereas homozygous α1–Na,K–ATPase−/− and α2–Na,K–ATPase−/− null mutations are uniformly lethal at birth (Flagella et al. 1999; Moseley et al. 2003). Homozygous mutants were, therefore, not used in this investigation.
Heterozygote NKCC1+/−, α1–Na,K–ATPase+/−, and α2–Na,K–ATPase+/− mutants generated were used for experimentation. These heterozygotes were also used for further interbreeding. Heterozygote crossbreeding between NKCC1+/− and α1–Na,K–ATPase+/− and α2–Na,K–ATPase+/– produced the double heterozygote mutants 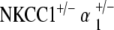 and
and 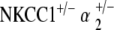 . Germline transmission for all mutant variations was verified by polymerase chain reaction genotyping of DNA from tail samples. All mice analyzed in this study were from F3 to F6 generations of the originally generated mutant mice.
. Germline transmission for all mutant variations was verified by polymerase chain reaction genotyping of DNA from tail samples. All mice analyzed in this study were from F3 to F6 generations of the originally generated mutant mice.
Several mice strains, including Black Swiss–129/SvJ, our original background strain used for targeted gene knockout, are known to have inherent age-related hearing loss (AHL; Zheng et al. 1999). To minimize intrinsic age-related hearing loss effects in our background strain, we performed backcrossing of NKCC1, α1-Na,K-ATPase, and α2-Na,K-ATPase mutant mice with Cast-AHL congenic strains, which have been shown to be free of such age-related hearing loss (Erway et al. 1996; Erway et al. 1993). In addition, we crossed our original background strain Black Swiss–129/SvJ with Cast-AHL to generate an equivalent non-mutated strain, which was immune to age-related hearing loss to facilitate comparison of auditory brainstem response (ABR) and EP data from each mutant group with a control.
Genotyping of all mice was achieved by polymerase chain reaction and Southern blot analysis of tail DNA, as described in previous experiments (Flagella et al. 1999).
Measurement of ABR
Control, NKCC1+/−, α1–Na,K–ATPase+/−, α2–Na,K–ATPase+/−, 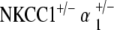 , and
, and 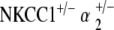 ge‐notyped mice were tested for auditory threshold by measurement of ABR at 5–10 weeks of age and were repeated every 10 weeks to age 90 to 105 weeks. Six to 12 mice per genotype were tested. Student’s t test analysis was used to determine significance of hearing threshold changes with age. ABR methodology was performed in similar fashion to previously published experiments by our group and is summarized below (Bautista et al. 2006; Dou et al. 2004; Flagella et al. 1999).
ge‐notyped mice were tested for auditory threshold by measurement of ABR at 5–10 weeks of age and were repeated every 10 weeks to age 90 to 105 weeks. Six to 12 mice per genotype were tested. Student’s t test analysis was used to determine significance of hearing threshold changes with age. ABR methodology was performed in similar fashion to previously published experiments by our group and is summarized below (Bautista et al. 2006; Dou et al. 2004; Flagella et al. 1999).
Mice were anesthetized with tribromoethanol (Avertin; Sigma-Aldrich Corp., St. Louis, MO) at 0.5 mg/g body weight injected intraperitoneally. Anesthetized mice were then placed in a sound attenuated chamber on a custom holder. Core body temperature was maintained at 36.5 ± 0.5°C using a homeothermic blanket control unit (Harvard Apparatus, Inc., Holliston, MA) and rectal probe feedback. Ground and recording electrodes were placed subcutaneously in the scalp and a calibrated transducer (Intelligent Hearing Systems, Miami, FL) placed in the right pinna. At a rate of 20 Hz, with intensity from 0–90 dB sound pressure level (SPL; rms for click stimuli) in 10 dB increments, 0.1 ms broadband clicks, and 3 ms pure tone pips at 8, 16, and 32 kHz were presented. ABR activity was extracted from 128–1,024 stimuli. Hearing threshold was defined as the minimum sound intensity that elicited a characteristic ABR waveform.
Measurement of distortion product otoacoustic emissions
NKCC1+/−, α1–Na,K–ATPase+/−, α2–Na,K–ATPase+/−, 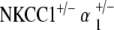 , and
, and 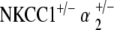 genotyped mice were tested for distortion product otoacoustic emissions (DPOAE) at 20–30 weeks of age and were repeated at 40–60 weeks of age. Three to five mice per genotype group were tested. DPOAE methodology was performed in similar fashion to previously published experiments by our group and is summarized below (Bautista et al. 2006; Dou et al. 2004).
genotyped mice were tested for distortion product otoacoustic emissions (DPOAE) at 20–30 weeks of age and were repeated at 40–60 weeks of age. Three to five mice per genotype group were tested. DPOAE methodology was performed in similar fashion to previously published experiments by our group and is summarized below (Bautista et al. 2006; Dou et al. 2004).
Mice were anesthetized with Avertin as for ABR measurements. After visual inspection to ensure adequate health of external and middle ear, mice were placed in a sound attenuated chamber and dual acoustic probe/microphone assembly (Etymotic Research, Inc., Elk Grove Village, IL) placed in the ear. Primary tones with an f2/f1 ratio of 1.25 were presented at equal sound pressure levels at 55, 65, and 75 dB SPL over 92 ms, and tones were routed to independent transducers and allowed to mix acoustically in the ear canal. Ear canal sound pressure level was sampled and synchronously averaged by digital signal processor for geometric mean frequencies below 20.1 kHz. Above 20.1 kHz, sampling input was automatically switched to dynamic signal analyzer for frequency analysis to avoid DSP aliasing artifacts above 22.1 kHz. Cubic (2f1 − f2) DPOAE levels and corresponding noise floor levels were calculated.
DPOAE levels were plotted as a function of the primary tones’ geometric mean frequency GM = (f1 × f2)0.5, from 5.6–48.5 kHz (f2 = 6.3–54.2 kHz) in 0.1 octave increments. DP grams were generated for each stimulus level and were evaluated for presence of DPOAE > 5 dB above noise floor.
Light microscopy
Histology was performed on the cochleae of NKCC1+/−, 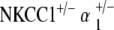 , and
, and 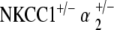 ge‐notyped mice at ages 20–30 weeks and 60–80 weeks. Three to five cochleae per genotype group at each age group were examined under light microscopy. Light microscope analysis was performed using previously described procedures and is described below (Kozel et al. 1998).
ge‐notyped mice at ages 20–30 weeks and 60–80 weeks. Three to five cochleae per genotype group at each age group were examined under light microscopy. Light microscope analysis was performed using previously described procedures and is described below (Kozel et al. 1998).
Mice were deeply anesthetized with sodium pentobarbital (Sigma-Aldrich Corp., St. Louis, MO) and transcardially perfused with 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.3. Temporal bones were harvested and perfused with the same fixative through oval and round windows and kept in fixative overnight. The temporal bones were decalcified in 0.1 M ethylenediaminetetraacetic acid (EDTA) solution for 1 week. Specimens were post-fixed in buffered 1% osmium tetroxide at room temperature for 1–2 h, dehydrated in graded ethanol solutions followed by propylene oxide, and embedded in Spurr resin. Serial 1- and 2-um sections were cut with a diamond knife and stained with toludine blue.
Scanning electron microscopy
Scanning electron microscopy was performed on the cochleae of NKCC1+/−, 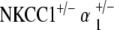 , and
, and 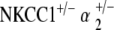 genotyped mice at ages 20–30 and 60–80 weeks. Three to five cochleae per genotype group at each age group were examined under electron microscopy.
genotyped mice at ages 20–30 and 60–80 weeks. Three to five cochleae per genotype group at each age group were examined under electron microscopy.
Mice were anesthetized with sodium pentobarbital and transcardially perfused with fixative with similar technique as for light microscopy specimens. Temporal bones were harvested, fixed, decalcified, and dehydrated as for histology. Prepared specimens were critical-point dried from liquid CO2, sputter-coated with gold–palladium, and examined in a scanning electron microscope (Philips Electronics NV, Eindhoven, The Netherlands).
Measurement of EP
Control, NKCC1+/−, α1–Na,K–ATPase+/−, α2–Na,K–ATPase+/−, 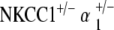 , and
, and 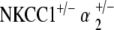 ge‐notyped mice underwent EP measurement at 60 weeks of age. Five to six mice per genotype group were measured for EP.
ge‐notyped mice underwent EP measurement at 60 weeks of age. Five to six mice per genotype group were measured for EP.
Mice were anesthetized with urethane (Sigma-Aldrich Corp., St. Louis, MO) at 2 mg/g body weight injected intraperitoneally. The temperatures of the mice were monitored and controlled as described above for ABR recordings. Anesthetized mice then underwent tracheostomy and tracheal cannulation. A ground electrode was sutured to cervical musculature, and the tympanic bulla was opened by ventral approach. Under a dissecting microscope, a small fenestration was made at the basal turn of the cochlea, and a glass recording microelectrode filled with 150-mM KCl and mounted on a micromanipulator was slowly passed through the lateral wall and into the StV. The recording electrode was connected to a current-clamp amplifier (Warner Instrument), and potential measurements were recorded throughout insertion and until a stable potential was identified.
RESULTS
NKCC1+/− single heterozygote mice develop progressive age-related hearing loss
ABR results from NKCC1+/− single heterozygote mice demonstrated an early hearing loss that progressed with age. We performed initial ABR at 5–10 weeks of age. At this early age, NKCC1+/− mice revealed ABR thresholds comparable to age-matched control mice for broadband, as well as across all tone pip stimuli. Heterozygote mice were retested at bi-weekly intervals, and subsequent results indicated worsening hearing function by progressively elevated ABR thresholds, with severe hearing loss of greater than 80 dB SPL thresholds (for click stimuli) at 51–60 weeks of age and profound hearing loss on the order of 100 dB SPL (for click stimuli) at 100+ weeks, with concomitant threshold elevations across all tone stimuli.
ABR thresholds were compared at advanced ages to the initial ABR threshold at 5–10 weeks, and threshold differences were found to be statistically significant as early as 21–30 weeks of age (p < 0.05), retaining statistical relevance throughout the remainder of life. Elevated thresholds were consistently observed across all stimulus frequencies: click burst, 8, 16, and 32 kHz (Fig. 1a).
FIG. 1.

NKCC1+/− single heterozygote mice exhibit progressive hearing loss with age without gross deterioration in cochlear or hair cell morphology. a ABR thresholds indicate initial mild hearing loss at 5–10 weeks of life with progressive deterioration with age, displayed at bi-weekly intervals to age 60 weeks (105 weeks for click stimuli). Progressive hearing loss occurs comparably across all tested stimulus frequencies: click, 8, 16, and 32 kHz. p < 0.05, statistically significant difference compared to baseline threshold at 5–10 weeks; error bars standard deviation. b Representative DP gram at 75 weeks, showing presence of DPOAE levels for 2f1 − f2 distortion products across the frequency range. 65 dB SPL DP gram is representative and is shown. DPOAE were present at 55 and 75 dB SPL as well (data not shown). c Histology of cochlear cross-section of representative animal at 21 weeks reveals gross conservation in cochlear morphology, with normal positioning and appearance of the Reissner’s membrane, preservation of the endolymphatic volume and scala media, dense and compact appearance of the stria vascularis, and preservation of the tunnel of Corti and organ of Corti. d Scanning electron microscopy of the organ Corti of a representative animal at 80 weeks appears grossly unremarkable, with normal morphology, spacing, and population density of inner and outer hair cell hair bundles. Sporadic hair cell loss is noted along the length of the cochlea (not shown)
DPOAE testing for these mice revealed persistent DPOAE levels above the noise floor even at late age. All tested mice revealed DPOAE present at all stimulus equilevels of 55, 65, and 75 dB SPL, and across all tested geometric mean frequencies of 5 to 50 kHz (Fig. 1b). These results suggest integrity of electromechanical transduction and competence of outer hair cell function throughout the cochlear duct despite declining hearing threshold indicated by the ABR results.
Cochlear specimens were harvested for processing and histologic review. Light microscopy revealed normal appearance to the cochlear architecture. There was normal positioning and appearance of the Reissner’s membrane; preservation of the endolymphatic volume and scala media; compact, dense appearance of the stria vascularis; and preservation of the architecture of the tunnel of Corti and organ of Corti (Fig. 1c). In contrast, previous work on the NKCC1−/− null mutant has demonstrated complete collapse of the scala media and atelectasis of Reissner’s membrane onto the lateral wall, vacuolation or spongy appearance to the stria vascularis, and abnormal architecture of the tunnel and organ of Corti (Flagella et al. 1999).
Scanning electron microscopy of the apical surface of the organ of Corti demonstrated normal morphology to the inner and outer hair cell bundles. Hair cell bundles retained their structure and orientation, and there was only sporadic hair cell loss along the length of the cochlea (Fig. 1d). Scanning microscopy was performed at later age (80 weeks).
α1–Na,K–ATPase+/− single heterozygote mice develop age-related hearing loss
The group of α1–Na,K–ATPase+/− heterozygote mice exhibited similar phenotypic patterns of hearing loss on ABR as did the NKCC1+/− mice. These mice demonstrated ABR thresholds at early age (5–10 weeks) comparable to those of the NKCC1+/− heterozygotes. This group’s hearing loss deteriorated with age more rapidly than the NKCC1+/− mice, with severe elevations in ABR threshold greater than 80 dB SPL (for click stimuli) as early as 31–40 weeks of age. This degree of hearing loss was not seen in NKCC1+/− mice until 51–60 weeks of age. Thus, we find in the α1–Na,K–ATPase+/− group a large jump from baseline hearing to severe hearing loss at mid-age, whereas in the NKCC1+/− mice, we find a gradually progressive drop in hearing function over time.
ABR thresholds were measured in this group at bi-weekly intervals, as with the NKCC1+/− counterparts. Effects on hearing were consistent across all stimulus frequencies: click, 8, 16, and 32 kHz. ABR thresholds at age were compared to the group’s ABR thresholds at 5–10 weeks. Thresholds measured as early as 31–40 weeks of age were shown to be statistically significantly different from the initial ABR threshold (p < 0.05). Although there was a slight drop off in ABR threshold from 31–40 weeks to 51–60 weeks, these comparative thresholds were not statistically significantly different (p > 0.05; Fig. 2a).
FIG. 2.

α1–Na,K–ATPase+/− single heterozygote mice exhibit progressive hearing loss with age. a ABR thresholds demonstrate initial mild hearing loss at 5–10 weeks of life with progressive deterioration with age, displayed at bi-weekly intervals to age 60 weeks. Progressive hearing loss occurs comparably across all tested stimulus frequencies: click, 8, 16, and 32 kHz. *p < 0.05, statistically significant difference compared to baseline threshold at 5–10 weeks; error bars standard deviation. b Representative DP-gram at 75 weeks, showing absence of DPOAE levels for 2f1 − f2 distortion products across the frequency range. 65 dB SPL DP gram is representative and is shown. DPOAE were absent at 55 and 75 dB SPL as well (data not shown)
DPOAE recordings for this genotype group displayed no significant DPOAE levels above noise floor at all stimulus levels used (55, 65, and 75 dB SPL; Fig. 2b). The lack of measurable DPOAE levels suggests failure of electromechanical, or reverse, transduction to produce intermodulation distortion robust enough for detection (Liberman et al. 2004). Presence of intermodulation distortion from stereociliary transduction by the use of higher level stimuli was not investigated in this study.
α2–Na,K–ATPase+/− single heterozygote mice develop age-related hearing loss
The group of α2–Na,K–ATPase+/− heterozygote mice exhibited phenotypic hearing responses comparable to that of their α1–Na,K–ATPase+/− heterozygote counterparts. Again, we found initial ABR thresholds at 5–10 weeks approximately equal to those of age-matched controls. Hearing loss progressed slightly at 11–20 weeks but deteriorated rapidly at 31–40 weeks to a threshold on the order of 90 dB SPL (click stimuli). This severe-to-profound hearing loss was maintained at 51–60 weeks, with ABR threshold on the order of 90 dB SPL (click stimuli). This pattern of hearing loss is similar in temporal pattern to that of the α1–Na,K–ATPase+/− group, with a large deterioration in ABR threshold at mid-age rather than a progressive deterioration in threshold over time as seen in the NKCC1+/− group.
ABR thresholds were measured at bi-weekly intervals, as with the NKCC1+/− and α1–Na,K–ATPase+/− single heterozygote groups. Effects on hearing were again consistent across all stimulus frequencies: click, 8, 16, and 32 kHz. ABR thresholds at each age interval were compared to the group’s initial ABR threshold at 5–10 weeks. Thresholds at 31–40 weeks and older were shown to be statistically significantly different from the 5- to 10-week threshold (p < 0.05) across all stimulus frequencies. Thresholds at 51–60 weeks were maintained from the 31- to 40-week interval (Fig. 3a).
FIG. 3.

α2–Na,K–ATPase+/− single heterozygote mice exhibit progressive hearing loss with age. a ABR thresholds again show an initial mild hearing loss at 5–10 weeks of life with significant and progressive deterioration with age, displayed at bi-weekly intervals to age 60 weeks. Progressive hearing loss occurs comparably across all tested stimulus frequencies: click, 8, 16, and 32 kHz. *p < 0.05, statistically significant difference compared to baseline threshold at 5–10 weeks; error bars standard deviation. b Representative DP gram at 75 weeks, showing absence of DPOAE levels for 2f1 − f2 distortion products across the frequency range. 65 dB SPL DP gram is representative and is shown. DPOAE were absent at 55 and 75 dB SPL as well (data not shown)
DPOAE recordings for the α2–Na,K–ATPase+/− heterozygote mice also did not demonstrate measurable intermodulation distortion above the noise floor (Fig. 3b). As for the α1–Na,K–ATPase+/− heterozygotes, the lack of measurable intermodulation distortion up to 75 dB SPL stimulus level implies failure of reverse transduction at the outer hair cells. Integrity of passive stereociliary transduction at higher level stimuli was not investigated.
NKCC1+/−α1+/− double heterozygote mice show delayed age-related hearing loss
ABR measurements of the 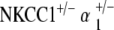 double heterozygote group demonstrate terminal hearing loss with age comparable to that of the single heterozygote groups NKCC1+/−, α1–Na,K–ATPase+/−, and α2–Na,K-ATPase+/−, but with a delay in onset of progression. These double heterozygotes demonstrated initial thresholds at 5–10 weeks of age equivalent to control mice across all frequencies. Progression of ABR threshold shift and hearing loss occurred fairly later in life compared with the single heterozygote groups. Thresholds at age were compared with the initial ABR threshold at 5–10 weeks, and statistically significant progression in threshold change (p < 0.05) did not occur until 61–70 weeks of age. This hearing loss then accelerated over time to terminate at a severe degree of hearing loss at 71–80 weeks, on the order of 100 dB SPL (click stimuli), comparable to the terminal threshold for all groups of single heterozygote mutants. These effects were seen across all stimulus frequencies: click, 8, 16, and 32 kHz (Fig. 4a).
double heterozygote group demonstrate terminal hearing loss with age comparable to that of the single heterozygote groups NKCC1+/−, α1–Na,K–ATPase+/−, and α2–Na,K-ATPase+/−, but with a delay in onset of progression. These double heterozygotes demonstrated initial thresholds at 5–10 weeks of age equivalent to control mice across all frequencies. Progression of ABR threshold shift and hearing loss occurred fairly later in life compared with the single heterozygote groups. Thresholds at age were compared with the initial ABR threshold at 5–10 weeks, and statistically significant progression in threshold change (p < 0.05) did not occur until 61–70 weeks of age. This hearing loss then accelerated over time to terminate at a severe degree of hearing loss at 71–80 weeks, on the order of 100 dB SPL (click stimuli), comparable to the terminal threshold for all groups of single heterozygote mutants. These effects were seen across all stimulus frequencies: click, 8, 16, and 32 kHz (Fig. 4a).
FIG. 4.

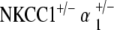 double heterozygote mice exhibit progressive hearing loss with age without gross deterioration in cochlear morphology. a ABR thresholds indicate initial mild hearing loss at 5–10 weeks of life with mild deterioration at 61–70 weeks, but with gross threshold shift at 71–80 weeks. Hearing loss occurs comparably across all tested stimulus frequencies: click, 8, 16, and 32 kHz. *p < 0.05, statistically significant difference compared to baseline threshold at 5–10 weeks; error bars standard deviation. b Representative DP gram at 39 weeks, showing presence of DPOAE levels for 2f1 − f2 distortion products at low frequencies but not at higher. Shown is the representative 65 dB SPL DP gram. Similar results were found at 55 and 75 dB SPL (data not shown). c Histology of cochlear cross-section of representative animal at 60 weeks reveals gross conservation in cochlear morphology, comparable to that seen in NKCC1+/−: normal appearance of Reissner’s membrane and scala media, dense and compact appearance of the stria vascularis, and preservation of the organ of Corti. d Scanning electron microscopy of the organ of Corti of a representative animal at 80 weeks appears grossly unremarkable, with normal morphology, spacing, and population density of inner and outer hair cell hair bundles. Sporadic hair cell loss is noted along the length of the cochlea (shown at top), comparable to NKCC1+/−
double heterozygote mice exhibit progressive hearing loss with age without gross deterioration in cochlear morphology. a ABR thresholds indicate initial mild hearing loss at 5–10 weeks of life with mild deterioration at 61–70 weeks, but with gross threshold shift at 71–80 weeks. Hearing loss occurs comparably across all tested stimulus frequencies: click, 8, 16, and 32 kHz. *p < 0.05, statistically significant difference compared to baseline threshold at 5–10 weeks; error bars standard deviation. b Representative DP gram at 39 weeks, showing presence of DPOAE levels for 2f1 − f2 distortion products at low frequencies but not at higher. Shown is the representative 65 dB SPL DP gram. Similar results were found at 55 and 75 dB SPL (data not shown). c Histology of cochlear cross-section of representative animal at 60 weeks reveals gross conservation in cochlear morphology, comparable to that seen in NKCC1+/−: normal appearance of Reissner’s membrane and scala media, dense and compact appearance of the stria vascularis, and preservation of the organ of Corti. d Scanning electron microscopy of the organ of Corti of a representative animal at 80 weeks appears grossly unremarkable, with normal morphology, spacing, and population density of inner and outer hair cell hair bundles. Sporadic hair cell loss is noted along the length of the cochlea (shown at top), comparable to NKCC1+/−
DPOAE measurements demonstrated the presence of DPOAE levels above the noise floor for low geometric mean frequencies from 5–20 kHz, but loss of DPOAE amplitude above 20 kHz (Fig. 4b). This finding suggests some preservation in reverse transduction at the cochlear duct apex, but with possible loss of some function at the base.
Histologic analysis of the cochlea was grossly unremarkable. There was good preservation of the volume of the scala media and endolymph space without collapse of Reissner’s membrane; compact, dense appearance to the stria vascularis; and normal architecture of the organ of Corti (Fig. 4c). These results are similar to those seen in the histology for the NKCC1+/− single heterozygote group.
Scanning electron microscopy of the apical surface of the organ of Corti appeared grossly unremarkable as well. There appeared to be normal morphology to the inner and outer hair cell bundles across the cochlear length, with only scattered loss of hair cells; this was comparable in comparison to the NKCC1+/− group (Fig. 4d).
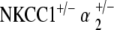 double heterozygote mice have relative preservation of hearing
double heterozygote mice have relative preservation of hearing
ABR threshold measurements of 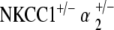 double heterozygotes reveal equivalent hearing as compared with control mice at 5–10 weeks of age. With age, we again see a progression in hearing loss beginning at 51–60 weeks, which is statistically significant compared to the group’s own baseline threshold (p < 0.05) (Fig. 5a). However, the magnitude of this hearing loss is in stark contrast to that seen with all other mutant groups. Terminal thresholds are on the order of 80–90 dB SPL for NKCC1+/−, α1–Na,K–ATPase+/−, and α2–Na,K–ATPase+/− single heterozygotes, as well as for the
double heterozygotes reveal equivalent hearing as compared with control mice at 5–10 weeks of age. With age, we again see a progression in hearing loss beginning at 51–60 weeks, which is statistically significant compared to the group’s own baseline threshold (p < 0.05) (Fig. 5a). However, the magnitude of this hearing loss is in stark contrast to that seen with all other mutant groups. Terminal thresholds are on the order of 80–90 dB SPL for NKCC1+/−, α1–Na,K–ATPase+/−, and α2–Na,K–ATPase+/− single heterozygotes, as well as for the 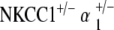 double heterozygote group. The terminal threshold of
double heterozygote group. The terminal threshold of 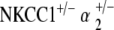 mice, on the other hand, is only 50 dB SPL, approximate to that of age-matched control mice (Fig. 6). This contrast in terminal hearing thresholds between the
mice, on the other hand, is only 50 dB SPL, approximate to that of age-matched control mice (Fig. 6). This contrast in terminal hearing thresholds between the 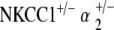 double heterozygote group and the other mutant groups is significant and preserved across all three tone pip stimuli. Of interest, at 30–40 weeks, the two double heterozygote groups both exhibit similar levels of hearing to that of the age-matched control group. However, after 1 year of age, the two groups diverge drastically, with the
double heterozygote group and the other mutant groups is significant and preserved across all three tone pip stimuli. Of interest, at 30–40 weeks, the two double heterozygote groups both exhibit similar levels of hearing to that of the age-matched control group. However, after 1 year of age, the two groups diverge drastically, with the 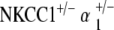 mice demonstrating age-related hearing loss as opposed to the
mice demonstrating age-related hearing loss as opposed to the 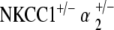 mice, whose hearing remains stable relative to control.
mice, whose hearing remains stable relative to control.
FIG. 5.

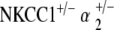 double heterozygote mice exhibit relative preservation in hearing function with age. a ABR thresholds indicate initial mild hearing loss at 5–10 weeks of life with minimal progression with age. *p < 0.05, statistically significant difference compared to baseline threshold at 5–10 weeks; error bars standard deviation. b Representative DP gram at 110 weeks, showing presence of DPOAE levels for 2f1 − f2 distortion products at all frequencies. Shown is the representative 65 dB SPL DP-gram. DPOAE were present at 55 and 75 dB SPL as well (data not shown). c Histology of cochlear cross-section of representative animal at 88 weeks reveals gross conservation in cochlear morphology, comparable to that seen in both NKCC1+/− and
double heterozygote mice exhibit relative preservation in hearing function with age. a ABR thresholds indicate initial mild hearing loss at 5–10 weeks of life with minimal progression with age. *p < 0.05, statistically significant difference compared to baseline threshold at 5–10 weeks; error bars standard deviation. b Representative DP gram at 110 weeks, showing presence of DPOAE levels for 2f1 − f2 distortion products at all frequencies. Shown is the representative 65 dB SPL DP-gram. DPOAE were present at 55 and 75 dB SPL as well (data not shown). c Histology of cochlear cross-section of representative animal at 88 weeks reveals gross conservation in cochlear morphology, comparable to that seen in both NKCC1+/− and 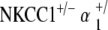 genotypes: normal appearance of Reissner’s membrane, scala media, stria vascularis, and organ of Corti. d Scanning electron microscopy of the organ of Corti of a representative animal at 80 weeks appears grossly unremarkable, as with NKCC1+/− and
genotypes: normal appearance of Reissner’s membrane, scala media, stria vascularis, and organ of Corti. d Scanning electron microscopy of the organ of Corti of a representative animal at 80 weeks appears grossly unremarkable, as with NKCC1+/− and 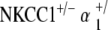 , with normal morphology, spacing, and population density of inner and outer hair cell hair bundles. Sporadic hair cell loss is noted along the length of the cochlea, comparable to NKCC1+/− and
, with normal morphology, spacing, and population density of inner and outer hair cell hair bundles. Sporadic hair cell loss is noted along the length of the cochlea, comparable to NKCC1+/− and 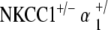
FIG. 6.

Comparison of ABR thresholds over time between control and single and double heterozygote mutant groups. At 5–10 weeks, all single and double heterozygote groups have thresholds comparable to those of the control strain. At 31–40 weeks, single heterozygote groups (NKCC1+/−, α1–Na,K–ATPase+/−, α1–Na,K–ATPase+/−) have already begun to display threshold shifts and hearing loss. However, both double heterozygote groups (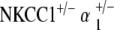 and
and 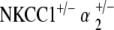 ) maintain stable hearing thresholds compared to the control. At 51–70 weeks,
) maintain stable hearing thresholds compared to the control. At 51–70 weeks, 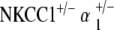 mice now display threshold shifts comparable to single heterozygotes, but
mice now display threshold shifts comparable to single heterozygotes, but 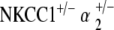 mice continue to maintain hearing levels stable and comparable to control. Graph displays results from click stimuli; 8, 16, and 32 kHz tone pip stimuli demonstrate comparable results (data not shown). *p < 0.05, statistically significant difference compared to control threshold at the same age
mice continue to maintain hearing levels stable and comparable to control. Graph displays results from click stimuli; 8, 16, and 32 kHz tone pip stimuli demonstrate comparable results (data not shown). *p < 0.05, statistically significant difference compared to control threshold at the same age
DPOAE response measurements were performed on 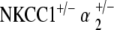 mice for stimulus equilevels of 55, 65, and 75 dB SPL, as with previous groups. DPOAE responses above the noise floor were evident and robust for all geometric mean frequencies tested, from 5–50 kHz (Fig. 5b). As with the NKCC1+/− single heterozygote mice, this finding suggests competency of reverse transduction and outer hair cell somatic motility.
mice for stimulus equilevels of 55, 65, and 75 dB SPL, as with previous groups. DPOAE responses above the noise floor were evident and robust for all geometric mean frequencies tested, from 5–50 kHz (Fig. 5b). As with the NKCC1+/− single heterozygote mice, this finding suggests competency of reverse transduction and outer hair cell somatic motility.
Histology of cochlear architecture is comparable to that seen in the NKCC1+/− single heterozygote and 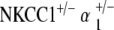 double heterozygote groups. Cross-sectional examination of the cochleae of representative specimens reveals normal appearance and positioning of the Reissner’s membrane, full volume to the scala media, a densely staining, compact stria vascularis, and normal appearance and configuration to the organ of Corti (Fig. 5c).
double heterozygote groups. Cross-sectional examination of the cochleae of representative specimens reveals normal appearance and positioning of the Reissner’s membrane, full volume to the scala media, a densely staining, compact stria vascularis, and normal appearance and configuration to the organ of Corti (Fig. 5c).
Scanning electron microscopy of the 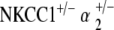 group also demonstrates normal stereociliary architecture. Inner and outer hair cell stereocilia are present and hair cells have normal spacing and appearance with only sporadic hair cell loss, consistent with the light microscopy of this group (Fig. 5d).
group also demonstrates normal stereociliary architecture. Inner and outer hair cell stereocilia are present and hair cells have normal spacing and appearance with only sporadic hair cell loss, consistent with the light microscopy of this group (Fig. 5d).
EP is preserved in 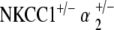 double heterozygote mice
double heterozygote mice
EP were recorded in the control group and all mutant groups at 60 weeks. Results were consistent with ABR threshold findings for each group, as measured EP amplitudes correlated well with the degrees of threshold shift observed between initial and terminal ABR measurements. The aged control mice group demonstrated a mean EP amplitude of 79 ± 4 mV, within the normal range for young wild-type mice, thus, signifying their immunity to age-related hearing loss. EP were significantly depressed but were not completely absent for the single heterozygote groups, as mean EP amplitudes were roughly half of normal range: NKCC1+/− = 45 ± 12 mV; α1–Na,K–ATPase+/− = 46 ± 7 mV; and α2–Na,K–ATPase+/− = 45 ± 14 mV. The mean EP for the 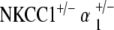 double heterozygote condition was also half of normal at 48 ± 6 mV. Curiously, the
double heterozygote condition was also half of normal at 48 ± 6 mV. Curiously, the 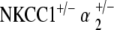 group again demonstrated preservation in cochlear function with a 60-week mean EP amplitude of 81 ± 4 mV (Fig. 7).
group again demonstrated preservation in cochlear function with a 60-week mean EP amplitude of 81 ± 4 mV (Fig. 7).
FIG. 7.

Measurement of EP in all genotype groups confirms functional data. EP were measured in all genotype groups at 60 weeks of age. Control mice at 60 weeks maintained potentials comparable to those seen in young wild types. NKCC1+/−, α1–Na,K–ATPase+/−, α1–Na,K–ATPase+/− single heterozygotes, and 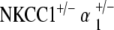 double heterozygote mice exhibit significantly diminished but still existent positive amplitude of EP compared to the control (*p < 0.05, **p < 0.005). The
double heterozygote mice exhibit significantly diminished but still existent positive amplitude of EP compared to the control (*p < 0.05, **p < 0.005). The 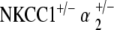 double heterozygote condition demonstrates potentials that are comparable to similarly aged control mice (p = 0.4) and in the normal range
double heterozygote condition demonstrates potentials that are comparable to similarly aged control mice (p = 0.4) and in the normal range
DISCUSSION
The remarkable dynamic range of the mammalian inner ear relies on the exceptional sensitivity of hair cells, which are critically dependent on the existence a unique, organ-specific, positive EP (Davis 1957; Hudspeth 1997). Processes that are associated with disruption of K+ transport and recycling within the inner ear—and therefore, with the generation of the EP—are associated with hearing loss (Carlisle et al. 1990; Salt and DeMott 1999). Recent work studying the knockout of specific K+ channel KCNJ10 (Kir4.1) demonstrates abolition of EP with profound hearing loss, whereas pharmacologic inhibition of KCNJ10 confers reduction in EP and concomitant reduction in hearing thresholds (Marcus et al. 2002; Nie et al. 2005; Wangemann et al. 2004). By far, the most clinically relevant process for neurosensory hearing loss in humans is age-related hearing loss. Age-related hearing loss, or presbycusis, affects one in three persons over 60 years of age and one in two persons over 85 years of age (Gratton and Vazquez 2003).
Histopathological evidence exists, from cadaveric evaluation of temporal bones of human patients with presbycusis, that age-related hearing loss can be described by four types of cochlear histopathology: (1) sensory presbycusis involving hair cell loss, (2) neural presbycusis involving loss of spiral ganglion cells and axons, (3) mechanical or conductive presbycusis thought to be conferred by stiffening of the basilar membrane, and (4) metabolic or strial presbycusis, whereby atrophy of the StV is thought to confer hearing loss (Schuknecht and Gacek 1993). Clinically, however, presbycusis is rarely found to present as one specific audiometric pattern correlating to one specific histopathologic process, but it is commonly considered to be an amalgamation of patterns—and hence, a combination of several processes (Shih 1994).
Considerable experimental evidence exists to suggest that age-related hearing loss may be associated with progressive metabolic and structural changes within the lateral wall of the cochlea. Animal models of age-related hearing loss demonstrate progressive alteration of biochemical and structural integrity of the cochlear lateral wall, including reduction in expression and activity of Na,K–ATPase, reduction in expression of NKCC1, involution and atrophy of the stria vascularis, degeneration of spiral ligament fibrocytes, segmental degeneration of capillary vasculature, and decline in EP associated with such changes (Gratton et al. 1996, 1997, 1995 Gratton and Schulte 1995; Hequembourg and Liberman 2001; McGuirt and Schulte 1994; Sakaguchi et al. 1998; Schulte and Adams 1989; Schulte and Schmiedt 1992; Schulte and Steel 1994; Spicer et al. 1997).
In this series of genetic experiments, these same key ion transport mechanisms were identified and targeted for gene deletion and systematically nulled, individually and in combination, to investigate effects on hearing, cochlear function and structure, and EP generation.
Homozygote deletion of NKCC1 has previously been studied and showed 28% incidence of death at the time of weaning. Those mice that survived demonstrated immediate profound hearing loss without measurable thresholds on ABR (Flagella et al. 1999). Homozygote deletion of α1–Na,K–ATPase and α2–Na,K–ATPase both were uniformly fatal at birth, indicating the critical role that the Na,K pump plays for the viability of mammalian organisms (Moseley et al. 2003; unpublished data).
Heterozygote mutations of these three critical transporters, on the other hand, are not partially or uniformly fatal, and they display audiometric characteristics analogous to that of mammalian forms of age-related hearing loss. Heterozygote mutation of NKCC1 individually demonstrates a mild to moderate hearing loss across all frequencies early in age that progresses with age. Heterozygote mutation of α1–Na,K–ATPase and α2–Na,K–ATPase individually show similar age-progressive hearing loss, comparable in severity to the NKCC1+/− heterozygote at later age but with an earlier onset of severity. In addition, terminal EP amplitude measurements demonstrate significant depression, but still, existence of a positive EP amplitude in each of the single heterozygote models. This finding is consistent with EP measurements obtained from quiet-aged mammals (Gratton et al. 1996, 1997). The normal appearance of the cochlear duct and organ of Corti from light and scanning electron microscopy imply that changes to hearing function as a result of these genetic manipulations are dynamic or metabolic in origin rather than static or structural.
Considered individually, each of these single heterozygote mutations can serve as a potential genetic and molecular biologic model for age-related hearing loss based on ABR threshold and EP data. Each model is not necessarily equivalent, however, as the earlier disruption in hearing thresholds in the Na,K–ATPase heterozygotes implies a relative primacy of Na,K–ATPase over NKCC1 in the preservation of hearing.
Intermodulation distortion from low-level stimuli is observed only in the NKCC1+/− single heterozygote model and not measurable in either of the Na,K–ATPase heterozygote models. This divergence between NKCC1 and Na,K–ATPase is not fully understood at this time. One potential mechanism for the effects of heterozygote mutation on DPOAE would involve depression in OHC function and subsequent reduction in DPOAE level secondary to depressed EP amplitude. However, as terminal EP amplitudes are comparable between the three heterozygote groups, there is no reason why the degree of intermodulation distortion produced would be favored for one group over the others. Conversely, as low-level intermodulation distortion is generated by reverse transduction at the OHC, it is plausible that neuronal pathways, rather than strial/lateral wall metabolism, are the dominant site of disruption causing this phenomena. Na,K–ATPase has been identified in high levels both in spiral ganglion neurons and in unmyelinated nerve fibers within the organ of Corti, whereas NKCC1 has been identified only weakly in satellite cells within the spiral ganglion (Crouch et al. 1997; Schulte and Adams 1989). This differential expression of Na,K–ATPase over NKCC1 within the cochlear neuronal pathways makes the latter proposition more plausible. It is evident from these data, however, that generation of low-level stimuli intermodulation distortion is more sensitive to disruption in Na,K–ATPase activity than NKCC1 activity.
Functional results from double heterozygote mutations of NKCC1 with either α1–Na,K–ATPase and α2–Na,K–ATPase demonstrate even more fascinating results. The 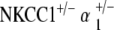 double heterozygote mice exhibit age-related ABR threshold shifts and hearing loss comparable to each of the single heterozygote conditions NKCC1+/−, α1–Na,K–ATPase+/−, and α2–Na,K–ATPase+/−, but with a delayed progression of severity compared to the single heterozygotes.
double heterozygote mice exhibit age-related ABR threshold shifts and hearing loss comparable to each of the single heterozygote conditions NKCC1+/−, α1–Na,K–ATPase+/−, and α2–Na,K–ATPase+/−, but with a delayed progression of severity compared to the single heterozygotes. 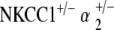 double heterozygote mice demonstrate an even more remarkable effect of ABR threshold stability and complete hearing preservation with age. The
double heterozygote mice demonstrate an even more remarkable effect of ABR threshold stability and complete hearing preservation with age. The 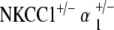 double heterozygote mice proceeded to a profound hearing loss after 1 year of age, whereas the
double heterozygote mice proceeded to a profound hearing loss after 1 year of age, whereas the 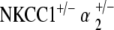 double heterozygote mice maintained ABR thresholds equivalent to age-matched controls at all times. Mean terminal EP amplitude of the
double heterozygote mice maintained ABR thresholds equivalent to age-matched controls at all times. Mean terminal EP amplitude of the 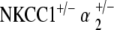 group was preserved, suggesting preservation of the mechanisms for EP generation within the cochlear lateral wall.
group was preserved, suggesting preservation of the mechanisms for EP generation within the cochlear lateral wall.
Intermodulation distortion was present in both double heterozygote models, only at low frequencies for 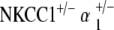 but throughout the frequency range for
but throughout the frequency range for 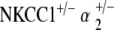 . This is contrasted with the complete absence of low-level stimuli DPOAE in α1–Na,K–ATPase+/− and α2–Na,K–ATPase+/− single heterozygotes.
. This is contrasted with the complete absence of low-level stimuli DPOAE in α1–Na,K–ATPase+/− and α2–Na,K–ATPase+/− single heterozygotes.
Modulation of the activity of NKCC1 simultaneously with Na,K–ATPase appears to confer a beneficial crosstalk or rescue effect within the cochlear metabolic architecture over that of either ion transporter alone: Reverse transduction and OHC function are reinstated, and age-related hearing loss is delayed. In addition, the specific combination of NKCC1 modulation with α2–Na,K–ATPase appears to confer the greatest advantage, as EP is preserved and age-related hearing loss is completely mitigated.
The results of these double heterozygote experiments have implications beyond modeling for age-related hearing loss, as they help to verify predictions of the roles of NKCC1, α1–Na,K–ATPase, and α2–Na,K–ATPase in K+ cycling within the inner ear. Based on the two cell model of EP generation, these transport proteins serve in balancing the K+ flux through the lateral wall, α2–Na,K–ATPase + NKCC1 in spiral ligament absorption from the perilymph at types II, IV, and V fibrocytes, and α1–Na,K–ATPase + NKCC1 in strial secretion back into the endolymph through the MC (Fig. 8). Downregulation of one mechanism causes disruption in EP generation and hearing loss, but coincident downregulation of both mechanisms—as is done in the double heterozygote models—results in stability of hearing, presumably from downregulated but balanced K+ flux and, thus, successful regulation of EP generation over time. Based on the single heterozygote experiments, Na,K–ATPase appears to be the more relevant rate-limiting factor over NKCC1, and based on the double heterozygote experiments, α2–Na,K–ATPase appears to be the more relevant rate-limiting factor over α1–Na,K–ATPase.
FIG. 8.

K+ cycling and ion transport in the inner ear—diagrammatic representation of the two-cell model for the generation of EP in the StV. We have targeted the NKCC1, α1–Na,K–ATPase, and α2–Na,K–ATPase ion transport proteins with gene targeting. Our results suggest a crosstalk between α2–Na,K–ATPase and NKCC1, and to a limited extent between α1–Na,K–ATPase and NKCC1, implicating these proteins as rate limiting steps in the influx and efflux of K+ through the StV, respectively
The development of this multiple targeted gene deletion framework continues to expand upon our knowledge and understanding of the complex biochemical machinery necessary for the development and maintenance of the proper intracochlear environment requisite for hearing and balance. It is clear that each of these key ion transporters is critical to the proper functioning of this machinery. The relative primacy of Na,K–ATPase over NKCC1 within this milieu, and the implication of spiral ligament α2–Na,K–ATPase as the rate-limiting element for K+ flux within the lateral wall, is proposed based on the functional results of the series of single and compound knockout experiments performed.
From these results, we gain a better understanding of the pathophysiology of strial-related mechanisms of hearing loss. Further, we identify multiple potential loci for intervention in certain forms of hearing loss imparted by ion transport defects, such as ototoxicity and age-related hearing loss. For example, it may be conceivable to counter the effects of a diuretic-induced ototoxicity at NKCC1 by administering a titrated dose of a cardiac glycoside to differentially inhibit Na,K–ATPase. As genetic identification of an individual’s hearing loss becomes reality, it may be possible to pharmacologically or genetically counterbalance an inherited predisposition to age-related hearing loss. Should a patient have an inherited defect in one of these ion transporters within the ear, it is conceivable that future therapy may involve genetic means of upregulating the gene for that transporter, or, perhaps more elegantly, downregulating the gene for its counterbalancing transporter. In a more general sense then, if inhibition or downregulation at one of these K+ transporters represents a molecular site of hearing loss, then deliberate inhibition at the counterbalancing site may provide a mechanism to preserve hearing or mitigating the hearing loss that would otherwise ensue.
Acknowledgment
We thank Dr. Hanqi Chu, Dr. Catherine Tamse, and Ellen Ingalla, MS for their assistance with select experiments. This research was supported by the National Institutes of Health grants R01 DC 07592 and RO1 DC 03826 (ENY), RO1 DC 006421 (AEV), and RO1 DK 50594 and RO1 HL 61974 (GES).
References
- Bautista DM, Jordt SE, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI, Julius D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124:1269–1282, 2006. [DOI] [PubMed]
- Carlisle L, Steel K, Forge A. Endocochlear potential generation is associated with intercellular communication in the stria vascularis: structural analysis in the viable dominant spotting mouse mutant. Cell Tissue Res. 262:329–337, 1990. [DOI] [PubMed]
- Casimiro MC, Knollmann BC, Yamoah EN, Nie L, Vary JC, Jr, Sirenko SG, Greene AE, Grinberg A, Huang SP, Ebert SN, Pfeifer K. Targeted point mutagenesis of mouse Kcnq1: phenotypic analysis of mice with point mutations that cause Romano–Ward syndrome in humans. Genomics 84:555–564, 2004. [DOI] [PubMed]
- Crouch JJ, Sakaguchi N, Lytle C, Schulte BA. Immunohistochemical localization of the Na–K–Cl co-transporter (NKCC1) in the gerbil inner ear. J. Histochem. Cytochem. 45:773–778, 1997. [DOI] [PubMed]
- Davis H. Biophysics and physiology of the inner ear. Physiol. Rev. 37:1–49, 1957. [DOI] [PubMed]
- Dou H, Vazquez AE, Namkung Y, Chu H, Cardell EL, Nie L, Parson S, Shin HS, Yamoah EN. Null mutation of alpha1D Ca2+ channel gene results in deafness but no vestibular defect in mice. J. Assoc. Res. Otolaryngol. 5:215–226, 2004. [DOI] [PMC free article] [PubMed]
- Erway LC, Willott JF, Archer JR, Harrison DE. Genetics of age-related hearing loss in mice: I. Inbred and F1 hybrid strains. Hear Res. 65:125–132, 1993. [DOI] [PubMed]
- Erway LC, Shiau YW, Davis RR, Krieg EF. Genetics of age-related hearing loss in mice. III. Susceptibility of inbred and F1 hybrid strains to noise-induced hearing loss. Hear Res. 93:181–187, 1996. [DOI] [PubMed]
- Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na–K–2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J. Biol. Chem. 274:26946–26955, 1999. [DOI] [PubMed]
- Gratton MA, Schulte BA. Alterations in microvasculature are associated with atrophy of the stria vascularis in quiet-aged gerbils. Hear. Res. 82:44–52, 1995. [DOI] [PubMed]
- Gratton MA, Vazquez AE. Age-related hearing loss: current research. Curr. Opin. Otolaryngol. Head Neck Surg. 11:367–371, 2003. [DOI] [PubMed]
- Gratton MA, Smyth BJ, Schulte BA, Vincent DA, Jr. Na,K–ATPase activity decreases in the cochlear lateral wall of quiet-aged gerbils. Hear. Res. 83:43–50, 1995. [DOI] [PubMed]
- Gratton MA, Schmiedt RA, Schulte BA. Age-related decreases in endocochlear potential are associated with vascular abnormalities in the stria vascularis. Hear Res. 102:181–190, 1996 [corrected and republished article originally printed in Hear. Res. 94(1–2):116–24, 1996 May]. [DOI] [PubMed]
- Gratton MA, Smyth BJ, Lam CF, Boettcher FA, Schmiedt RA. Decline in the endocochlear potential corresponds to decreased Na,K–ATPase activity in the lateral wall of quiet-aged gerbils. Hear. Res. 108:9–16, 1997. [DOI] [PubMed]
- Hequembourg S, Liberman MC. Spiral ligament pathology: a major aspect of age-related cochlear degeneration in C57BL/6 mice. J. Assoc. Res. Otolaryngol. 2:118–129, 2001. [DOI] [PMC free article] [PubMed]
- Hudspeth AJ. How hearing happens. Neuron 19:947–950, 1997. [DOI] [PubMed]
- Ichimiya I, Adams JC, Kimura RS. Immunolocalization of Na+, K(+)–ATPase, Ca(++)–ATPase, calcium-binding proteins, and carbonic anhydrase in the guinea pig inner ear. Acta. Otolaryngol. 114:167–176, 1994. [DOI] [PubMed]
- Kaplan MR, Mount DB, Delpire E. Molecular mechanisms of NaCl cotransport. Annu. Rev. Physiol. 58:649–668, 1996. [DOI] [PubMed]
- Kikuchi T, Kimura RS, Paul DL, Adams JC. Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat. Embryol. 191:101–118, 1995. [DOI] [PubMed]
- Kozel PJ, Friedman RA, Erway LC, Yamoah EN, Liu LH, Riddle T, Duffy JJ, Doetschman T, Miller ML, Cardell EL, Shull GE. Balance and hearing deficits in mice with a null mutation in the gene encoding plasma membrane Ca2+–ATPase isoform 2. J. Biol. Chem. 273:18693–18696, 1998. [DOI] [PubMed]
- Kuijpers W, Bonting SL. The cochlear potentials. II. The nature of the cochlear endolymphatic resting potential. Pflugers Arch. 320:359–372, 1970. [DOI] [PubMed]
- Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, Washington K, Argani P, Goldenring JR, Coffey RJ, Feinberg AP. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. J. Clin. Invest. 106:1447–1455, 2000. [DOI] [PMC free article] [PubMed]
- Liberman MC, Zuo J, Guinan JJ, Jr. Otoacoustic emissions without somatic motility: can stereocilia mechanics drive the mammalian cochlea? J. Acoust. Soc. Am. 116:1649–1655, 2004. [DOI] [PMC free article] [PubMed]
- Lingrel J, Moseley A, Dostanic I, Cougnon M, He S, James P, Woo A, O’connor K, Neumann J. Functional roles of the alpha isoforms of the Na,K–ATPase. Ann. N.Y. Acad. Sci. 986:354–359, 2003. [DOI] [PubMed]
- Marcus DC, Wu T, Wangemann P, Kofuji P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am. J. Physiol. Cell. Physiol. 282:C403–C407, 2002. [DOI] [PubMed]
- Mcguirt JP, Schulte BA. Distribution of immunoreactive alpha- and beta-subunit isoforms of Na,K–ATPase in the gerbil inner ear. J. Histochem. Cytochem. 42:843–853, 1994. [DOI] [PubMed]
- Mizuta K, Adachi M, Iwasa KH. Ultrastructural localization of the Na–K–Cl cotransporter in the lateral wall of the rabbit cochlear duct. Hear. Res. 106:154–162, 1997. [DOI] [PubMed]
- Moseley AE, Lieske SP, Wetzel RK, James PF, He S, Shelly DA, Paul RJ, Boivin GP, Witte DP, Ramirez JM, Sweadner KJ, Lingrel JB. The Na,K-ATPase alpha 2 isoform is expressed in neurons, and its absence disrupts neuronal activity in newborn mice. J. Biol. Chem. 278:5317–5324, 2003. [DOI] [PubMed]
- Nie L, Feng W, Diaz R, Gratton MA, Doyle KJ, Yamoah EN. Functional consequences of polyamine synthesis inhibition by l-alpha-difluoromethylornithine (DFMO): cellular mechanisms for DFMO-mediated ototoxicity. J. Biol. Chem. 280:15097–15102, 2005. [DOI] [PubMed]
- Offner FF, Dallos P, Cheatham MA. Positive endocochlear potential: mechanism of production by marginal cells of stria vascularis. Hear. Res. 29:117–124, 1987. [DOI] [PubMed]
- Rybak LP, Morizono T. Effect of furosemide upon endolymph potassium concentration. Hear. Res. 7:223–231, 1982. [DOI] [PubMed]
- Sakaguchi N, Crouch JJ, Lytle C, Schulte BA. Na-K-Cl cotransporter expression in the developing and senescent gerbil cochlea. Hear. Res. 118:114–122, 1998. [DOI] [PubMed]
- Salt AN, Demott JE. Physical processes underlying homeostasis of cochlear endolymph. In: Reid A (ed) Intracranial and Inner Ear Physiology and Pathophysiology. London, Whurr Publishers, 1999.
- Salt AN, Melichar I, Thalmann R. Mechanisms of endocochlear potential generation by stria vascularis. Laryngoscope 97:984–991, 1987. [DOI] [PubMed]
- Schuknecht HF, Gacek MR. Cochlear pathology in presbycusis. Ann. Otol. Rhinol. Laryngol. 102:1–16, 1993. [DOI] [PubMed]
- Schulte BA, Adams JC. Distribution of immunoreactive Na+,K+–ATPase in gerbil cochlea. J. Histochem. Cytochem. 37:127–134, 1989. [DOI] [PubMed]
- Schulte BA, Schmiedt RA. Lateral wall Na,K–ATPase and endocochlear potentials decline with age in quiet-reared gerbils. Hear. Res. 61:35–46, 1992. [DOI] [PubMed]
- Schulte BA, Steel KP. Expression of alpha and beta subunit isoforms of Na,K–ATPase in the mouse inner ear and changes with mutations at the Wv or Sld loci. Hear Res. 78:65–76, 1994. [DOI] [PubMed]
- Shih L. Cochlear Hearing Loss. In: Jackler RK and Brackmann DE (eds) Neurotology. St. Louis, MO, Mosby Year Book, Inc., pp. 619–627, 1994.
- Shugyo A, Mori N, Matsunaga T. A comparison of the reduction in the K+ activity of the scala media produced by furosemide and ouabain. Eur. Arch. Otorhinolaryngol. 248:79–81, 1990. [DOI] [PubMed]
- Spicer SS, Schulte BA. The fine structure of spiral ligament cells relates to ion return to the stria and varies with place-frequency. Hear. Res. 100:80–100, 1996. [DOI] [PubMed]
- Spicer SS, Gratton MA, Schulte BA. Expression patterns of ion transport enzymes in spiral ligament fibrocytes change in relation to strial atrophy in the aged gerbil cochlea. Hear. Res. 111:93–102, 1997. [DOI] [PubMed]
- Takeuchi S, Ando M. Inwardly rectifying K+ currents in intermediate cells in the cochlea of gerbils: a possible contribution to the endocochlear potential. Neurosci. Lett. 247:175–178, 1998. [DOI] [PubMed]
- Takeuchi S, Ando M, Kakigi A. Mechanism generating endocochlear potential: role played by intermediate cells in stria vascularis. Biophys. J. 79:2572–2582, 2000. [DOI] [PMC free article] [PubMed]
- Von Bekesy G. DC potentials and energy balance of the cochlear partition. J. Acoust. Soc. Am. 22:576–582, 1950.
- Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2:30, 2004. [DOI] [PMC free article] [PubMed]
- Xia A, Kikuchi T, Hozawa K, Katori Y, Takasaka T. Expression of connexin 26 and Na,K–ATPase in the developing mouse cochlear lateral wall: functional implications. Brain Res. 846:106–111, 1999. [DOI] [PubMed]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear. Res. 130:94–107, 1999. [DOI] [PMC free article] [PubMed]