

Abstract
Exposure to intense noise can damage the stereocilia of sensory hair cells in the inner ear. Since stereocilia play a vital role in the transduction of sound from a mechanical stimulus into an electrical one, this pathology is thought to contribute to noise-induced hearing loss. Mice homozygous for null mutations in either the myosin VIIa (Myo7a) or cadherin 23 (Cdh23) genes are deaf and have disorganized stereocilia bundles. We show that mice heterozygous for a presumed null allele of Cdh23 (Cdh23v) have low- and high-frequency hearing loss at 5–6 weeks of age, the high-frequency component of which worsens with increasing age. We also show that noise-induced hearing loss in 11–12-week-old Cdh23v heterozygotes is two times greater than for wild-type littermates. Interestingly, these effects are dependent upon the genetic background on which the Cdh23v mutation is carried. Noise-induced hearing loss in 11–12-week-old mice heterozygous for a null allele of Myo7a (Myo7a4626SB) is not significantly different from wild-type littermates. CDH23 is the first gene known to cause deafness in the human population to be linked with predisposition to noise-induced hearing loss.
Keywords: noise-induced hearing loss, progressive hearing loss, waltzer, shaker1, mouse mutant, stereocilia, cadherin 23, myosin VIIa, genetic predisposition
INTRODUCTION
Deafness is an extremely common disorder within the aging population with approximately 40% of people over the age of 60 suffering a hearing loss greater than 25 dB (Davis 1989). Genetic predisposition to environmental insults (e.g., noise and ototoxic compounds) has been shown to contribute to age-related deafness. For example, the A1555G substitution in 12S rRNA predisposes individuals to aminoglycoside-induced deafness, and in mice susceptibility to noise-induced hearing loss varies considerably between strains suggesting a genetic origin (Prezant et al. 1993; Erway et al. 1996; Yoshida et al. 2000). Indeed, the loss of function of several genes has been shown to increase susceptibility to noise-induced hearing loss in mice. These genes include Cu/Zn-superoxide dismutase (Sod1) and glutathione peroxidase (Gpx1), which both encode enzymes involved in reducing the levels of reactive oxygen species, Slc1a3, which encodes a glutamate transporter, and plasma membrane calcium ATPase isoform 2 (Atp2b2), which encodes a calcium pump (Ohlemiller et al. 1999, 2000; Hakuba et al. 2000; Kozel et al. 2002).
Exposure to intense noise damages a variety of cell types within the inner ear (Saunders et al. 1985; Wang et al. 2002). Damage to the sensory hair cell includes the splaying, fusion, and sporadic loss of stereocilia (Saunders et al. 1985; Wang et al. 2002). These hairlike structures on the apical surface of hair cells are normally organized in precise bundles that are deflected by the vibration of sound causing extracellular links that run between adjacent stereocilia to be stretched. Links at the tip of the stereocilia are believed to gate transduction channels (Hudspeth 1989). Thus, stereocilia are essential for normal sound transduction.
It is therefore not surprising that deafness in several mouse mutants is associated with stereocilia defects (reviewed by Steel and Kros 2001). Waltzer and shaker1 mutants both have disorganized stereocilia bundles caused by mutations in cadherin 23 (Cdh23) and myosin VIIa (Myo7a), respectively (Gibson et al. 1995; Self et al. 1998; Di Palma et al. 2001; Holme and Steel 2002). CDH23 is mutated in Usher’s syndrome type 1D (USH1D) and MYO7A in USH1B patients (Weil et al. 1995; Bolz et al. 2001; Bork et al. 2001). Patients with USH1 are congenitally deaf, have vestibular dysfunction, and develop retinitis pigmentosa (Smith et al. 1994). Both genes have also been shown to underlie nonsyndromic forms of deafness (Liu et al. 1997a,b; Weil et al. 1997; Bork et al. 2001). Recently, Myo7a and Cdh23 were both shown to bind to harmonin suggesting that these molecules form a functional complex within the stereocilia (Boeda et al. 2002). Cdh23 may form links between adjacent stereocilia, with Myo7a anchoring the whole complex to the actin core (Di Palma et al. 2001; Kros et al. 2002).
Since Cdh23 and Myo7a are required for the proper organization of the stereocilia bundle and these structures are damaged by intense noise, we have explored the possibility that mice heterozygous for recessive mutations in these genes are predisposed to noise-induced hearing loss. Our findings show that mice heterozygous for a presumed null mutation of Cdh23 are predisposed to noise-induced hearing loss and provide evidence for the existence of a modifier(s) of Cdh23 or a hypomorphic allele of Cdh23. We found no evidence to suggest that mice heterozygous for a null mutation of Myo7a are predisposed to noise-induced hearing loss. This is the first report linking a gene that is known to cause deafness in the human population with predisposition to noise-induced hearing loss.
MATERIALS AND METHODS
Mice
All mice were housed and used in accordance with UK Home Office regulations. The Cdh23v mutation (waltzer, v) arose spontaneously on a heterogeneous genetic background. We obtained this allele from Harwell, UK, and have since outcrossed and intercrossed twice to CBA/Ca. The genetic background of Cdh23v mice used in this study was approximately 75% CBA/Ca and 25% unknown heterogeneous background. The Myo7a4626SB mutation (shaker1, sh1) arose in a N-ethyl-N-nitrosourea-mutagenized BALBc male and was repeatedly backcrossed to the BS inbred strain (Rinchik et al. 1990). This stock has since been outcrossed once to CBA/Ca and maintained since by intercrossing. The genetic background of Myo7a4626SB mice used in this study was approximately 50% CBA/Ca, 50% BS, and some BALBc. Both the mutations are predicted to be functionally null (Hasson et al. 1997b; Mburu et al. 1997; Wilson et al. 2001). Mice were genotyped for the Cdh23v and Myo7a4626SB mutations using the molecular assays described by Holme and Steel (2002).
Noise exposure
Mice were exposed to noise unanesthetized and unrestrained within a wire cage suspended in a wooden sound box. The base of the box was 58.4 cm square and the roof 45.7 cm square. The height of the front of the box was 70 cm, but only 55.9 cm at the rear. Thus, no two sides of the box were parallel. The wire cage was divided by a wire mesh partition and one mouse placed in each division. The cage was positioned so that the sound pressure within the cage varied by less than 0.5 dB SPL at 97 dB SPL. The exposure stimulus was generated by a custom-made white noise source (Institute of Hearing Research, Nottingham, UK), filtered to a bandwidth of 8–16 kHz (Kemo high pass VBF/8 and low pass VBF/23, Beckenham, UK), amplified (Marantz 1072, Roselle, IL), and delivered to a compression driver (JBL 2446H, Northridge, CA) connected to a flat front biradial horn (JBL 2380A, Northridge, CA) secured to the roof of the sound box. Sound exposure levels were measured at the same point within the cage before each exposure, using a 1/8-in. Bruel and Kjaer microphone and amplifier and adjusted to 103 dB SPL.
Ten-to-11-week-old mice were exposed to 8–16 kHz at an intensity of 103 dB SPL for 2 h. Auditory function and hair cell pathology was then assessed a week later and compared to nonexposed littermates that had remained in their normal environment throughout. A one-week recovery time was used to minimize temporary effects and has been used by other investigators (Yoshida et al. 2000). To control for any effects moving the mice to the sound box for 2 h might have, a small group (n = 6) of 10–11-week-old +/Cdh23v mice were placed in the sound box (two at a time) for 2 h with the sound source turned off and then returned to their normal environment. A week later their compound action potential (CAP) thresholds at all frequencies tested were not significantly different from 11–12-week-old +/Cdh23v mice (n = 14) that had remained throughout within their normal environment (p > 0.05, t-test, data not shown).
Auditory physiology
Auditory function was assessed by measuring compound action potential (CAP) thresholds. Mice were anesthetized with urethane (intraperitoneal injection of 2 mg/g), the middle ear cavity opened, and a recording electrode placed on the round window. Tone-burst stimuli of 15 ms duration, 1 ms rise-fall time, and a 100 ms interstimulus interval were delivered through a closed and calibrated sound system. The stimulus intensity was varied in 2 dB SPL increments to obtain the threshold for CAP detection. Threshold was defined as the lowest intensity for which a CAP response of 2 µV or more could be detected in the waveforms by eye. CAP thresholds were determined for 3, 6, 9, 12, 15, 18, 24, and 30 kHz. Note that mice were genotyped only after CAP thresholds had been measured.
Threshold data were collected for three experimental groups. Group 1 comprised exposed and nonexposed mice (11–12 weeks of age) generated by crossing +/Myo7a4626SB with +/Cdh23v mice (sample size shown in Table 1). Genetic factors from both the shaker1 and waltzer stock contribute to the genetic background of these mice. Group 2 comprised exposed and nonexposed mice (11–12 weeks of age) generated by crossing +/Cdh23v with +/Cdh23v mice (sample size shown in Table 1). Genetic factors from the shaker1 stock do not contribute to the genetic background of these mice. Group 3 comprised nonexposed mice at 5–6 weeks, 11–12 weeks, and 23–24 weeks of age (sample size shown in Table 2). These mice were generated by crossing +/Myo7a4626SB with +/Cdh23v mice, so genetic factors from both the shaker1 and the waltzer stock contribute to the genetic background of these mice. The 11–12-week-old data in this group were also used in group 1. Analysis of variance (ANOVA) was performed separately for each experimental group after confirming that the data satisfied the normal distribution. p values for comparison of the adjusted means (adjusted for variables in the model) were determined by post hoc analysis.
Table 1.
Number of 11–12-week-old mice used for physiology, hair cell counts, and stereocilia counts
| Physiology | Hair cell counts | Stereocilia counts | |||||
|---|---|---|---|---|---|---|---|
| Group | Genotype | Nonexposed | Exposed | Nonexposed | Exposed | Nonexposed | Exposed |
| 1a | +/+ +/+ | 14 | 11 | 6 | 4 | 5 | 5 |
| +/sh1 +/+ | 9 | 20 | 5 | 5 | 5 | 5 | |
| +/+ +/v | 9 | 10 | 5 | 5 | 5 | 6 | |
| +/sh1 +/v | 13 | 7 | 5 | 5 | 5 | 5 | |
| 2b | +/+ | 9 | 11 | – | – | – | – |
| +/v | 20 | 15 | – | – | – | – | |
aMice in experimental group 1 generated by crossing +/Myo7a4626SB with +/Cdh23v mice.
bMice in experimental group 2 generated by crossing +/Cdh23v with +/Cdh23v mice.
Table 2.
Number of nonexposed mice in experimental group 3 used for physiology
| Age (weeks) | |||
|---|---|---|---|
| Genotype | 5–6 | 11–12 | 23–24 |
| +/+ | 9 | 23 | 13 |
| +/v | 9 | 22 | 9 |
Mice were generated by crossing +/Myo7a4626SB with +/Cdh23v mice. +/+ +/+ and +/sh1 +/+ genotypes were merged and designated +/+. +/+ +/v and +/sh1 +/v genotypes were merged and designated +/v.
Scanning electron microscopy
Following electrophysiology, cochleas were fixed for approximately 12 h in 2.5% glutaraldehyde at 4°C and then dissected to expose the organ of Corti. Cochleas were processed for scanning electron microscopy (SEM), using the osmium tetroxide–thiocarbohydrazide (OTOTO) method as described by Hunter-Duvar (1978) and Self et al. (1998), and coated in gold. Specimens were examined using a Phillips XL30 scanning electron microscope. Hair cell counts were made from regions centered at 14%, 33%, 58%, and 77% of the total length of the cochlear duct from the base. At each location approximately 150 µm of organ of Corti was sampled. The number of mice in each category for which hair cell counts were performed is shown in Table 1. Stereocilia counts were made from outer hair cells (OHCs) in the innermost row at 55–60% of the total length of the cochlear duct from the base. The mean number of stereocilia and splayed stereocilia in the outermost (tallest) rank was calculated for each mouse by sampling 5–10 cells. Splayed stereocilia were defined as stereocilia that did not appear to be in contact with the middle rank. The number of mice in each category for which stereocilia counts were performed is shown in Table 1. Hair cell and stereocilia count data were analyzed separately assuming the Poisson distribution and using a log–linear model. p values for comparison of the adjusted means (adjusted for variables in the model) were determined by post hoc analysis.
Results
Effects of genotype and age in mice not exposed to noise
Compound action potential (CAP) thresholds are raised at low and high frequencies in +/Cdh23v heterozygotes
Mice carrying either a single mutant copy of Myo7a (sh1) or Cdh23(v) and mice carrying mutations in both these genes (double heterozygotes) were generated by crossing +/Myo7a4626SB with +/Cdh23v mice. Genetic factors from both the shaker1 and the waltzer stocks will therefore contribute to the genetic background of these mice. At 11–12 weeks of age, CAP thresholds for +/+ +/+ and +/sh1 +/+ mice did not significantly differ from one another at any of the frequencies tested (p > 0.05; Fig. 1A). The interanimal variability for these two genotypes was relatively low; at 6 kHz thresholds were within 12 dB for both genotypes and at 24 kHz thresholds were within 28 dB for +/+ +/+ and 20 dB for +/sh1 +/+ mice (Fig. 1B, C).
Figure 1.
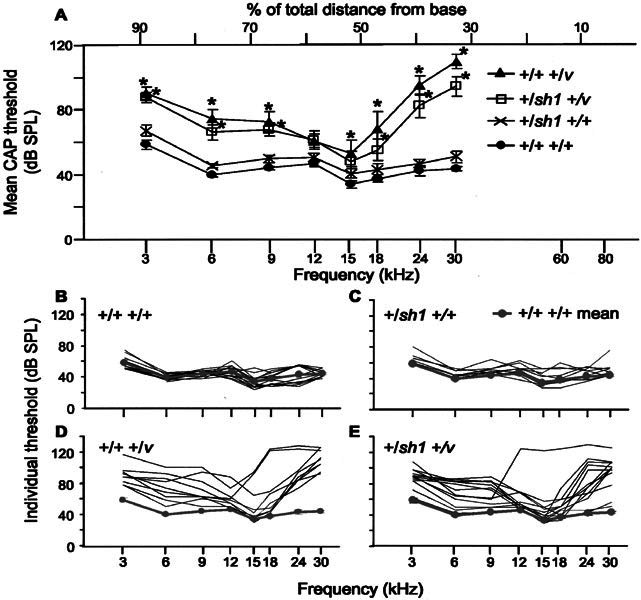
CAP thresholds for 11–12-week-old nonexposed mice. A. Mean CAP thresholds for +/+ +/+, +/sh1 +/+, +/+ +/v, and +/sh1 +/v mice plotted against frequency scaled according to the frequency place map described by Ehret (1975). * signifies frequencies at which thresholds were significantly different from those of +/+ +/+ mice (p < 0.05). Error bars represent SEM. CAP thresholds for +/+ +/v mice were significantly raised compared with +/+ +/+ mice at both low (3 kHz, +31 dB, p = 0.002; 6 kHz, +36 dB, p < 0.001; and 9 kHz, +29 dB, p = 0.004) and high frequencies (15 kHz, +19 dB, p = 0.047; 18 kHz, +30 dB, p = 0.001; 24 kHz, +53 dB, p < 0.001; and 30 kHz, +66 dB, p < 0.001). Similarly, for +/sh1 +/v mice, thresholds were significantly raised at low (3 kHz, +29 dB, p = 0.002, 6 kHz, +27 dB, p = 0.003; and 9 kHz, +24 dB, p =0.007) and high frequencies (18 kHz, +17 dB, p = 0.041; 24 kHz, +40 dB, p < 0.001; and 30 kHz, +51 dB, p < 0.001) compared with +/+ +/+ mice. B–E. CAP thresholds for individual +/+ +/+ (B), +/sh1 +/+ (C), +/+ +/v (D), and +/sh1 +/v (E) mice. For reference, the mean CAP threshold for +/+ +/+ mice has been plotted in grey. Mice heterozygous for Cdh23v show greater interanimal variability than mice wild type at this locus.
In contrast, CAP thresholds for +/+ +/v mice were significantly raised compared with +/+ +/+ mice at both low (3, 6, and 9 kHz) and high frequencies (15, 18, 24, and 30 kHz; Fig. 1A). Thresholds at 12 kHz did not significantly differ between these two genotypes (p > 0.05; Fig. 1A). Similarly, for +/sh1 +/v mice thresholds were significantly raised at low (3, 6, and 9 kHz) and high frequencies (18, 24, and 30 kHz) compared with +/+ +/+ mice (Fig. 1A). Again, thresholds at middle frequencies (12 and 15 kHz) did not differ significantly between these two genotypes (p > 0.05; Fig. 1A). CAP thresholds for +/+ +/v mice did not significantly differ from those of +/sh1 +/v mice at any of the frequencies tested (p > 0.05).
In contrast to mice wild type at the v locus, interanimal variability was extremely large for +/v mice (Fig. 1D, E). At 6 kHz, thresholds varied by as much as 50 dB for +/+ +/v and 42 dB for +/sh1 +/v mice. At 24 kHz, thresholds varied by as much as 54 dB for +/+ +/v and 86 dB for +/sh1 +/v mice. Indeed, Figure 1E shows that the thresholds for some individual +/sh1 +/v mice were very similar to the mean +/+ +/+ threshold.
Hearing loss in Cdh23v heterozygotes progressively worsens with age
To establish whether or not hearing loss in +/v mice worsens with increasing age, we next examined mice at 5–6 and 23–24 weeks of age. Because double heterozygotes did not significantly differ from +/+ +/v mice and there was no significant difference between +/sh1 +/+ and +/+ +/+ mice at 11–12 weeks of age, these groups were merged to increase statistical power. For mice wild type at the v locus, there was no significant change in CAP thresholds over time at any of the frequencies tested (p > 0.1; Fig. 2B). The low- and high-frequency hearing loss observed in +/v mice at 11–12 weeks of age was already evident at 5–6 weeks of age as shown by the significantly raised CAP thresholds at 3, 6, 24, and 30 kHz in this group compared with +/+ mice (Fig. 2A). For the oldest age group (23–24 weeks of age), CAP thresholds in +/v mice were significantly greater than those of +/v mice in the youngest age group (5–6 weeks of age) at 15, 18, 24, and 30 kHz, demonstrating that the high-frequency hearing loss worsens with increasing age (Fig. 2C). Interestingly, no significant increase in thresholds was observed at low frequencies (3 and 6 kHz) between 5–6 and 23–24-week-old +/v mice (Fig. 2C).
Figure 2.
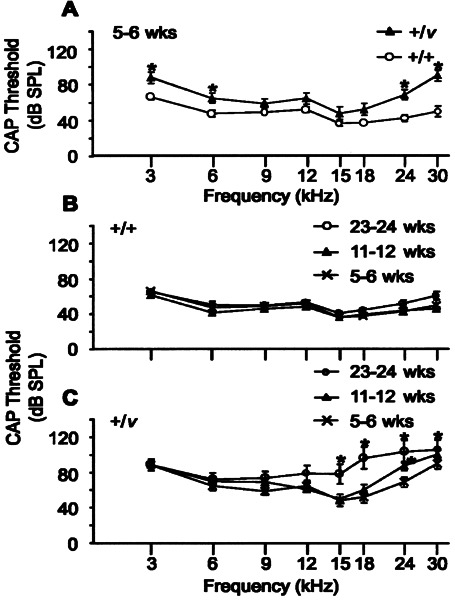
Effect of age on CAP thresholds for nonexposed mice. Data for +/+ +/v and +/sh1 +/v mice were merged and designated +/v, and data for +/sh1 +/+ and +/+ +/+ mice merged and designated +/+. A. Mean CAP thresholds for +/v and +/+ mice aged between 5 and 6 weeks. * signifies frequencies at which thresholds for +/v mice were significantly greater than those of +/+ mice (p < 0.05). Error bars represent SEM. CAP thresholds were significantly raised at 3 kHz (+22 dB, p = 0.0103), 6 kHz (+17 dB, p = 0.0284), 24 kHz (+26 dB, p = 0.001), and 30 kHz (+40 dB, p < 0.0001) in +/v compared with +/+ mice. B, C. Mean CAP thresholds for +/+ (B) and +/v (C) mice aged either 5–6 weeks, 11–12 weeks, or 23–24 weeks of age. * signifies frequencies at which thresholds were significantly greater than those of 5–6-week-old mice (p < 0.05). Error bars represent SEM. CAP thresholds for +/+ mice did not change with age. In contrast, CAP thresholds in 23–24-week-old +/v mice were significantly greater than those of 5–6-week-old +/v mice at 15 kHz (+30 dB, p = 0.0002), 18 kHz (+44 dB, p < 0.0001), 24 kHz (+34 dB, p < 0.0001), and 30 kHz (+16 dB, p = 0.0491) demonstrating that the high-frequency hearing loss worsens with increasing age. No significant increase in thresholds was observed at the low frequencies (3 kHz, p = 0.943 and 6 kHz, p = 0.3677) between 5–6- and 23–24-week-old +/v mice.
Basal hair cells are missing in Cdh23v heterozygotes
Hair cell counts were made at four locations along the duct: centered at 14%, 33%, 58%, and 77% of the total length of the duct from the base. According to the frequency place map described by Ehret (1975), these locations respond best to 60, 30, 12, and 6 kHz, respectively. In 11–12-week-old mice there was no significant difference in the number of inner (IHCs) or outer hair cells (OHCs) between +/+ +/+ and +/sh1 +/+ mice at any of these locations (p > 0.l; Fig. 3A, B). This is consistent with these genotypes having similar CAP thresholds (Fig. 1A). In +/+ +/v mice, there were fewer IHCs and OHCs than in +/+ +/+ mice at the 14% (60 kHz) region (Fig. 3A, C). In +/sh1 +/v mice, the number of IHCs, at this region were also reduced compared with +/+ +/+ mice but not by a significant amount (p = 0.26; Fig. 3A, D). However, the number of OHCs in all three rows were significantly reduced in +/sh1 +/v compared with +/+ +/+ mice at this region (Fig. 3A, D). For both genotypes, OHCs were more severely affected than IHCs, with OHCs in the innermost row the most vulnerable. The loss of hair cells at this location is consistent with the high-frequency hearing loss observed in these mice (Fig. 1A). However, because of technical constraints, it was not actually possible to measure CAP thresholds for 60 kHz. The highest frequency for which CAP thresholds were measured, 30 kHz, corresponds to the 33% region. At this location there was no significant reduction in the number of IHCs in +/+ +/v and +/sh1 +/v mice compared with +/+ +/+ mice (p > 0.1; Fig. 3A, C, and D). However, the number of OHCs in rows 1 and 2 were reduced significantly in +/+ +/v compared with +/+ +/+ mice. The 23% reduction in the number of OHCs in row 3 was not significant (p = 0.08; Fig. 3A, C). The reduction in OHC numbers in +/sh1 +/v compared with the number in +/+ +/+ mice was not significant (Fig. 3A, D). The loss of OHCs at the 33% region correlates with the raised CAP thresholds observed at 30 kHz in +/+ +/v mice (Fig. 1A). At the 58% (12 kHz) region, there was no significant reduction in either the number of IHCs or OHCs in +/+ +/v and +/sh1 +/v mice compared with +/+ +/+ mice, which correlates with the observation that CAP thresholds did not significantly differ between these genotypes at 12 kHz (Fig. 3A, C, D, Fig. 1A). At the 77% (6 kHz) region, there was also no significant reduction in either the number of IHCs or OHCs in +/+ +/v and +/sh1 +/v mice compared with +/+ +/+ mice despite the fact that CAP thresholds at 6 kHz were significantly raised (Fig. 3A, C, D, Fig. 1A). Therefore, hair cell loss cannot account for the low-frequency hearing loss observed in these mice. Furthermore, no defects in the arrangement of hair cells within in the organ of Corti, their surfaces, or stereocilia were observed by scanning electron microscopy at this region (data not shown).
Figure 3.
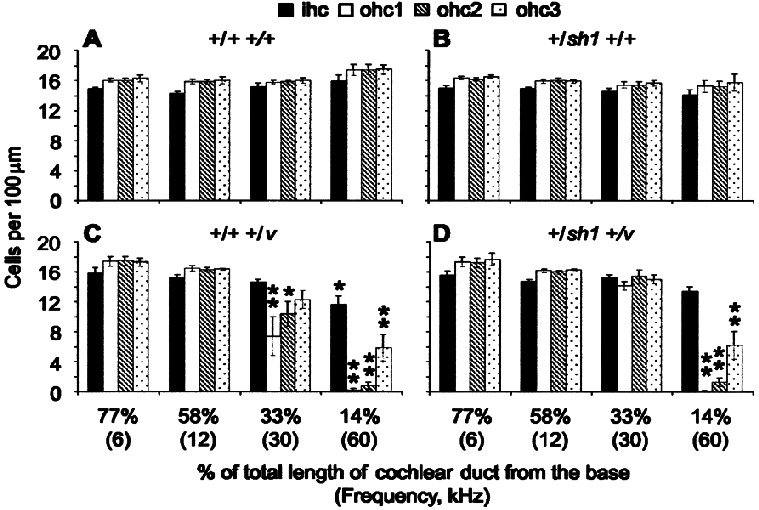
Hair cell counts for nonexposed mice. Histograms showing the mean number of IHCs and OHCs in rows 1, 2, and 3 per 100 µm. OHC row 1 is defined as the row nearest the IHCs and 3 the furthest. Counts were made for +/+ +/+ (A), +/sh1 +/+ (B), +/+ +/v (C), and +/sh1 +/v (D) mice at 14%, 33%, 58%, and 77% of the total length of the cochlear duct from the base. The frequencies that each of these locations responds best to are shown in brackets. * signifies a significant reduction in cell number compared with the number of cells in +/+ +/+ mice (*p < 0.05, **p < 0.001). Error bars represent SEM. ihc: inner hair cell; ohc1: outer hair cell in row 1; ohc2: outer hair cell in row 2; ohc3: outer hair cell in row 3.
Hearing loss in Cdh23v heterozygotes is dependent upon genetic background
Since +/v mice exhibiting a progressive hearing loss were generated by crossing mice with different genetic backgrounds (Cdh23v: 75% CBA/Ca and 25% unknown background: Myo7a4626SB: 50% CBA/Ca, 50% BS, and some BALBc background), we next investigated whether the hearing loss in +/v mice is dependent upon genetic background. Therefore, CAP thresholds were measured for +/+ and +/v mice with the mutation carried on the waltzer genetic background (75% CBA/Ca and 25% unknown) with no genetic contribution from the shaker1 stock. At all frequencies tested, the CAP thresholds of +/+ and +/v mice did not differ significantly at 11–12 weeks of age (p > 0.05; Fig. 4). This suggests that the genetic background on which the Myo7a4626SB mutation is carried contains a modifier(s) that interacts with the single copy of the Cdh23v mutant allele to cause progressive hearing loss. Alternatively, this background may carry a hypomorphic allele of Cdh23.
Figure 4.
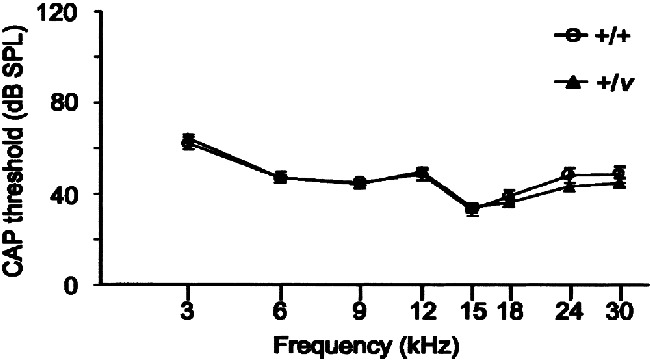
Effect of genetic background on CAP thresholds of 11–12-week-old nonexposed mice. Mean CAP thresholds for +/+ and +/v mice with no genetic contribution from the sh1 background. At all frequencies tested there were no significant differences in CAP thresholds between +/+ and +/v mice (p > 0.05). Error bars represent SEM.
Effect of exposure to noise
Heterozygotes for Cdh23v are predisposed to noise-induced hearing loss on the waltzer/shaker1 genetic background
To test whether mice heterozygous for either Cdh23v or Myo7a4626SB or double heterozygotes are more susceptible to noise-induced hearing loss than wild-type mice, we exposed all four genotypes to noise for 2 h (bandwidth of 8–16 kHz and delivered at an intensity of 103 dB SPL). Mice were generated by crossing +/sh1 and +/v mice. Genetic factors from both the shaker1 and waltzer stock will therefore contribute to the genetic background of these mice. As we have shown, +/v mice have a progressive hearing loss on this genetic background. Mice were all 10–11 weeks old at the time of exposure. One week after exposure, their auditory function and hair cell pathology were assessed and compared with those of their littermates that were not exposed to noise. At 12 kHz, the frequency at the center of the bandwidth of noise used, thresholds were significantly raised in +/+ +/+ (p = 0.043), +/+ +/v (p = 0.002), and +/sh1 +/v (p < 0.001) but not in +/sh1 +/+ (p > 0.05) mice exposed to noise compared with nonexposed mice (Fig. 5A). There was no significant difference in threshold shift between +/+ +/+ and +/sh1 +/+ at this frequency, nor for any of the other frequencies tested (p > 0.l; Fig. 5A). In contrast, the threshold shifts for +/+ +/v and +/sh1 +/v mice at 12 kHz were significantly greater than those for +/+ +/+ mice (p = 0.028 and p = 0.017; Fig. 5A). In addition to this frequency, the threshold shift at 15 kHz in +/+ +/v mice was significantly greater than the shift observed in +/+ +/+ mice (p = 0.034; Fig. 5A). In +/sh1 +/v mice, the threshold shifts at 15 and 18 kHz were significantly greater than those observed at these frequencies in +/+ +/+ mice (p = 0.004 and p = 0.004, respectively; Fig. 5A). A comparison of threshold shifts between +/+ +/v and +/sh1 +/v mice revealed no significant differences at any of the frequencies tested (p > 0.1; Fig. 5A).
Figure 5.
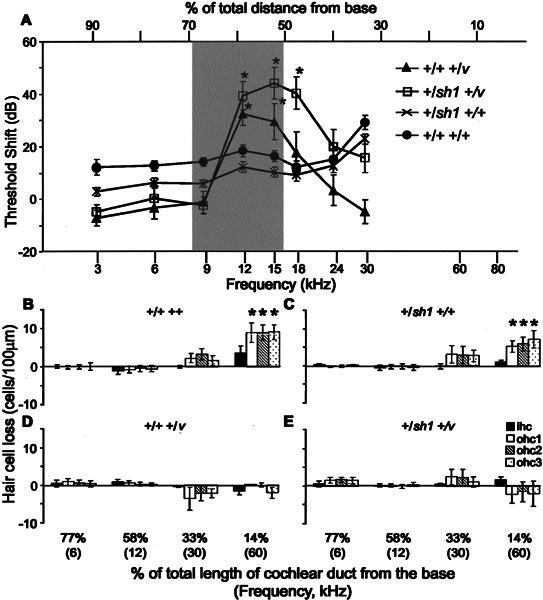
Effect of noise exposure on CAP thresholds and hair cell numbers. A. Mean threshold shift a week after exposure to 8–16 kHz delivered at 103 dB SPL for 2 h for 11–12-week-old +/+ +/+, +/sh1 +/+, +/+ +/v, and +/sh1+/v mice. Threshold shifts were calculated by subtracting the mean threshold for exposed mice from that of nonexposed mice with the same genotype. They are plotted against frequency scaled according to the frequency place map described by Ehret (1975). Frequencies at which threshold shifts differ significantly (p < 0.05) from those of +/+ +/+ mice are signified by *. The shaded box marks the frequency of noise used to expose these mice. Error bars represent SEM. B–E. Histograms showing the mean loss of IHCs and OHCs a week after noise exposure for 11–12-week-old +/+ +/+ (B),+/sh1 +/+ (C), +/+ +/v (D), and +/sh1 +/v (E) mice. Hair cell loss was measured at 14%, 33%, 58%, and 77% of the total length of the cochlear duct from the base. The frequencies that each of these locations responds best to are shown in brackets. Hair cell loss was calculated by subtracting the mean number of cells in exposed mice from the mean number of cells in nonexposed control mice. * denotes a significant (p ≤ 0.01) increase in hair cell loss. Note that there was no significant loss of hair cells at the 58% (12 kHz) region of either +/+ +/v or +/sh1 +/v mice despite a large threshold shift (approximately 35 dB) at this frequency. Error bars represent SEM. ihc: inner hair cell; ohc1: outer hair cell in row 1, nearest the IHCs; ohc2: outer hair cell in row 2; ohc3: outer hair cell in row 3, furthest from the IHCs.
In addition to raised thresholds corresponding to the bandwidth of noise to which the mice were exposed (8–16 kHz), thresholds at 30 kHz were also raised in +/+ +/+ and +/sh1 +/+ mice (p = 0.001 and p = 0.01 respectively; Fig. 5A). No significant threshold shifts were observed for +/+ +/v and +/sh1 +/v mice for this frequency (p > 0.05; Fig. 5A), but it should be noted that these mice had approximately 35 dB of hearing loss at this frequency compared with +/+ +/+ mice without being exposed to noise (Fig. 1A).
Individual thresholds for +/+ +/v and +/sh1 +/v mice exposed to noise varied by approximately 74 dB at 12 kHz, considerably more than the approximate 40 dB of variation observed at this frequency for mice wild type at the v locus. Indeed, 2 out of 17 individuals actually had thresholds lower than the +/+ +/+ mean (data not shown). This large interanimal variation is similar to the large variation in thresholds observed for this genotype without exposure to noise. However, it is not known if individual mice with low thresholds before noise exposure also have low thresholds after noise exposure because of the design of this study.
Effect of noise exposure on hair cells
One week after noise exposure hair cell loss was evident in both +/+ +/+ and +/sh1 +/+ mice. In +/+ +/+ mice, the number of OHCs per 100 µm was reduced by 50% (p < 0.0001) in each of the three rows at the 14% (60 kHz) region (Fig. 5B). There was also an indication that the number of IHCs was reduced following noise exposure in this region, but this proved not to be significant (p = 0.10; Fig. 5B). At the 14% (60 kHz) region of +/sh1 +/+ mice exposed to noise, the number of OHCs in row 1 was reduced by 30% (p = 0.0104), in row 2 by 40% (p = 0.0036) and in row 3 by 40% (p = 0.0022) compared with nonexposed mice (Fig. 5C). Similar to +/+ +/+ mice, there was no significant reduction in the number of IHCs at this position along the cochlear duct following noise exposure (p = 0.73; Fig. 5C). Unfortunately, we are unable to correlate hair cell loss in this region of the duct with CAP threshold shifts because it is not possible to measure thresholds at 60 kHz due to technical constraints.
At 30 kHz (33%), +/+ +/+ and +/sh1 +/+ mice exhibited threshold shifts of 29 dB and 23 dB, respectively (Fig. 5A). Although not significant, there was a 10% (p > 0.1) reduction in the number of OHCs in all three rows of these mice at this position (Fig. 5B, C). No hair cell loss was observed in either +/+ +/+ or +/sh1 +/+ mice at the 58% region which responds best to 12 kHz despite this being the frequency at the center of the bandwidth of noise exposed to these mice (Fig. 5B, C).
Hair cell counts for +/+ +/v and +/sh1 +/v mice exposed to noise showed that at 14% (60 kHz) and 33% (30 kHz) noise exposure did not significantly increase the amount of hair cell loss inherently associated with these regions (Fig. 5D, E). +/+ +/v and +/sh1 +/v mice exhibited a relatively large threshold shift of approximately 35 dB at 12 kHz (Fig. 5A), but no hair cell loss was observed at the 58% region which responds best to this frequency (Fig. 5D, E).
Stereocilia damage in noise-exposed mice
Closer examination of the surface of OHCs at the 58% (12 kHz) region by SEM revealed that their stereocilia were damaged. Stereocilia damage was seen in all exposed mice irrespective of genotype. The stereocilia on OHCs of nonexposed mice were arranged tightly in three parallel ranks that formed a V-shaped pattern (Fig. 6A). In noise-exposed mice, gaps were observed within the ranks suggesting that stereocilia were missing (Fig. 6B, arrowhead). We also observed splayed stereocilia that leaned outward from the bundle so that their tips were not in contact with the adjacent rank (Fig. 6B, arrow). All three rows of OHCs were affected and, for an individual cell, the outermost (tallest) rank of stereocilia appeared to be the most severely affected. We quantified this damage by counting the number of stereocilia and the number of splayed stereocilia in the outermost rank of OHCs positioned in the row closest to the IHCs (row 1). There was a significant reduction (p < 0.0001 using a log–linear model for the group as a whole) in the number of stereocilia per rank for all genotypes from approximately 33 in nonexposed mice to 29 in exposed mice (Fig. 6C). Similarly, there was a significant increase (p < 0.0001 using a log–linear model for the group as a whole) in the number of splayed stereocilia per rank from approximately 0.7% of stereocilia being splayed in nonexposed mice to approximately 6.0% being splayed in exposed mice (Fig. 6D). Although +/+ +/v and +/sh1 +/v mice had a twofold increase in noise-induced threshold shift at 12 kHz compared with +/+ +/+ mice, neither the amount of stereocilia loss nor the increase in number of splayed stereocilia at the location that this frequency maps to differed significantly between these genotypes (p > 0.1).
Figure 6.
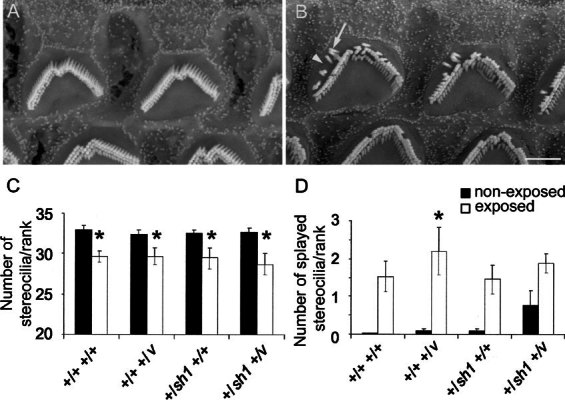
Effect of noise exposure on hair cell stereocilia. A, B. Scanning electron micrographs of the surface of OHCs, in the outermost row (row 3), 58% of the total length of the cochlear duct from the base. This region responds best to 12 kHz according to the frequency place map described by Ehret (1975). A. Nonexposed +/sh1 +/v mouse, 11–12 weeks of age. Stereocilia are upright and arranged in a tight V-shaped bundle. B. +/sh1+/v mouse, 11–12 weeks of age, a week after being exposed to 8–16 kHz delivered at 103 dB SPL for 2 h. Gaps within the ranks of stereocilia are present (arrowhead) as are splayed stereocilia that lean outward from the bundle (arrow). Scale bar = 2 µm. C, D. Histograms showing the mean number of stereocilia (C) and splayed stereocilia (D) in the outermost (tallest) rank. Only OHCs positioned in the row nearest the IHCs and 58% of the total length of the cochlear duct from the base were analyzed. Counts were made for nonexposed (black bars) and noise-exposed (white bars) +/+ +/+, +/+ +/v, +/sh1 +/+, and +/sh1 +/v mice a week after exposure. * indicates a significant difference (p < 0.05) between exposed and nonexposed mice. At the genotype level, only +/+ +/v mice had a significant increase in splayed stereocilia after noise exposure. However, the effect of noise on stereocilia splaying was highly significant for the group as a whole (p < 0.0001 using a log–linear model). Neither the amount of stereocilia loss nor the increase in number of splayed stereocilia differed significantly between genotypes (p > 0.1). Error bars represent the SEM.
For all genotypes, the stereocilia damage observed at the 58% (12 kHz) region following noise exposure was not observed at the other three locations (77%, 6 kHz; 33%, 30 kHz; and 14%, 60 kHz) of the cochlear duct studied (data not shown).
The stereocilia on IHCs positioned at 58% (12 kHz) did not appear abnormal at a gross level one week after noise exposure. However, we were unable to perform a quantitative analysis of IHC stereocilia because it was difficult to assign stereocilia to specific ranks or to judge whether they were splayed from our preparations.
Increased susceptibility to noise-induced hearing loss in Cdh23v heterozygotes is dependent upon genetic background
Since the hearing loss associated with Cdh23v heterozygotes is dependent upon the genetic background on which the mutation is carried, we next investigated whether this was also the case for increased susceptibility to noise-induced hearing loss. Mice were generated by crossing Cdh23v heterozygotes from the original waltzer stock, so that the mutation is on a genetic background (75% CBA/Ca and 25% unknown) on which it does not cause hearing loss in heterozygotes. At between 10 and 11 weeks of age, these mice were exposed to noise as previously described and their CAP thresholds compared with nonexposed littermates one week after exposure. Noise-exposed wild-type mice had significantly raised thresholds at 12, 15, and 30 kHz but not at 3, 6, 9, 18, or 24 kHz compared with nonexposed mice (Fig. 7). Similarly, noise-exposed +/v mice had significantly raised thresholds at 9, 12, 15, 18, 24, and 30 kHz but not at 3 or 6 kHz compared with nonexposed mice (Fig. 7). However, these threshold shifts did not significantly differ between +/+ and +/Cdh23v mice at any of the frequencies tested (Fig. 7). This demonstrates that the increased susceptibility to noise-induced hearing loss associated with Cdh23v heterozygotes is indeed dependent upon the genetic background on which the mutation is carried.
Figure 7.
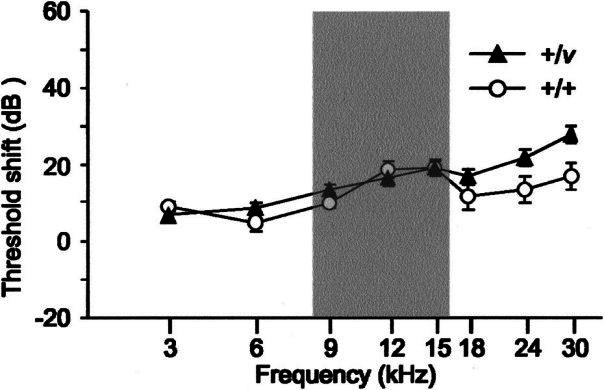
Effect of genetic background on CAP threshold shifts. Graph shows the mean shift in CAP threshold a week after exposure to 8–16 kHz delivered at 103 dB SPL for 2 h for 11–12-week-old +/+ and +/v mice with no genetic contribution from the sh1 background. Threshold shifts were calculated by subtracting the mean threshold for exposed mice from those of nonexposed mice. For +/+ mice, threshold shifts were significant at 12 kHz (+18 dB, p = 0.008), 15 kHz (+19 dB, p = 0.006), and 30 kHz (+17 dB, p = 0.016) but not at 3, 6, 9, 18, or 24 kHz. Similarly, for +/v mice threshold shifts were significant at 9 kHz (+13 dB, p = 0.012), 12 kHz (+16 dB, p = 0.002), 15 kHz (+19 dB, p < 0.001), 18 kHz (+17 dB, p = 0.002), 24 kHz (+21 dB, p < 0.001), and 30 kHz (+28 dB, p < 0.001) but not at 3 or 6 kHz compared with nonexposed mice. Threshold shifts did not significantly differ between +/+ and +/Cdh23v-mice at any of the frequencies tested (3, 9, 12, 15, 18, 24, and 30 kHz; p > 0.1). The shaded box marks the frequency of noise exposed to these mice. Error bars represent SEM.
DISCUSSION
Exposure to loud noise is known to damage the stereocilia of hair cells (Robertson 1982; Wang et al. 2002). These structures are essential for mechanotransduction of sound (Hudspeth 1989). In the present study, we explored the possibility that mice heterozygous for recessive mutations in genes known to be required for the formation and maintenance of the stereocilia bundle are predisposed to noise-induced hearing loss. Myo7a and Cdh23 are mutated in shaker1 and waltzer mice, respectively, causing deafness and disorganization of the stereocilia bundle in homozygotes (Gibson et al. 1995; Self et al. 1998; Di Palma et al. 2001; Holme and Steel 2002). Our findings show that Myo7a4626SB heterozygotes have no hearing loss at 11–12 weeks of age and are no more susceptible to noise-induced hearing loss than wild-type littermates. The Cdh23v mutation was studied on two different genetic backgrounds. Cdh23v heterozygotes with a genetic background contributed fully from the waltzer stock have no hearing loss at 11–12 weeks of age and are no more susceptible to noise-induced hearing loss than wild-type littermates. In contrast, Cdh23v heterozygotes with a mixed genetic background derived from both the shaker1 and waltzer stocks have hearing loss at both low and high frequencies, which is progressive at high frequencies, and are more susceptible to noise-induced hearing loss than wild-type littermates. This suggests that the shaker1 stock contains a hypomorphic allele of Cdh23 or a modifier(s) that interacts with the Cdh23v allele to cause progressive hearing loss and increased susceptibility to noise-induced hearing loss.
Noise-induced hearing loss
Exposure to narrow-band noise typically raises hearing thresholds at frequencies corresponding to the exposure stimulus (tonotopic) and at frequencies higher than the stimulus (Yoshida et al. 2000; Wang et al. 2002). In our study, exposure to noise with a bandwidth of 8–16 kHz raised CAP thresholds in Cdh23 wild-type mice by approximately 16 dB at 12 kHz and approximately 26 dB at 30 kHz, which is consistent with the expected pattern of hearing loss. The amount of hearing loss observed at these frequencies, following the same noise exposure conditions (8–16 kHz, 103 dB SPL for 2 h) and recovery time (1 week) as used in our study, has been shown to vary considerably depending upon the strain of mouse used (Yoshida et al. 2000). For example, CAP thresholds for 10–11-week-old CBA/Ca mice were raised by approximately 50 dB at 12 kHz and 30 kHz, while thresholds for 129/SvEv mice were raised by only about 10 dB at these frequencies (Yoshida et al. 2000). The amount of hearing loss observed in the mice used in our study which have a heterogeneous background falls between these two strains.
Although thresholds were raised at 12 kHz following noise exposure, we observed no hair cell loss in the corresponding region of the cochlear duct to account for this hearing loss. However, the stereocilia projected by OHCs at this region were damaged and this could account for the hearing loss at this frequency. This correlation between stereocilia damage and threshold shift has been previously observed for both OHCs and IHCs (Robertson 1982; Liberman 1987; Wang et al. 2002). In mice, tonotopic damage to IHC stereocilia is seen immediately after noise exposure (8–16 kHz, ≥106 dB SPL for 2 h) and there is no evidence of recovery over a 2-week period (Wang et al. 2002).
In the extreme base of the cochlea we did observe OHC loss at regions corresponding to 60 and 30 kHz, though the latter was not significant. Therefore, hair cell loss could account for hearing loss at higher frequencies. Again, this pattern of hair cell loss (in the base, away from the region most sensitive to the frequency used to expose) has been previously observed for mice exposed to narrow-band noise at levels ≥100 dB and <116 dB SPL (Wang et al. 2002). It is not clear why cells in the extreme base should be lost when the noise stimulus contains little energy at these high frequencies. For example, the noise stimulus used in our study contains <30 dB of sound pressure at 60 kHz (measured with a bandwidth of 375 Hz; data not shown). However, the observation that basal hair cells from 1–2-day-old mice are more susceptible to the ototoxic effects of aminoglycosides invitro than apical cells suggests that these cells may be intrinsically more vulnerable to environmental insults (Richardson and Russell 1991). Furthermore, the majority of damaged cochleas, whether from genetic or environmental insult, show a greater tendency for basal turn hair cell loss than apical turn, supporting the suggestion that basal hair cells are intrinsically vulnerable.
At 12 kHz, Cdh23v heterozygote mice, with a mixed genetic background derived from both the waltzer and the shaker1 stocks, have a twofold greater shift in CAP thresholds following noise exposure than wild-type mice. However, we detected no qualitative or quantitative differences in damage to stereocilia of OHCs in the first row at the corresponding region of the cochlear duct to account for this difference. Also, although not quantified, there did not appear to be any gross difference in damage to stereocilia of IHCs or OHCs in the second and third rows. This suggests that stereocilia damage may not be the only cause of hearing loss at this frequency. Loss of type IV fibrocytes in the spiral ligament and limbus and shrinkage of the stria vascularis have been reported in mice 2 weeks after exposure to narrow-band noise (Wang et al. 2002). Differential damage to these cell types in our exposed mice may therefore contribute to the difference in threshold shift between Cdh23v heterozygotes and wild-type mice. This would imply that Cdh23 functions not only in hair cells but also in other cell types of the inner ear. Alternatively, abnormal Cdh23 function in the hair cell could have secondary effects on other cell types. A better understanding of the expression pattern of Cdh23 in the inner ear and pathology associated with noise-exposed Cdh23v heterozygotes is needed to clarify this issue.
Age-related hearing loss
Predisposition to noise-induced hearing loss is not the only characteristic associated with Cdh23v heterozygotes with a mixed genetic background derived from both the waltzer and the shaker1 stocks. These mice also have hearing loss at low and high frequencies that is already present at 5–6 weeks of age (the youngest age group studied). The high-frequency component of this hearing loss progressively worsens with increasing age. In the extreme base of the cochlear duct at a region corresponding to 60 kHz, extensive OHC loss and, to a lesser extent, IHC loss are observed in 11–12-week-old mice. This degeneration lessens moving away from the base, but OHCs are still missing at the region corresponding to 30 kHz. No hair cell loss is observed in the apex at a region corresponding to 6 kHz. Thus, hair cell loss can account for the high-frequency hearing loss observed in these mice, but not the low-frequency hearing loss.
C57BL/6 mice exhibit a similar pattern of high and low-frequency hearing loss that progressively worsens from 6 weeks of age (Hequembourg and Liberman 2001). Interestingly, the low-frequency hearing loss progresses at a slower rate than the high-frequency hearing loss. We studied a relatively small age range (18 weeks) which might explain why we did not observe a worsening of the low-frequency hearing loss in Cdh23v heterozygotes. C57BL/6 mice lose hair cells in the base from 13 weeks of age and in the apex from 30 weeks of age (Hequembourg and Liberman 2001). This pattern of hair cell loss (basal cells lost first) and the fact that the low-frequency hearing loss in mice younger than 30 weeks of age cannot be accounted for by hair cell loss is also similar to our observations in Cdh23v heterozygotes. Examination of the spiral ligament in C57BL/6 mice showed a decrease in the number of type IV fibrocytes in the apex of mice as young as 6 weeks of age and it has been proposed that this damage is the primary cause of hearing loss (Hequembourg and Liberman 2001). Therefore, fibrocyte degeneration in the spiral ligament of Cdh23v heterozygotes may also explain why we observed raised thresholds at 6 kHz but no associated hair cell degeneration.
Role of Cdh23 in predisposition to noise-induced hearing loss and progressive deafness
Cadherin 23 is a member of a superfamily of transmembrane proteins that mediate cell–cell adhesion. In Cdh23v homozygote mutants, the stereocilia bundle is disorganized and it has been proposed that Cdh23 may be a component of one of the extracellular filaments that crosslink stereocilia (Holme and Steel 2002). These links are thought to be important in holding the stereocilia bundle together. Boeda et al. (2002) recently reported that Cdh23 could not be detected in stereocilia of mice older than post natal day 30, suggesting that Cdh23 could be involved in crosslinking only developing stereocilia. However, we do detect a phenotypic effect of a presumed 50% reduction in the amount of functional Cdh23 in the adult cochlea. It may be that Cdh23 elsewhere in the hair cell mediates the effects we see or that only small amounts of Cdh23 protein are present in mature stereocilia, below the detection limits of the technique used by Boeda et al. (2002). Alternatively, in Cdh23v heterozygotes, the stereocilia bundle may not attain maximum strength because of the proposed transient links being compromised during development. This may sensitize the bundle to the effects of noise exposure and aging in adulthood.
The stereocilia disorganization observed in Myo7a4626SB homozygote mice is remarkably similar to that observed in Cdh23v mutants (Holme and Steel 2002). Recently, Myo7a has been shown to interact with harmonin which in turn interacts with Cdh23 (Boeda et al. 2002; Siemens et al. 2002). All three proteins are present in the stereocilia, thus potentially placing Cdh23 and Myo7a in the same functional complex (Hasson et al. 1997a; Boeda et al. 2002). Therefore, it is surprising that only Cdh23v heterozygotes show increased susceptibility to noise-induced hearing loss and not Myo7a4626SB heterozygotes. Presuming a 50% reduction in Cdh23 and Myo7a protein levels in the respective heterozygotes, it suggests that this putative complex can tolerate a reduction in the level of Myo7a more than a reduction in Cdh23. Alternatively, Cdh23 may function in another cell type of the inner ear as previously discussed.
Potential modifier(s) of Cdh23 that cause predisposition to noise-induced hearing loss and progressive hearing loss
The ahl locus, responsible for age-related hearing loss and increased susceptibility to noise-induced hearing loss in C57BL/6 mice, has been mapped to chromosome 10 (Erway et al. 1996; Johnson et al. 1997, 2000; Davis et al. 2001). The position of this locus overlaps that of Cdh23 and it has been proposed that they may be allelic (Bryda et al. 2001). Our results show that hearing loss and increased susceptibility to noise-induced hearing loss in Cdh23v heterozygotes are dependent upon a genetic contribution from the shaker1 background. Fifty percent of the shaker1 background is derived from the BS inbred strain, which was, in turn, derived from several common inbred strains at Oak Ridge National Laboratory. Therefore, ahl may segregate in our shaker1 stock. If Cdh23 and ahl are indeed allelic, then it could explain why we observe hearing loss and increased susceptibility to noise-induced hearing loss in Cdh23v heterozygotes with a mixed genetic background derived from both the shaker1 and the waltzer stocks.
Alternatively, the shaker1 stock may contain one or a small number of modifiers that interact with the single-mutant Cdh23v allele to cause hearing loss and predisposition to noise-induced hearing loss. A mutation in the mitochondrial DNA (mtDNA) of the A/J strain of mice has been shown to exacerbate the hearing loss in mice homozygous for ahl (Johnson et al. 2001). However, the Cdh23v modifier(s) are not of mitochondrial origin since hearing loss and increased susceptibility to noise-induced hearing loss were observed in Cdh23v heterozygotes with mtDNA derived from either the shaker1 or waltzer stock (data not shown). The modifier(s) is also not sex linked since male and female +/Cdh23v mice were equally affected (data not shown).
Polymorphisms in autosomal genes associated with noise-induced hearing loss could cause hearing loss and increased susceptibility to noise-induced hearing loss in Cdh23v heterozygotes. These include genes encoding neurotrophic and growth factors (Keithley et al. 1998; Shoji et al. 2000; Zhai et al. 2002), enzymes involved in reducing the levels of reactive oxygen species (Ohlemiller et al. 1999, 2000), proteins involved in the uptake of glutamate following release at the IHC–auditory nerve synapse (Hakuba et al. 2000), and components of the olivocochlear efferent system (Maison et al. 2002), all of which have been shown to influence susceptibility to noise-induced hearing loss. Perhaps the most relevant gene in the context of Cdh23 function is Atp2b2. This gene is mutated in deafwaddler mice causing deafness in homozygotes (Street et al. 1998). Kozel et al. (2002) recently demonstrated that deafwaddler heterozygotes are more susceptible to noise-induced hearing loss than their wild-type littermates. A modifier of this gene (mdfw), causes hearing loss in deafwaddler heterozygotes, has been mapped to the same chromosomal location as ahl and Cdh23, suggesting that all three may be allelic (Noben-Trauth et al. 1997; Bryda et al. 2001; Zheng and Johnson 2001). Atp2b2 encodes a plasma membrane Ca2+–ATPase pump that removes Ca2+ from the stereocilia (Street et al. 1998; Yamoah et al. 1998). It is conceivable that disruption of Ca2+ homeostasis in the stereocilia could affect Cdh23 function since the homophilic interaction between cadherins is Ca2+ dependent (Leckband and Sivasankar 2000). Thus, any polymorphisms in Atp2b2 may be good candidates as modifiers of waltzer.
Acknowledgements
This work was supported by the MRC, Defeating Deafness, and the European Commission (contract number: QLRT-1999-00988). We thank Jabulani Sithole and Miguel Goncalves for statistical advice. We are also grateful to Charles Liberman for helpful advice on noise exposure conditions, and Dave Bullock and Angela Lucas for technical assistance.
References
- 1.Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. EMBO J. 2002;21(24):6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, del C-Salcedo Cabrera M, Vila MC, Molina OP, Gal A, Kubisch C. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat. Genet. 2001;27(1):108–112. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- 3.Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Riazuddin S, Bitner–Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. AM. J. Hum. Genet. 2001;68(l):26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bryda EC, Kim HJ, Legare ME, Frankel WN, Noben–Trauth K. High-resolution genetic and physical mapping of modifier-of-deafwaddler (mdfw) and Waltzer (Cdh23v). Genomics. 2001;73(3):338–342. doi: 10.1006/geno.2001.6538. [DOI] [PubMed] [Google Scholar]
- 5.Davis AC. The prevalence of hearing impairment and reported hearing disability among adults in Great Britain. Int. J. Epidemiol. 1989;8(4):911–917. doi: 10.1093/ije/18.4.911. [DOI] [PubMed] [Google Scholar]
- 6.Davis RR, Newlander JK, Ling X, Cortopassi GA, Krieg EF, Erway LC. Genetic basis for susceptibility to noise-induced hearing loss in mice. Hear. Res. 2001;155(1–2):82–90. doi: 10.1016/S0378-5955(01)00250-7. [DOI] [PubMed] [Google Scholar]
- 7.Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben–Trauth K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat. Genet. 2001;27(1):103–107. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- 8.Ehret G. Masked auditory thresholds, critical ratios, and scales of the basilar membrane of the housemouse (Mus musculus). J. Comp. Physiol. 1975;103:329–341. [Google Scholar]
- 9.Erway LC, Shiau YW, Davis RR, Krieg EF. Genetics of age-related hearing loss in mice. III. Susceptibility of inbred and F1 hybrid strains to noise-induced hearing loss. Hear. Res. 1996;93(1–2):181–187. doi: 10.1016/0378-5955(95)00226-X. [DOI] [PubMed] [Google Scholar]
- 10.Gibson F, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SD. A type VII myosin encoded by the mouse deafness gene shaker-1. Nature. 1995;374:62–64. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- 11.Hakuba N, Koga K, Gyo K, Usami SI, Tanaka K. Exacerbation of noise-induced hearing loss in mice lacking the glutamate transporter GLAST. J. Neurosci. 2000;20(23):8750–8753. doi: 10.1523/JNEUROSCI.20-23-08750.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasson T, Gillespie PG, Garcia JA, MacDonald RB, Zhao Y, Yee AG, Mooseker MS, Corey DP. Unconventional myosins in inner-ear sensory epithelia. J. Cell. Biol. 1997a;137(6):1287–1307. doi: 10.1083/jcb.137.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasson T, Walsh J, Cable J, Mooseker MS, Brown SD, Steel KP. Effects of shaker-1 mutations on myosin-VIIa protein and mRNA expression. Cell Motil. Cytoskeleton. 1997b;37(2):127–138. doi: 10.1002/(SICI)1097-0169(1997)37:2<127::AID-CM5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 14.Hequembourg S, Liberman MC. Spiral ligament pathology: a major aspect of age-related cochlear degeneration in C57BL/6 mice. J. Assoc. Res. Otolaryngol. 2001;2(2):118–129. doi: 10.1007/s101620010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holme RH, Steel KP. Stereocilia defects in waltzer (Cdh23), shaker1 (Myo7a) and double waltzer/shaker1 mutant mice. Hear. Res. 2002;169(1–2):13–23. doi: 10.1016/S0378-5955(02)00334-9. [DOI] [PubMed] [Google Scholar]
- 16.Hudspeth AJ. How the ear’s works work. Nature. 1989;341:397–404. doi: 10.1038/341397a0. [DOI] [PubMed] [Google Scholar]
- 17.Hunter–Duvar IM. A technique for preparation of cochlear specimens for assessment with the scanning electron microscope. Acta Otolaryngol. Suppl. 1978;351:3–23. doi: 10.3109/00016487809122718. [DOI] [PubMed] [Google Scholar]
- 18.Johnson KR, Erway LC, Cook SA, Willott JF, Zheng QY. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear. Res. 1997;114(1–2):83–92. doi: 10.1016/S0378-5955(97)00155-X. [DOI] [PubMed] [Google Scholar]
- 19.Johnson KR, Zheng QY, Bykhovskaya Y, Spirina O, Fischel–Ghodsian N. A nuclear–mitochondrial DNA interaction affecting hearing impairment in mice. Nat. Genet. 2001;27(2):191–194. doi: 10.1038/84831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000;70(2):171–180. doi: 10.1006/geno.2000.6377. [DOI] [PubMed] [Google Scholar]
- 21.Keithley EM, Ma CL, Ryan AF, Louis JC, Magal E. GDNF protects the cochlea against noise damage. Neuroreport. 1998;9(10):2183–2187. doi: 10.1097/00001756-199807130-00007. [DOI] [PubMed] [Google Scholar]
- 22.Kozel PJ, Davis RR, Krieg EF, Shull GE, Erway LC. Deficiency in plasma membrane calcium ATPase isoform 2 increases susceptibility to noise-induced hearing loss in mice. Hear. Res. 2002;164(1–2):231–239. doi: 10.1016/S0378-5955(01)00420-8. [DOI] [PubMed] [Google Scholar]
- 23.Kros CJ, Marcotti W, van Netten SM, Self TJ, Libby RT, Brown SD, Richardson GP, Steel KP. Reduced climbing and increased slipping adaptation in cochlear hair cells of mice with Myo7a mutations. Nat. Neurosci. 2002;5(1):41–47. doi: 10.1038/nn784. [DOI] [PubMed] [Google Scholar]
- 24.Leckband D, Sivasankar S. Mechanism of homophilic cadherin adhesion. Curr. Opin. Cell Biol. 2000;12(5):587–592. doi: 10.1016/S0955-0674(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 25.Liberman MC. Chronic ultrastructural changes in acoustic trauma: serial-section reconstruction of stereocilia and cuticular plates. Hear. Res. 1987;26(1):65–88. doi: 10.1016/0378-5955(87)90036-0. [DOI] [PubMed] [Google Scholar]
- 26.Liu XZ, Walsh J, Mburu P, Kendrick–Jones J, Cope MJ, Steel KP, Brown SD. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet. 1997a;16(2):188–190. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- 27.Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SD. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet. 1997b;17(3):268–269. doi: 10.1038/ng1197-268. [DOI] [PubMed] [Google Scholar]
- 28.Maison SF, Luebke AE, Liberman MC, Zuo J. Efferent protection from acoustic injury is mediated via alpha9 nicotinic acetylcholine receptors on outer hair cells. J. Neurosci. 2002;22(24):10838–10846. doi: 10.1523/JNEUROSCI.22-24-10838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mburu P, Liu XZ, Walsh J, Saw D Jr, Cope MJ, Gibson F, Kendrick–Jones J, Steel KP, Brown SD. Mutation analysis of the mouse myosin VIIA deafness gene. Genes Funct. 1997;1(3):191–203. doi: 10.1046/j.1365-4624.1997.00020.x. [DOI] [PubMed] [Google Scholar]
- 30.Noben–Trauth K, Zheng QY, Johnson KR, Nishina PM. mdfw: a deafness susceptibility locus that interacts with deaf waddler (dfw). Genomics. 1997;44(3):266–272. doi: 10.1006/geno.1997.4869. [DOI] [PubMed] [Google Scholar]
- 31.Ohlemiller KK, McFadden SL, Ding DL, Flood DG, Reaume AG, Hoffman EK, Scott RW, Wright JS, Putcha GV, Salvi RJ. Targeted deletion of the cytosolic Cu/Zn-superoxide dismutase gene (Sod1) increases susceptibility to noise-induced hearing loss. Audiol. Neurootol. 1999;4(5):237–246. doi: 10.1159/000013847. [DOI] [PubMed] [Google Scholar]
- 32.Ohlemiller KK, McFadden SL, Ding DL, Lear PM, Ho YS. Targeted mutation of the gene for cellular glutathione peroxidase (Gpx1) increases noise-induced hearing loss in mice. J. Assoc. Res. Otolaryngol. 2000;1(3):243–254. doi: 10.1007/s101620010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, Shohat M, Fischel–Ghodsian N. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 1993;4(3):289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- 34.Richardson GP, Russell IJ. Cochlear cultures as a model system for studying aminoglycoside induced ototoxicity. Hear. Res. 1991;53(2):293–311. doi: 10.1016/0378-5955(91)90062-E. [DOI] [PubMed] [Google Scholar]
- 35.Rinchik EM, Carpenter DA, Selby PB. A strategy for fine-structure functional analysis of a 6- to 11-centimorgan region of mouse chromosome 7 by high-efficiency mutagenesis. Proc. Natl. Acad. Sci. USA. 1990;87(3):896–900. doi: 10.1073/pnas.87.3.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robertson D. Effects of acoustic trauma on stereocilia structure and spiral ganglion cell tuning properties in the guinea pig cochlea. Hear. Res. 1982;7(l):55–74. doi: 10.1016/0378-5955(82)90081-8. [DOI] [PubMed] [Google Scholar]
- 37.Saunders JC, Dear SP, Schneider ME. The anatomical consequences of acoustic injury; A review and tutorial. J. Acoust. Soc. Am. 1985;78(3):833–860. doi: 10.1121/1.392915. [DOI] [PubMed] [Google Scholar]
- 38.Self T, Mahony M, Fleming J, Walsh J, Brown SD, Steel KP. Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development. 1998;125(4):557–566. doi: 10.1242/dev.125.4.557. [DOI] [PubMed] [Google Scholar]
- 39.Shoji F, Miller AL, Mitchell A, Yamasoba T, Altschuler RA, Miller JM. Differential protective effects of neurotrophins in the attenuation of noise-induced hair cell loss. Hear. Res. 2000;146(1–2):134–142. doi: 10.1016/S0378-5955(00)00106-4. [DOI] [PubMed] [Google Scholar]
- 40.Siemens J, Kazmierczak P, Reynolds A, Sticker M, Littlewood–Evans A, Muller U. The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc. Natl. Acad. Sci. USA. 2002;99(23):14946–14951. doi: 10.1073/pnas.232579599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, Moller CG, Pelias MZ, Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am. J. Med. Genet. 1994;50(1):32–38. doi: 10.1002/ajmg.1320500107. [DOI] [PubMed] [Google Scholar]
- 42.Steel KP, Kros CJ. A genetic approach to understanding auditory function. Nat. Genet. 2001;27(2):143–149. doi: 10.1038/84758. [DOI] [PubMed] [Google Scholar]
- 43.Street VA, McKee–Johnson JW, Fonseca RC, Tempel BL, Noben–Trauth K. Mutations in a plasma membrane Ca2+-ATPase gene cause deafness in deafwaddler mice. Nat. Genet. 1998;19(4):390–394. doi: 10.1038/1284. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Hirose K, Liberman MC. Dynamics of noise-induced cellular injury and repair in the mouse cochlea. J. Assoc. Res. Otolaryngol. 2002;3(3):248–268. doi: 10.1007/s101620020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, Kelly PM, Kimberling WJ, Wagenaar M, Levi–Acobas F, Larget–Piet D, Munnich A, Steel KP, Brown SDM, Petit C. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–61. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- 46.Weil D, Kussel P, Blanchard S, Levy G, Levi–Acobas F, Drira M, Ayadi H, Petit C. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat. Genet. 1997;16(2):191–193. doi: 10.1038/ng0697-191. [DOI] [PubMed] [Google Scholar]
- 47.Wilson SM, Householder DB, Coppola V, Tessarollo L, Fritzsch B, Lee EC, Goss D, Carlson GA, Copeland NG, Jenkins NA. Mutations in Cdh23 cause nonsyndromic hearing loss in waltzer mice. Genomics. 2001;74(2):228–233. doi: 10.1006/geno.2001.6554. [DOI] [PubMed] [Google Scholar]
- 48.Yamoah EN, Lumpkin EA, Dumont RA, Smith PJ, Hudspeth AJ, Gillespie PG. Plasma membrane Ca2+-ATPase extrudes Ca2+ from hair cell stereocilia. J. Neurosci. 1998;18(2):610–624. doi: 10.1523/JNEUROSCI.18-02-00610.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida N, Hequembourg SJ, Atencio CA, Rosowski JJ, Liberman MC. Acoustic injury in mice: 129/SvEv is exceptionally resistant to noise-induced hearing loss. Hear. Res. 2000;141(1–2):97–106. doi: 10.1016/S0378-5955(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 50.Zhai SQ, Cheng JC, Wang JL, Yang WY, Gu R, Jiang SC. Protective effect of basic fibroblast growth factor on auditory hair cells after noise exposure. Acta Otolaryngol. 2002;122(4):370–373. doi: 10.1080/00016480260000030. [DOI] [PubMed] [Google Scholar]
- 51.Zheng QY, Johnson KR. Hearing loss associated with the modifier of deaf waddler (mdfw) locus corresponds with age-related hearing loss in 12 inbred strains of mice. Hear. Res. 2001;154(1–2):45–53. doi: 10.1016/S0378-5955(01)00215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]