

Abstract
The synthesis of a new inhibitor, N-phosphonacetyl-L-isoasparagine (PALI), of Escherichia coli aspartate transcarbamoylase (ATCase) is reported, as well as structural studies of the enzyme·PALI complex. PALI was synthesized in 7 steps from β-benzyl L-aspartate. The KD of PALI was 2 μM. Kinetics and small-angle X-ray scattering experiments showed that PALI can induce the cooperative transition of ATCase from the T to the R state. The X-ray structure of the enzyme·PALI complex showed 22 hydrogen bonding interactions between the enzyme and PALI. The kinetic characterization and crystal structure of the ATCase·PALI complex also provides detailed information regarding the importance of the α-carboxylate for the binding of the substrate aspartate.
Introduction
Aspartate transcarbamoylase (ATCase, EC 2.1.3.2) from Escherichia coli catalyzes the committed step in de novo pyrimidine nucleotide biosynthesis, the condensation of carbamoyl phosphate (CP) and L-aspartate to yield N-carbamoyl-L-aspartate and Pi. The binding of substrates is ordered where the binding of CP is required for the binding of Asp.1 In E. coli, ATCase is allosterically regulated homotropically by Asp and heterotropically by the end products of the pyrimidine and purine pathways. CTP and UTP, the end products of the pyrimidine biosynthetic pathway, inhibit ATCase.2 However, UTP only inhibits the enzyme in the presence of CTP.3 ATP, an end product of the purine biosynthetic pathway, activates the enzyme.2
N-phosphonacetyl-L-aspartate (PALA), a bisubstrate analog of ATCase, inhibits both the E. coli and mammalian enzymes.4,5 Since ATCase is involved in the biosynthesis of pyrimidine nucleotides, which are required for cell division, PALA has been tested as a potential anti-cancer agent. PALA was found to block the growth of colonic cancer cells, Lewis cell carcinoma, and melanoma, leading to clinical trials of PALA as an anticancer drug.6,7 As a means to produce even more effective inhibitors of ATCase various modifications of PALA have been synthesized.8,9 However, none have been found with significantly enhanced activity compared to PALA.8 Because of the large negative charge on PALA at physiological pH attempts have been made to encapsulate it into liposomes for delivery.10,11
E. coli ATCase is a dodecamer composed of six chains (Mr 34,000 each) grouped into two catalytic trimers, and six chains (Mr 17,000 each) grouped into three regulatory dimers. Three active sites are located in the catalytic trimer, where each active site is shared between the adjacent catalytic chains. Each catalytic chain contains two structural domains, the carbamoyl phosphate (CP) domain (residues 1–135 and 292–310) and the aspartate (ASP) domain (residues 136–291), which contain the binding sites for CP and Asp, respectively. The regulatory chain is composed of two domains, the allosteric (AL) domain (residues 1–100) and the zinc (ZN) domain (residues 100–153), which contain the binding sites for nucleotides and zinc, respectively.
In E. coli ATCase, the binding of PALA induces large conformational changes in the enzyme from the low activity, low affinity T-state, to the high activity, high affinity R-state. Upon the binding of PALA, the enzyme undergoes a rotation of 10° and an expansion of 11 Å along the three-fold axis in addition there is a 15° rotation of the regulatory dimers around their respective 2-fold axes.12 Small-angle X-ray scattering (SAXS) experiments in solution also show an expansion of the enzyme upon the addition of PALA.13 The mechanism is ordered with CP binding before Asp and N-carbamoyl-L-aspartate leaving before Pi. Upon the binding of CP local conformational changes occur, the 50’s loop (residues 50–55) moves toward the CP molecule when compared to the Tapo structure, and the binding pocket for Asp is formed.14 The binding of Asp to the enzyme·CP complex induces the two domains of the catalytic chain to close bringing both substrates into close proximity.14 The binding of PALA induces the same conformational change as do the natural substrates.13
The α-carboxylate of PALA interacts with the guanidinium group of Arg167 in the active site of ATCase. However, mutation of Arg167 to Gln does not effect the binding of PALA to the enzyme. The charge interaction between Arg167 and the α-carboxylate led us to believe that a derivate of PALA with reduced charge could bind with similar affinity to the enzyme and induce the allosteric transition. To test this hypothesis we synthesized the α-amide derivative of PALA, which would reduce the charge of the analog and at the same time enhance its lipophilicity. Here we report the synthesis, inhibition kinetic data, and X-ray crystal structure of a derivative of PALA, N-phosphonacetyl-L-isoasparagine, PALI.
Results
Synthesis of N-phosphonacetyl-L-isoasparagine (1, PALI)
PALI was synthesized from commercially available β-benzyl L-aspartate (98% ee) (2) using modified literature procedures as shown in Figure 1. After protecting the amino group using Boc chemistry, reaction of β-benzyl N-tert-butoxycarbonyl-L-aspartate (3) with ethyl chloroformate and aqueous ammonia afforded the amide (4). Then the TFA mediated Boc deprotection (5) and subsequent N-acylation using chloroacetic anhydride and pyridine gave the β-benzyl N-chloroacetyl-L-isoasparaginate (6) with a yield of 80%. Reaction of 6 with triethyl phosphite under reflux condition afforded the corresponding phosphonate ester (7) in 95% yield. The β-benzyl N-phosphonacetyl-L-isoasparginate (8) was obtained in quantitative yield by the deprotection of phosphonate esters using trimethylsilylbromide. Then the final benzyl ester was deprotected using 10% Pd on activated carbon in a H2 atmosphere at room temperature affording N-phosphonacetyl-L-isoasparagine (1, PALI).
Figure 1.

Scheme for the synthesis of PALI. (a) di-tert-butyl dicarbonate, NaHCO3, H2O/THF (1:1), rt, overnight, (b) (i) Et3N, ethyl chloroformate, CH2Cl2/dioxane, (ii) 28% aqu.NH3, rt, 4 h, (c) CH2Cl2/TFA (1:1), rt, 4 h, (d) chloroacetic anhydride, pyridine, CH2Cl2, rt, 4 h, (e) triethyl phosphite, 150 °C, 8 h, (f) trimethyl silylbromide, acetonitrile, rt, overnight, (g) 10% Pd/C, H2, THF/EtOH, rt, overnight.
Binding of PALI to ATCase
Due to the cooperative binding of aspartate and severe substrate inhibition, kinetic experiments to obtain KI values for PALI were not performed with ATCase. Therefore, to measure the affinity of PALI for ATCase a pyrene-labeled version of the ATCase holoenzyme was used.15 The fluorescence intensity of this pyrene-labeled enzyme is related to the fraction of the active sites filled. Dissociation constants of 0.69 μM and 2.0 μM were calculated for PALA and PALI, respectively. A comparison of the standard free energies of PALA and PALI reveal a difference (|ΔGºPALA − ΔGºPALI|) in energy of 1.0 kcal/mol, where ΔGº = −RT ln (1/KD).
At a subsaturating concentration of Asp and saturating concentration of CP, addition of PALA dramatically increases the activity of ATCase. However, at higher concentrations of PALA the activity decreases towards zero. The increase in activity, at low PALA concentrations, results from the binding of PALA inducing the transition from the low-activity low-affinity T state to the high-activity high-affinity R state.4 Under identical conditions, PALI also activates ATCase. As seen in Figure 2, the extent of the activation induced by PALI is almost the same as PALA, furthermore, the concentration of PALI necessary to cause maximum activity is almost identical to that for PALA indicating that the affinity of the two inhibitors for the enzyme are similar.
Figure 2.
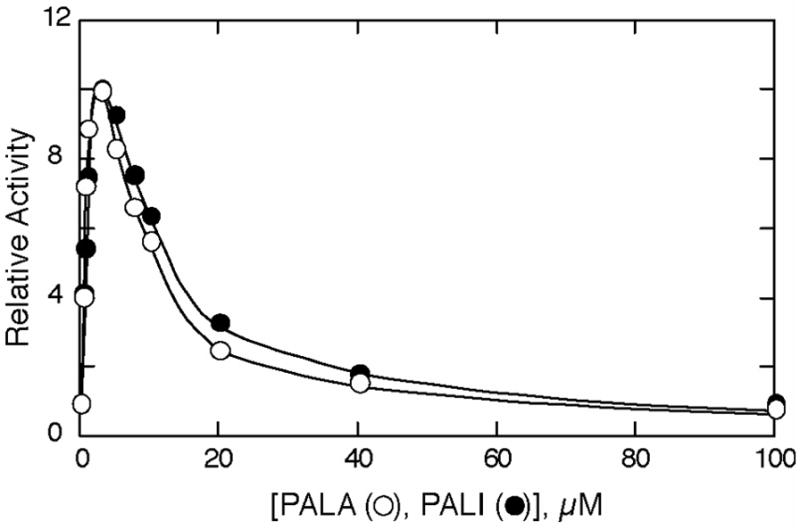
Influence of PALA (○) and PALI (●) on the active site of ATCase. Colorimetric assays were carried out at 25 °C in 50 mM Tris acetate buffer (pH 8.3) at saturating concentrations of carbamoyl phosphate (4.8 mM) and subsaturating concentrations of aspartic acid (2.4 mM). Relative activity represents the ratio of activity in the absence or presence of inhibitor to that in the absence of inhibitor.
Small-angle X-ray Scattering
The T and R structures of ATCase are easily distinguished by SAXS.16 In order to determine if the binding of PALI converts ATCase from the T to R state, SAXS was utilized. Shown in Figure 3 are the X-ray scattering patterns of ATCase in the absence of ligands and in the presence of 2 mM PALA and in the presence of 2 mM PALI. The addition of PALI caused almost the identical change in the scattering pattern as did PALA, signifying that PALI induces the allosteric transition to the R state, and the R-state structure of the enzyme in the presence of PALA or PALI are virtually identical.
Figure 3.

Small-angle X-ray scattering of wild-type ATCase. The SAXS pattern in the absence of ligand (thick line), in the presence of 2 mM PALA (thin line) and in the presence of 2 mM PALI (dotted line). The X-ray scattering experiments were carried out in 50 mM Tris acetate buffer, 2 mM 2-mercaptoethanol, pH 8.3.
Structure of the ATCase in the Presence of PALI
To better understand how PALI interacts with ATCase, the three-dimensional structure of the enzyme·PALI complex was determined by X-ray crystallography. Diffraction quality crystals of the enzyme·PALI complex grew to an average size of 0.3 × 0.3 × 0.2 mm within one week. The RPALI crystals diffracted to a maximal resolution of 2.3 Å in the P321 space group. The unit cell dimensions of these RPALI crystals (a = b = 120.32, c = 154.51) varied slightly from the unit cell dimensions of the RPALA crystals (a = b = 122.24, c = 156.36)17 in the same space group, see Table 1.
Table 1.
Data collection and Refinement Summary of the RPALI Structure
| Data collection | |
| Space group | P321 |
| Resolution (Å) | 30-2.3 (2.38-2.30) |
| Total no. of reflections | 435,756 |
| No. of unique reflections | 57,901 |
| Redundancy | 7.53 (7.50) |
| Completeness (%) | 99.9 (100.0) |
| Unit cell (Å) | a = b = 120.32, c = 154.51 |
| Angles (deg) | α = β = 90, γ = 120 |
| Average I/σ | 12.1 |
| Rmergea (%) | 10.5 |
| Refinement | |
| Resolution (Å) | 30.0-2.1 |
| R | 0.206 |
| Rfree | 0.250 |
| No. of waters | 477 |
| Average B factor (Å2) | 38.03 |
| RMS deviations | |
| Bonds (Å) | 0.006 |
| Angles (°) | 1.31 |
Values in parentheses are for the highest resolution shell
The quaternary conformation of the RPALI structure PALI was determined using the method of Stieglitz et al.,18 which measures the vertical separation between the centers of mass of the upper and lower catalytic trimers and compares this separation to those of known T- and R-state structures. The vertical separation between the upper and lower trimers of the RPALI structure was 55.40 Å, which compares to 45.40 Å for the Tapo structure and 56.10 Å for the RPALA structure. These results indicate that the structure of the enzyme in the presence of PALI is in the R-state conformation. The RMS deviation for both the catalytic and regulatory chains between the RPALI and RPALA structures was 0.59 Å and 0.82 Å for the α-carbon and side chain atoms, respectively.
Several domain motions occur during the T to R transition, most notable are the movements in the 80’s (residues 73–88) and 240’s (residues 230–245) loops. In the Tapo structure, the 80’s loop is positioned approximately 5 Å from the active site and is partially disordered. In the R state the 80’s loop from the adjacent catalytic chain moves approximately 5 Å into the active site, forming the catalytically competent active site.19 The structure of the 80’s loop in the RPALI structure is ordered and the electron density is well defined. In the T state the 240’s loop of C1 interacts with the 240’s loop of C4 and the R4 regulatory chain, forming C1-C4 and C1-R4 interactions. In the RPALI structure the C1-C4 and C1-R4 interactions that stabilize the T state are lost.
Active Site of the RPALI Structure
The active site of the RPALI structure is shown in Figure 4. The electron density around PALI is well defined and confirms the proper placement of the inhibitor in the active site. Lys84 and Ser80 from the C2 chain are in their R-state conformation interacting with PALI. The conformation of PALI is virtually identical in the C1 and C6 catalytic chains.
Figure 4.
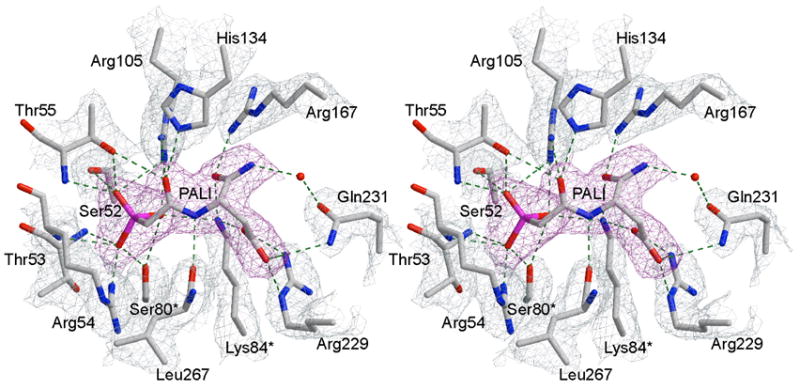
Stereoview of the active site with PALI bound. The 2Fo–Fc simulated annealing electron density map, contoured at 1.0 σ, with PALI (magenta) omitted from the map calculation. The refined positions of the active site residues are overlaid onto the 2FO-FC electron density map (sky blue), contoured at 1.2 σ. The hydrogen bonding interactions between the inhibitor and enzyme are shown as dotted lines.
The positioning of PALI is similar to that of PALA in the RPALA structure, see below. Residues that interact with PALI include Ser52, Thr53, Arg54, Thr55, Arg105, His134 (CP domain), Arg167, Arg229, and Gln231 (ASP domain) from the C1 catalytic chain, as well as to Ser80 and Lys84 from the adjacent C2 catalytic chain. Comparison of the C1 and C6 active sites show that the two sites are identical, with the exception of the guanidinium group of Arg54. Arg54NE is shifted 1.27 Å towards the inhibitor in C6 when compared to C1, and interacts with a phosphonate oxygen atom of PALI.
Comparison of the RPALI and RPALA active sites. Figure 5 shows an overlay of the RPALA and RPALI structures in the active site region. A list of the hydrogen bonding interactions and distances between the enzyme and the inhibitors, PALA and PALI, are summarized in Table 2. All residues that interact with PALA17 are also observed to interact with PALI. The binding of the PALI is very similar to that of PALA. The most noticeable difference occurs at the α-carboxylate where a hydrogen bonding interaction between the inhibitor and Arg167NE is lost in the RPALI structure. In addition, a water molecule, not observed in the RPALA structure, is coordinated to the nitrogen of the α-amide and Gln231OE1. The volumes of the RPALI and RPALA active sites were determined by CASTp,20 and found to be similar, 323.8 Å3 and 337.9 Å3, respectively.
Figure 5.
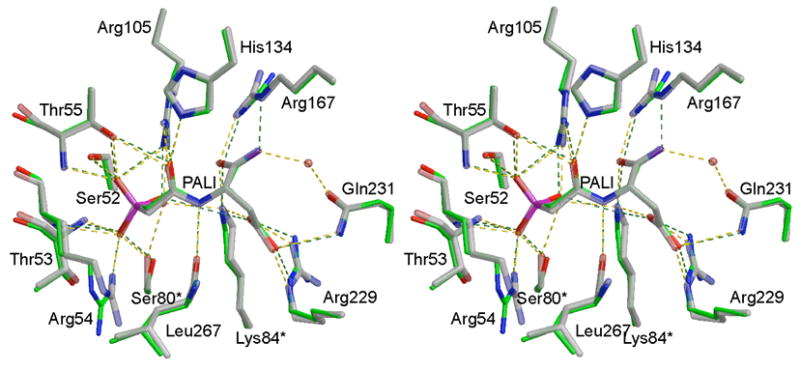
Comparison of the structures of (a) RPALA shown in green and (b) RPALI shown in grey. Notice the lack of a hydrogen bond between the α-amide of PALI and the guanadinium group of Arg167. The structure of the RPALA enzyme was overlaid onto the 37 RPALI structure using the program SEQUIOA.
Table 2.
Hydrogen bond distances between PALA/PALI and the active site residues of ATCase
| Residue | Atom | Inhibitor atom | PALA Distance (Å) | PALI distance (Å) |
|---|---|---|---|---|
| Ser52 | OG | OP3 | 2.52 | 2.69 |
| Thr53 | N | OP2 | 2.81 | 2.80 |
| Arg54 | N | OP2 | 3.08 | 2.95 |
| Arg54 | NH1 | OP2 | 3.21 | 2.36 |
| Thr55 | N | OP3 | 2.76 | 2.83 |
| Thr55 | OG1 | OP3 | 2.71 | 2.64 |
| Thr55 | OG1 | O1 | 3.14 | 2.97 |
| Arg105 | NH1 | OP3 | 3.28 | 3.25 |
| Arg105 | NH1 | OP1 | 2.58 | 2.77 |
| Arg105 | NH2 | O1 | 2.67 | 2.83 |
| His134 | NE2 | O1 | 2.51 | 2.77 |
| Arg167 | NE | O2 | 2.71 | — |
| Arg167 | NH2 | O3 | 3.00 | 2.80 |
| Arg229 | NE | O5 | 2.68 | 2.78 |
| Arg229 | NH2 | O4 | 2.89 | 2.91 |
| Arg229 | NH2 | O5 | — | 3.24 |
| Gln231 | NE2 | O5 | 3.11 | 3.22 |
| Leu267 | O | N2 | 3.06 | 2.68 |
| Ser80 | OG | OP2 | 2.91 | 2.65 |
| Ser80 | OG | OP1 | — | 3.08 |
| Lys84 | NZ | OP1 | 3.02 | 2.85 |
| Lys84 | NZ | O3 | 3.08 | 3.06 |
| Lys84 | NZ | O4 | 3.00 | 2.60 |
The interaction of PALA and PALI for their respective active sites was further investigated using the program X-Score.21 X-Score calculates the binding affinity of a ligand for a given enzyme of known structure. The scoring for the ligand-enzyme affinity is based on van der Waals interactions, hydrogen bonding, deformation effect and the hydrophobic effect. The scoring functions reveals that the van der Waals and hydrophobic interactions are greater in the PALA-enzyme complex then the PALI-enzyme complex. However, the hydrogen bonding contributions are greater in the enzyme-PALI complex. The scores, given in pKd units, for the enzyme-PALA and the enzyme-PALI complex are 6.11 and 6.02, respectively. The slightly lower affinity of PALI given by X-Score is consistent with the observed KD values determined by fluorescence.
Discussion
In order to reduce the charge on the known ATCase inhibitor, PALA, the α-carboxylate was converted into an amide. This modification was selected because site- specific mutagenesis experiments revealed that the removal of the side chain that interactions the α-carboxylate of PALA does not influence PALA binding. PALI binds remarkably similar to PALA. For example, upon the binding of PALA, or the natural substrates CP and Asp, the enzyme undergoes the T- to R-structural transition,13 which can be easily observed by small-angle X-ray scattering. The T to R transition brings the CP and ASP domains in close proximity and correctly orients catalytically important residues for catalysis.17 Small-angle X-ray scattering experiments showed that PALI can not only induce the T to R transition but also the structure observed in the presence of PALI was indistinguishable from that observed in the presence of PALA. The ability of PALI to induce the T to R transition was functionally verified by the activation of ATCase by low concentrations of PALI at subsaturating concentrations of Asp and saturating concentrations of CP. The X-ray structure of the enzyme·PALI complex also indicates the PALI induced the T to R transition. The vertical separation between the upper and lower catalytic trimers of 55.40 Å is almost identical to that observed for the RPALA structure (56.10 Å). Finally, analysis of the enzyme·PALI structure reveals that the repositioning of side chains upon the binding of PALI during the T to R transition is identical to that of PALA.
The structure of the ATCase in the presence of PALI was determined to investigate the exact mode of binding of the inhibitor to the enzyme. The active site of ATCase is highly electropositive in nature. PALA binds to the enzyme through a series of hydrogen bonding interactions, where every hydrogen bond donor/acceptor of PALA makes at least 1 hydrogen bonding interaction with the enzyme.17 PALI, with the α-carboxylate of PALA changed to an amide, loses a charged hydrogen bond between the α-carboxylate of PALI and the side chain of Arg167. The loss of a single hydrogen bond between a charged acceptor and an uncharged donor should account for approximately 4 kcal/mol of binding energy, whereas the loss of a non-charged hydrogen bond should account for approximately 0.5–1.5 kcal/mol of binding energy.22,23 Counting the hydrogen bonding interactions of the RPALI structure reveals 22 hydrogen bonds as opposed to the RPALA structure that contains 21 hydrogen bonds. The loss of the charged hydrogen bond to the α-carboxylate of PALI leads to a 2.7-fold reduction in the binding affinity.
A mutant version of ATCase, R167Q, has been characterized in our laboratory.24 Arg167 makes two hydrogen bonds with the α-carboxylate in PALA and the tetrahedral intermediate,17 neutralizing the negative charge on the carboxylate. The mutation of Arg167 to Gln results in an enzyme with reduced activity yet there is no alteration in the binding affinities for Asp or PALA. Upon mutation of Arg167 to glutamine one hydrogen bond is lost between the enzyme and the negatively charged α-carboxylate of PALA (or Asp). However, a charged-uncharged hydrogen bond at position 167 between the amide nitrogen and the carboxylic acid still exists. In the case of PALI the replacement of the negatively charged α-carboxylate with an amide leads to a loss of a charged hydrogen bond between the enzyme and PALI. As a result the affinity of PALI is decreased due to the loss of the charged hydrogen bond unlike the loss of the uncharged hydrogen bond in the case of R167Q.
If the difference in binding were solely related to the loss of a hydrogen bond between a charged hydrogen bond acceptor and an uncharged hydrogen bond donor, the ΔG between the two inhibitors should be approximately 4 kcal/mol. However the difference in ΔG between PALA and PALI is only 1.0 kcal/mol. In the RPALI structure the presence of two additional hydrogen bonds adds approximately 1.5 kcal/mol of binding energy, per hydrogen bond, to the enzyme-inhibitor complex. In addition, four hydrogen bonds in the RPALI are shorter than in the RPALA by at least 0.25 Å, potentially leading to stronger hydrogen bonding interactions. The increased hydrogen bonding compensates for the loss of the charged hydrogen bond between Arg167 and the α-carboxylate of PALA. In addition, the X-Score results support the conclusion that the hydrogen bonding interactions in the enzyme-PALI complex are greater than the PALA enzyme complex, resulting in a 2-fold loss of binding affinity of PALI for the active site of ATCase.
In this report we characterize the binding of a new potent inhibitor of ATCase, PALI, using fluorescence, SAXS and X-ray crystallography. Even though PALI has less charge than PALA, PALI still binds with high affinity. The binding of PALI induces the T to R transition of ATCase and is able to convert unoccupied active sites into their high-activity high-affinity functional state. The X-ray structure of the enzyme·PALI complex reveals that residues in the active site rearrange to form the catalytically active enzyme. These conformational changes afford high affinity binding for the inhibitor to the enzyme. PALI can induce the allosteric transition even though there is the loss of a charged hydrogen bonding interaction between the enzyme and inhibitor. The loss of this charge is not detrimental to the activity of PALI due to additional hydrogen bonding interactions between the enzyme-inhibitor complex. The kinetic characterization and crystal structure of the ATCase·PALI complex also provides detailed information regarding the importance of the α-carboxylate for the binding of both PALA and the substrate aspartate.
Experimental Procedures
Materials
L-aspartate, N-carbamoyl-L-aspartate, potassium dihydrogen phosphate, sodium azide and uracil were obtained from Sigma Chemical Co. (St. Louis, Missouri). Tris was purchased from ICN (Costa Mesa, California). Dialysis buttons (50 μL) and Aqua Sil were purchased from Hampton Research. Dialysis buttons were siliconized according to the supplier’s instructions. All commercially available starting materials and solvents were reagent grade or better and used without further purification. Thin-layer chromatography was performed on Merck silica gel 60 plates with fluorescent indicator and column chromatography was carried out on silica gel, purchased from Fisher Scientific.
Enzyme Preparation
Wild-type E. coli ATCase and the C47A/A241C mutant ATCase were expressed and purified as described previously.25 The C47A/A241C enzyme was labeled with pyrene as previously described.15 The concentration of the purified holoenzyme was determined by absorption at 280 nm with an extinction coefficient of 0.59 cm2/mg or by using the Bio-Rad version of Bradford’s dye binding assay26 using wild-type ATCase as the standard. The purity of the enzyme was determined using SDS-PAGE27 and non-denaturing PAGE.28,29
Preparation of β-Benzyl N-tert-butoxycarbonyl-L-aspartate (3).30
To a stirred solution of β-benzyl L-aspartate (2) (4.46 g, 20 mmol) and sodium bicarbonate (3.36 g, 40 mmol) in 60 mL water, di-tert-butyl dicarbonate (5.89 g, 27 mmol) in 60 mL THF was added at 0°C and the reaction mixture was stirred overnight at room temperature. After the completion of the reaction, THF was removed by distillation under reduced pressure and the residue extracted with ethyl acetate after acidifying the mixture using 10% citric acid. The organic layer was washed with brine, water and dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure to afford β-benzyl N-tert-butoxycarbonyl-L-aspartate (3) in quantitative yield. This crude material was used for the next reaction step without further purification. 1H NMR (400 MHz, dmso-d6) δ 1.36 (s, 9H), 2.59 (dd, J = 7.4 Hz, 15.0 Hz, 1H), 2.72 (dd, J = 6.0 Hz, 15.2 Hz, 1H), 4.05 (m, 1H), 5.05 (m, 2H), 6.46 (d, J = 6.8 Hz, 1H), 7.29–7.38 (m, 5H); 13C NMR (100 MHz, dmso-d6) δ 28.23, 37.74, 51.68, 65.23, 77.60, 127.52, 127.60, 128.13, 136.14, 154.70, 170.70, 172.45; ESI-MS (m/z) 346.0 [M + Na]+
Preparation of β-Benzyl N-tert-butoxycarbonyl-L-isoasparaginate (4).31
To a solution of β-benzyl N-tert-butoxycarbonyl-L-aspartate (6.5 g, 20 mmol) and triethylamine (2.06, 20.4 mmol) in a mixture of anhydrous of dichloromethane (100 mL) and dioxane (120 mL), ethyl chloroformate (2.4 g, 20.4 mmol) was added at −5 to −10 °C and the mixture was stirred at this temperature for 30 min. Then, 6 mL of a 28% aqueous ammonia solution was added to the reaction and the mixture stirred for 4 hours at room temperature. The reaction mixture was then poured into 100 mL dichloromethane and the mixture was washed with hydrochloric acid (0.5 N, 3 × 50 mL). The organic layer was separated, washed with water and concentrated under reduced pressure to give crude product. The crude product was further purified by precipitation from a mixture of ethyl acetate-hexane. Yield 5.56 g (86 %). 1H NMR (400 MHz, dmso-d6) δ 1.37 (s, 9H), 2.57 (dd, J = 8.6 Hz, 16.2 Hz, 1H), 2.75 (dd, J = 5.4 Hz, 15.8 Hz, 1H), 4.25–4.31 (m, 1H), 5.05–5.12 (m, 2H), 7.04 (d, J = 8.4 Hz, 1H), 7.10 (s, br, 1H), 7.27 (s, br, 1H), 7.31–7.37 (m, 5H); 13C NMR (100 MHz, DMSO-d6) δ 28.19, 36.40, 50.80, 65.49, 78.14, 93.80, 127.54, 127.71, 128.15, 135.87, 154.87, 169.95, 172.30; ESI-MS (m/z) 345.0 [M + Na]+
Preparation of β-Benzyl L-isoasparaginate (5).31
To a solution of β-benzyl N-tert-butoxycarbonyl-L-isoasparaginate (537 mg, 1.67 mmol) in 10 mL of dichloromethane, 10 mL of trifluoroacetic acid was added at 0 °C and the mixture was stirred for 4 hours at room temperature. The solvent and TFA were removed by distillation under reduced pressure and the residue obtained was triturated with anhydrous diethyl ether. The precipitate was dried under nitrogen to afford β-benzyl L-isoasparaginate (5) as the trifluoroacetic acid salt. Yield= 505 mg (90%). 1H NMR (400 MHz, dmso-d6) δ 2.89 (dd, J = 7.8 Hz, 17.4 Hz, 1H), 3.0 (dd, J = 4.4 Hz, 17.2 Hz, 1H), 4.05–4.08 (m, 1H), 5.12–5.19 (m, 2H), 7.34–7.40 (m, 5H), 7.65 (s, br, 1H), 7.87 (s, br, 1H), 8.2 (s, br, 3H); 13C NMR (100 MHz, dmso-d6) δ 35.22, 48.76, 66.24, 127.90, 128.00, 128.27, 135.45, 168.77, 168.92; ESI-MS (m/z) 245.09 [M + Na]+
Preparation of β-Benzyl N-chloroacetyl-L-isoasparaginate (6)
To a mixture of β-benzyl L-isoasparaginate trifluoroacetic acid salt (5.12 g, 15 mmol) and pyridine (6.05 mL, 75 mmol) in 125 mL anhydrous dichloromethane, chloroacetic anhydride (5.65g, 33 mmol) was added portion wise while keeping the temperature 0 °C. After the addition was complete, the reaction mixture was stirred for 4 hours at room temperature. Then the mixture was washed with water (2 × 50 mL), saturated sodium bicarbonate (3 × 50 mL) and again with water (1 × 50 mL). The organic layer was collected, dried over Na2SO4, and concentrated under reduced pressure to afford the crude product. This was purified by repeated precipitation using hexane from a saturated solution of crude mixture in dichloromethane. Yield = 3.6 g (80 %). 1H NMR (400 MHz, CDCl3) δ 2.73 (dd, J = 6.8 Hz, 17.2 Hz, 1H), 3.07 (dd, J = 4.0 Hz, 17.2 Hz, 1H), 4.02–4.10 (m, 2H), 4.81–4.86 (m, 1H), 5.17 (s, 2H), 5.51 (s, br, 1H), 6.45 (s, br, 1H), 7.33–7.40 (m, 5H), 7.85 (d, J = 6.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 35.87, 42.63, 49.40, 67.38, 128.43, 128.61, 128.72, 135.15, 166.34, 171.51, 171.64; ESI-MS (m/z) 320.9 [M + Na]+
Preparation of β-Benzyl N-(Diethoxyphosphinoyl)acetyl-L-isoasparaginate (7)
A mixture of β-benzyl N-chloroacetyl-L-isoasparaginate (3.6 g, 12 mmol) and triethyl phosphite (20 mL) were heated at 150 °C for 8 h and then excess triethyl phosphite was removed by distillation under reduced pressure to give crude product as a colorless viscous liquid. The pure product (7) was obtained by silica column chromatography using a mixture of ethyl acetate/methanol (9.5:0.5). Yield = 4.6 g (95 %). 1H NMR (400 MHz, CDCl3) δ 1.33 (t, J = 7.0 Hz, 6H), 2.76–3.02 (m, 3H), 3.11 (dd, J = 5.4 Hz, 17.4 Hz, 1H), 4.09–4.19 (m, 4H), 4.93–4.98 (m, 1H), 5.07–5.14 (m, 2H), 6.21 (s, br, 1H), 7.28–7.32 (m, 5H), 7.50 (s, br, 1H), 7.62 (d, J = 9.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 16.63, 35.74, 36.22 (d, JCP = 127 Hz), 49.30, 62.97, 63.75, 66.98, 128.26, 128.37, 128.61, 135.48, 164.22, 171.59, 173.22; 31P NMR (121 MHz, CDCl3) δ 22.77; ESI-MS (m/z) 423.13 [M + Na]+. Anal. (C17H25N2O7P), C, H, N.
Preparation of β-Benzyl N-phosphonacetyl-L-isoasparaginate (8)
To a solution of β-benzyl N-(diethoxyphosphinoyl)acetyl-L-isoasparaginate (400 mg, 1 mmol) in anhydrous acetonitrile (10 mL), trimethyl silylbromide was added (560 mL, 5.6 mmol) at 0 °C and then the mixture was stirred overnight at room temperature. The solvents were evaporated under vacuum and 5 mL of water was added. The mixture was stirred for 1 hour at room temperature, washed with dichloromethane and lyophilized to give pure product 8 in quantitative yield. 1H NMR (400 MHz, D2O) δ 2.62–2.78 (m, 2H), 2.85 (dd, J = 8.2 Hz, 16.6 Hz, 1H), 2.95 (dd, J = 5.1 Hz, 16.4 Hz, 1H), 4.72 (m, 1H), 5.15–5.37 (m, 2H), 7.36–7.40 (m, 5H); 31P NMR (121 MHz, D2O) δ 15.98; ESI-MS (m/z) 367.06 [M + Na]+
Preparation of N-phosphonacetyl-L-isoasparagine (1)
To a solution of β-benzyl N-phosphonacetyl-L-isoasparaginate (275 mg, 0.8 mmol) in THF/ethanol (25 mL/15 mL), add 10% Pd/C (132 mg) and mixture was stirred overnight under a hydrogen atmosphere. After the completion of reaction, Pd was removed by filtration and the filtrate was concentrated under reduced pressure to give a viscous colorless liquid. This was dissolved in 10 mL water, washed with dichloromethane and the aqueous phase was lyophilized to give pure product 1 as a hygroscopic solid. Yield = 186 mg (92%). 1H NMR (400 MHz, D2O) δ 2.78–2.99 (m, 4H), 4.72–4.79 (m, 1H); 31P NMR (121 MHz, D2O) δ 17.95; ESI-MS (m/z) 253.02 [M−H]−. Anal. (C6H11N2O7P·H2O·0.3 EtOH), C, H, N.
Methods
Synthesis
All moisture-sensitive reactions were performed under a nitrogen atmosphere with oven-dried glassware.
Characterization
1H and 13C NMR spectra were recorded on a Varian 400 or 300 spectrometer. Proton chemical shifts are reported in ppm (δ) relative to internal tetramethylsilane (TMS, δ 0.0) or with the solvent reference relative to TMS employed as the internal standard (CDCl3, δ 7.26 ppm; DMSO-d6, δ 2.50). Data reported as follows: chemical shift (multiplicity [singlet (s), doublet (d), triplet (t) and multiplet (m)], coupling constants [Hz], integration). 13C NMR spectra were recorded on a Varian 400 (100 MHz) spectrometer with complete proton decoupling. The chemical shifts were reported relative to CDCl3 (77.0 ppm) or DMSO-d6 (39.52 ppm) for solution in CDCl3 and DMSO-d6, respectively. 31P NMR spectra were recorded on a Varian 300 (121 MHz) spectrometer using 85% H3PO4 as an external standard. Mass spectra were obtained at the Mass Spectrometry Facilities of Boston College.
Fluorescence Measurements
Fluorescence emission spectra were recorded at 20 ± 1 °C in 50 mM Tris acetate buffer, pH 8.3, on a Jasco FP-6300 spectrofluorometer using pyrene-labeled ATCase. The excitation wavelength was 338 nm; emission spectra were collected between 360–560 nm; the excitation and emission bandwidths were set to 5 nm. To approximate the degree of quaternary structure change for the enzyme, the intensity of the fluorescence signal FT at 377 nm was measured as the population of T-state enzyme.15 Saturation curves were determined by the incremental addition of PALA or PALI to the pyrene-labeled ATCase (1 μM enzyme) until there was no further change in fluorescence. The data were normalized by dividing each data point by FT in the absence of ligand.
Small-angle X-ray Scattering
Small-angle X-ray Scattering curves were collected for the wild-type holoenzyme in the absence of ligands and in the presence of PALA or PALI, according to Stieglitz et. al.32 Solution X-ray scattering experiments were conducted at the Beam Line 4–2, at the Stanford Synchrotron Radiation Laboratory, Menlo Park, CA.
Crystallization and Freezing of Crystals
The purified enzyme was crystallized by microdialysis, using 50 μL wells. The enzyme solution, at 18 mg/mL, was dialyzed against the crystallization buffer, which consisted of 50 mM maleic acid, 1 mM PALI, and 3 mM sodium azide at pH 5.7. Crystals formed within three days with average dimensions of 0.3 × 0.3 × 0.2 mm. Crystals were soaked in a solution of 30% 2-methyl-2,4-pentanediol in crystallization buffer for approximately 1 minute prior to freezing in liquid nitrogen.
X-ray Data Collection and Processing
The diffraction data were collected using a Rigaku/MSC R-axis IV++ detector while X-rays were generated using a Rigaku/MSC RU-200 rotating-anode generator operating at 50 kV and 100 mA at the Boston College Crystallographic Facility. The diffraction data, collected to 2.3 Å, were integrated, scaled and averaged using the program d*TREK (Rigaku/MSC).33
Structural Refinement
The data were refined using the coordinates of wild-type E. coli ATCase complexed with PALA (PDB entry 1DO9), as the initial model.17 Prior to refinement all waters and ligands were removed. The structure was refined using CNS,34 and refinement statistics are summarized in Table 1. Initially rigid body refinement was carried out followed by simulated annealing. After rigid body, simulated annealing, energy minimization, and B-factor refinement initial maps were analyzed. Trouble areas of the structure, the N-termini of the regulatory chains (residues 1–11), the 50’s loop of the regulatory chains, and residues 65–72 of the R6 regulatory chain, were manually rebuilt using XtalView.35 The N-termini of the regulatory chains were highly disordered even after several rounds of rebuilding, therefore residues 1–9 of the regulatory chains were omitted from the structure. After rebuilding was complete PALI was fit into the Fo-Fc electron density. Placement of the PALI was verified using simulated annealing omit maps.
Waters were added to the structure using CNS and XtalView on the basis of the Fo-Fc electron density maps at or above the 2.8 σ level. Waters were checked and retained only when they were able to form hydrogen bonds. The model was checked for errors using PROCHECK.36 The details of the data processing and refinement are given in Table 1.
Supplementary Material
Acknowledgments
This work was supported by Grant GM26237 from the National Institutes of Health. The Stanford Synchrotron Radiation Laboratory (SSRL) is operated by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Biology Resource is supported by the National Institutes of Health, National Center for Research Resources (P41RR01209), and by the Department of Energy, Office of Biological and Environmental Research.
Abbreviations
- ATCase
Escherichia coli aspartate transcarbamoylase
- PALA
N-phosphonacetyl-L-aspartate
- PALI
N-phosphonacetyl-L-isoasparagine or N-phosphonacetyl-L-α-asparagine
- CP
carbamoyl phosphate
- C1/C6
the two catalytic chains of ATCase in the asymmetric unit of the crystal
- R1/R6
the two regulatory chains of ATCase in the asymmetric unit of the crystal
- RPALA
structure of the wild-type ATCase complexed with PALA (PDB entry 1D09)
- RPALI
structure of the wild-type ATCase complexed with PALI
- Tapo
structure of the wild-type ATCase in the T state (PDB entry 1ZA1)
- C chain
catalytic chain
- R chain
regulatory chain
- SAXS
small-angle X-ray scattering
- R167Q
the mutant ATCase in which Arg167 of the catalytic chain has been replaced by Gln
Footnotes
Coordinates for the aspartate transcarbamoylase complex with N-phosphonacetyl-L-isoasparagine has been deposited in the Protein Data Bank (entry 2H3E).
References
- 1.Hsuanyu Y, Wedler FC. Kinetic mechanism of native Escherichia coli aspartate transcarbamylase. Arch Biochem Biophys. 1987;259:316–330. doi: 10.1016/0003-9861(87)90498-x. [DOI] [PubMed] [Google Scholar]
- 2.Gerhart JC, Pardee AB. Enzymology of control by feedback inhibition. J Biol Chem. 1962;237:891–896. [PubMed] [Google Scholar]
- 3.Wild JR, Loughrey-Chen SJ, Corder TS. In the presence of CTP, UTP becomes an allosteric inhibitor of aspartate transcarbamylase. Proc Natl Acad Sci USA. 1989;86:46–50. doi: 10.1073/pnas.86.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins KD, Stark GR. Aspartate transcarbamylase: Interaction with the transition state analogue N-(phosphonacetyl)-L-aspartate. J Biol Chem. 1971;246:6599–6605. [PubMed] [Google Scholar]
- 5.Yoshida T, Stark GR, Hoogenraad J. Inhibition by N-(phosphonacetyl)-L-aspartate of aspartate transcarbamylase activity and drug-induced cell proliferation in mice. J Biol Chem. 1974;249:6951–6955. [PubMed] [Google Scholar]
- 6.Tsuboi KK, Edmunds HN, Linda K. Selective inhibition of pyrimidine biosynthesis and effect on proliferative growth of colonic cancer cells. Cancer Res. 1977;37:3080–3087. [PubMed] [Google Scholar]
- 7.Johnson RK, Inouye T, Goldin A, Stark GR. Antitumor activity of N-(phosphonacetyl)-L-aspartic acid, a transition-state inhibitor of aspartate transcarbamylase. Cancer Res. 1976;36:2720–2725. [PubMed] [Google Scholar]
- 8.Pfund E, Lequeux T, Masson S, Vazeux M, Cordi A, Pierre A, Serre V, Herve G. Efficient synthesis of fluorothiosparfosic acid analogues with potential antitumoral activity. Bioorg Med Chem. 2005;13:4921–4928. doi: 10.1016/j.bmc.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 9.Lindell SD, Turner RM. Synthesis of potential inhibitors of the enzyme aspartate transcarbamoylase. Tetrahedron Let. 1990;31:5381–5384. [Google Scholar]
- 10.Kim J-S, Heath TD. Antitumor efficacy of liposomal N-(phosphonacetyl)-L-aspartic acid in C-26 tumor bearing Balb/c mice. Yakche Hakhoechi. 2000;30:39–45. [Google Scholar]
- 11.Kim J-S, Heath TD. Therapeutic potency of N-(phosphonoacetyl)-L-aspartic acid in liposome in established tumor bearing mice. Yakche Hakhoechi. 2000;30:127–131. [Google Scholar]
- 12.Ke H-M, Lipscomb WN, Cho Y, Honzatko RB. Complex of N-phosphonacetyl-L-aspartate with aspartate carbamoyltransferase: X-ray refinement, analysis of conformational changes and catalytic and allosteric mechanisms. J Mol Biol. 1988;204:725–747. doi: 10.1016/0022-2836(88)90365-8. [DOI] [PubMed] [Google Scholar]
- 13.Fetler L, Tauc P, Vachette P. Carbamyl phosphate modifies the T quaternary structure of aspartate transcarbamylase, thereby facilitating the structural transition associated with cooperativity. J Appl Cryst. 1997;30:781–786. [Google Scholar]
- 14.Wang J, Stieglitz KA, Cardia JP, Kantrowitz ER. Structural basis for ordered substrate binding and cooperativity in aspartate transcarbamoylase. Proc Natl Acad Sci U S A. 2005;102:8881–8886. doi: 10.1073/pnas.0503742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West JM, Tsuruta H, Kantrowitz ER. A Fluorescent Probe-Labeled Aspartate Transcarbamoylase that Monitors the Allosteric Conformational State. J Biol Chem. 2004;279:945–951. doi: 10.1074/jbc.M304018200. [DOI] [PubMed] [Google Scholar]
- 16.Hervé G, Moody MF, Tauc P, Vachette P, Jones PT. Quaternary structure changes in aspartate transcarbamylase studied by X-ray solution scattering; signal transmission following effector binding. J Mol Biol. 1985;185:189–199. doi: 10.1016/0022-2836(85)90190-1. [DOI] [PubMed] [Google Scholar]
- 17.Jin L, Stec B, Lipscomb WN, Kantrowitz ER. Insights into the mechanism of catalysis and heterotropic regulation of E. coli aspartate transcarbamoylase based upon a structure of enzyme complexed with the bisubstrate analog N-phosphonacetyl-L-aspartate at 2.1 Å. Proteins: Struct Funct Genet. 1999;37:729–742. [PubMed] [Google Scholar]
- 18.Stieglitz K, Stec B, Baker DP, Kantrowitz ER. Monitoring the transition from the T to the R state in E. coli aspartate transcarbamoylase by X-ray crystallography: Crystal structures of the E50A mutant in four distinct allosteric states. J Mol Biol. 2004;341:853–868. doi: 10.1016/j.jmb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Krause KL, Voltz KW, Lipscomb WN. Structure at 2.9-Å resolution of aspartate carbamoyltransferase complexed with the bisubstrate analogue N-(phosphonacetyl)-L-aspartate. Proc Natl Acad Sci USA. 1985;82:1643–1647. doi: 10.1073/pnas.82.6.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 22.Street IP, Armstrong CR, Withers SG. Hydrogen bonding and specificity. Fluorodeoxy sugars as probes of hydrogen bonding in the glycogen phosphorylase-glucose complex. Biochemistry. 1986;25:6021–6027. doi: 10.1021/bi00368a028. [DOI] [PubMed] [Google Scholar]
- 23.Fersht A. The hydrogen bonding in molecular recognition. TIBS. 1987;12:301–304. [Google Scholar]
- 24.Stebbins JW, Zhang Y, Kantrowitz ER. The importance of residues Arg-167 and Gln-231 in both the allosteric and catalytic mechanisms of Escherichia coli aspartate transcarbamylase. Biochemistry. 1990;29:3821–3827. doi: 10.1021/bi00468a003. [DOI] [PubMed] [Google Scholar]
- 25.Nowlan SF, Kantrowitz ER. Superproduction and rapid purification of E. coli aspartate transcarbamoylase and its catalytic subunit under extreme derepression of the pyrimidine pathway. J Biol Chem. 1985;260:14712–14716. [PubMed] [Google Scholar]
- 26.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 28.Davis BJ. Disc electrophoresis-II Method and application to human serum proteins. Ann NY Acad Sci. 1964;121:404–427. doi: 10.1111/j.1749-6632.1964.tb14213.x. [DOI] [PubMed] [Google Scholar]
- 29.Ornstein L. Disc Electrophoresis. I. Background And Theory. Ann NY Acad Sci. 1964;121:321–349. doi: 10.1111/j.1749-6632.1964.tb14207.x. [DOI] [PubMed] [Google Scholar]
- 30.Asano Y, Umezaki M, Li Y-F, Tsubota S, Lubbehusen TL. Isolation of microorganisms which utilize acidic D-amino acid oligomers. J Molec Catal B: Enzymatic. 2001;12:53–59. [Google Scholar]
- 31.Ariyoshi Y, Shiba T, Kaneko T. Synthetic studies of bacitracin. II. Synthesis of a cyclic dodecapeptide, an analogue of bacitracin A. Bull Chem Soc Jpn. 1967;40:2648–2654. doi: 10.1246/bcsj.40.2648. [DOI] [PubMed] [Google Scholar]
- 32.Stieglitz KA, Dusinberre KJ, Cardia JP, Tsuruta H, Kantrowitz ER. Structure of the E. coli Aspartate Transcarbamoylase Trapped in the Middle of the Catalytic Cycle. J Mol Biol. 2005;352:478–486. doi: 10.1016/j.jmb.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 33.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Cryst. 1999;D55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 37.Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, Tainer JA. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol. 1999;288:427–439. doi: 10.1006/jmbi.1999.2697. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.