

Abstract
Greater than 40% of breast cancer patients treated with tamoxifen exhibit de novo or acquired tumor resistance. Recent clinical evidence indicates that loss of expression of HER4 is an independent marker for tamoxifen resistance. In direct corroboration with clinical observations, suppression of HER4 expression in the tamoxifen sensitive MCF-7 and T47D breast tumor cell lines resulted in resistance to tamoxifen induced apoptosis. Furthermore, HER4 expression was lost in three independent MCF-7 models of acquired tamoxifen resistance. The HER4 intracellular domain (4ICD) is an independently signaling nuclear protein which functions as a potent ERα coactivator. In addition, mitochondrial 4ICD functions as a proapoptotic BH3-only protein. Tamoxifen disrupts an estrogen driven interaction between ERα and 4ICD while promoting mitochondrial accumulation of the 4ICD BH3-only protein. BCL-2 inhibition of tamoxifen induced apoptosis and tamoxifen activation of BAK independent of BAX further supports a role for 4ICD during tamoxifen induced apoptosis. Finally, reintroduction of HER4, but not HER4 with a mutated BH3 domain, restores tamoxifen sensitivity to tamoxifen resistant TamR cells in a xenograph model. Clinically, breast cancer patients with tumor expression of nuclear 4ICD responded to tamoxifen therapy with no clinical failures reported after 14 years of follow-up, whereas 20% patients lacking nuclear 4ICD expression succumbed to their disease within 10 years of diagnosis. Our identification of the HER4/4ICD BH3-only protein as a critical mediator of tamoxifen action provides a clinically important role for 4ICD in human cancer and reveals a potential tumor marker to predict patient response to tamoxifen therapy.
Keywords: EGFR-family, Endocrine Therapy, BCL-2 family, Estrogen Receptor, Coactivators, Breast Cancer
INTRODUCTION
The selective estrogen receptor (ERα) modulator tamoxifen has been used as a single agent for the treatment of ERα(+) breast cancers since 1971. Currently, tamoxifen is the most commonly prescribed antiestrogen therapy and is uniquely effective in the treatment of premenopausal woman with ERα(+) breast tumors. Unfortunately, de novo and acquired tumor resistance to tamoxifen are serious clinical problems. In fact, nearly 30% of patients with ERα(+) breast tumors fail to respond to tamoxifen and up to 40% of initial responders relapse and succumb to their disease 1-3. Because the exact molecular basis for the mechanism of tamoxifen action remains unclear, clinicians lack reliable clinicopathological indicators to identify patients likely to fail tamoxifen therapy.
Elegant structural studies have demonstrated that tamoxifen binding to ERα not only results in competitive inhibition of estrogen binding but tamoxifen also alters ERα structure thereby occluding coactivator interactions with ERα essential for estrogen stimulated tumor cell proliferation 4. As predicted by this mechanism of action, tamoxifen treatment of ERα(+) tumors results in cytostatic effects reducing breast tumor cell proliferation in part by arresting cell cycle progression 5.
Less clear however is a mechanism to explain the clinical observation that tumors from patients undergoing tamoxifen treatment exhibit increased levels of apoptosis 6, 7. In addition, tamoxifen induced tumor regression observed in multiple preclinical xenograph models cannot be explained by cytostatic effects alone suggesting a role for apoptosis in these models as well 7. In multiple experimental systems tamoxifen induces cytochrome c release from mitochondria 8, 9, the essential and committed step of the mitochondrial or intrinsic apoptotic pathway. Although tamoxifen alters the expression of multiple effectors of the intrinsic apoptotic cascade consistent with increased cell-killing, including suppression of anti-apoptotic BCL-2 and upregulation of pro-apoptotic BAX 10-12, the contribution of these alterations to tamoxifen activity in breast tumors requires clinical verification.
Recent clinical studies have revealed that tumor expression of the receptor tyrosine kinase HER4/ERBB4 (referred to here as HER4) improves the overall survival of breast cancer patients with ERα(+) tumors 13-15 raising the possibility that HER4 influences patient response to endocrine therapy. HER4 is a unique member of the EGFR-family and undergoes proteolytic processing at the cell surface to release a HER4 intracellular domain (4ICD) 16 that independently regulates multiple divergent activities in breast tumor cells 17-19. For example, nuclear 4ICD functions as a potent ERα coactivator, directly interacting with ligand associated ERα at promoters of estrogen response genes and contributing to estrogen stimulated proliferation of breast tumor cells 19. In non-malignant breast epithelium nuclear 4ICD regulates differentiation and lactation in part through transcriptional coactivation of the mammary differentiation factor, STAT5A 20-24.
When excluded from the nucleus, however, cytosolic 4ICD accumulates within mitochondria 17, 18 and triggers breast tumor cell apoptosis through the activity of an intrinsic cell-killing BCL-2 homology 3 (BH3) domain 17. This novel activity for a receptor tyrosine kinase has been confirmed clinically where we have shown that cytosolic 4ICD but not membrane localized HER4 is strongly associated with increased breast tumor apoptosis 17. Importantly, HER4 regulation of gene expression and apoptosis are mediated by 4ICD released from the cell surface following proteolytic processing of HER4 17-19, 21, 24. The molecular mechanisms regulating these divergent 4ICD activities are poorly understood. Here we show for the first time that 4ICD is an important effector of tamoxifen induced apoptosis of breast tumor cells. Our results are consistent with a mechanism of tamoxifen action involving tamoxifen disruption of the growth promoting 4ICD/ERα coactivator complex thereby unleashing the cell-killing activity of an untethered 4ICD BH3-only protein.
MATERIALS AND METHODS
Plasmids
The HER4 constructs used in this study were all derived from the HER4 JM-a/Cyt1 isoform. The HER4 expression plasmids pHER4-EGFP and pHER4muBH3-EGFP have been described previously 17, 24 The plasmid pERα-flag expressing a flag tagged version of human ERα was provided by Dr Brian Rowen (Tulane University). The BCL-2 expression plasmid pcDNA-BCL2 was provided by the late Dr Stanley Korsmeyer (Harvard University).
To generate a plasmid expressing the HER2 oncogenic isoform HER2Δ16 25 the region of HER2 harboring an exon 16 deletion was amplified by RT-PCR from BT474 RNA and subcloned into pLXSN-HER2 26 harboring wild-type human HER2 to generate the clone pLXSN-HER2Δ16 using standard procedures. The exon 16 deletion was confirmed by sequencing. The plasmids pcDNA3-HER2 and pcDNA3-HER2Δ16 were generated by subcloning the 4.5 kb Hind III fragment containing the HER2 ORF from pLXSN-HER2 or pLXSN-HER2Δ16, respectively, into the same sites of pcDNA3 (Invitrogen).
Cell Lines
The T47D and MCF-7 human breast cancer cell lines were purchased from the American Type Cell Culture and maintained according to the manufacturer’s recommendations. The tamoxifen resistant MCF-7 variants TamR and LCC2 have been described previously 27, 28. The LCC2 cell line provided by Dr. Robert Clarke (Georgetown University) and maintained in Modified Eagle’s Medium with 10% fetal bovine serum. The MCF-7/HER2Δ16 cell line was obtained by stably transfecting MCF-7 cells with pcDNA3-HER2Δ16 (Invitrogen) using Fugene6 (Roche). Individual transfected colonies were selected in Geneticin (Invitrogen) and isolated using cloning cylinders. The TamR/Vector, TamR/HER4, and TamR/muBH3 cell lines were generated by stably transfecting the TamR cell line with pEGFPN3 (Clontech), pHER4-EGFP 24, or pHER4muBH3-EGFP 17, respectively. Individual transfected cell colonies were selected in kanamycin and isolated using cloning cylinders.
Suppression of HER4 expression
To suppress expression of endogenous HER4, T47D or MCF-7 cells were transfected with erbB-4/HER-4 siRNA SMARTpool using siIMPORTER transfection reagent (Upstate Biotechnology) according to the manufacturer’s instructions. Cells similarly transfected with Nonspecific Negative Control Pool (Upstate Biotechnology) were analyzed as RNAi controls.
Assays for apoptosis
Apoptosis was quantitated visually using a Nikon fluorescent microscope to determine the percentage of cells with condensed chromatin following DAPI staining. All samples were prepared in duplicate and each experiment was repeated at least three times. Significant differences between data sets was determined using the paired student t-test. Apoptosis in histological sections from paraffin embedded xenograph tumors was determined using the ApopTag Plus Peroxidase In Situ Apoptosis Detection Kit (Chemicon International) exactly as described by the manufacturer. Data is represented as the mean percentage of TUNEL positive cells and standard error from at least three individual tumors from each treatment group.
Immunoprecipitation and western blot analysis
The HEK-293T cells were maintained in phenol red-free DMEM containing 5% charcoal-stripped FBS during the entire immunoprecipitation experiment. Co-immunoprecipitation of ERα and 4ICD was performed by transfecting 5 × 105 HEK 293T cells cultured in 100 mm dishes with pERα-flag and/or pHER4-EGFP using Lipofectamine with Plus reagent (Invitrogen) exactly as described by the manufacturer. At 24 hrs post-transfection cells were treated with 100 pM 17-β-estradiol alone or in combination with 5 mM tamoxifen for 1 hour. Cell lysates were prepared and 1 mg of lysate was immunoprecipitated using α-Flag Agarose (Sigma) or rabbit α-HER4 (Cell Signaling) as described previously 29.
Immunoprecipitates and total cell lysates were analyzed by western blot as described elsewhere 29 with primary antibodies HER4 (Cell Signaling), Flag M2 (Sigma), α-tubulin (Upstate Biotechnology), TOM40 (Santa Cruz Biotechnology), BAK (Upstate Biotechnology), and BAX (Upstate Biotechnology).
Analysis of isolated mitochondria for 4ICD localization and BAK or BAX dimerization
T47D cells were cultured in phenol red-free RPMI containing 5% charcoal-stripped FBS for two days and then stimulated for 1 hr with 50 ng/ml of heregulin-β1 (HRG; R&D Systems), 12 hrs with 1 μM staurosporine (STS; Sigma), or 1 hr with 100 pM 17-β-estradiol alone or in combination with 5 mM tamoxifen,. Mitochondria were isolated and analyzed by western blot for 4ICD localization or for BAK and BAX dimerized activation as described elsewhere 17.
Tumor formation in nude mice
Four to five week old NU/NU immune compromised female mice (Charles-River Laboratories) were implanted subcutaneously in the scapular region with a 60 day release 0.72 mg estradiol pellet (Innovative Research of America) using a precision trochar. Seven days later the mice were injected subcutaneously at two rear flank sites with each injection site containing 5 × 106 cells in 100 μl of sterile PBS mixed with 100 μl of Matrigel Basement Membrane Matrix (BD Biosciences). Tumors were allowed to develop for 15 days at which time mice were randomized and implanted with a 60 day release 5 mg tamoxifen pellet (Innovative Research of America) or a 60 day release 5 mg placebo (Innovative Research of America). All mice were anesthetized using Isofluorane/Oxigene prior to surgery. Tumor size was measured every three days using a digital caliper and tumor volume was calculated. Statistical analysis of the data set was performed using the Kruskal-Wallis Test 30.
Immunohistochemical analysis of xenograph tumors
Two hours prior to sacrifice all mice were injected IP with 10 μl/g body weight of BrdU Cell Labeling Reagent (Amersham). Tumors were excised, fixed overnight in 4% paraformaldehyde, and embedded in paraffin using standard histological procedures. Immunohistochemical analysis of proliferating cells by BrdU incorporation and apoptosis by TUNEL assay have been described in detail elsewhere 17, 31.
Patient population and HER4 immunohistochemistry
A total of 791 archived formalin fixed paraffin-embedded breast tumors from patients diagnosed at Massachusetts General Hospital (Boston, MA) between 1976 and 1983 were used for this study. Follow-up for a mean of 15.6 years was used to determine patient outcome following surgery and adjuvant intervention. Additional clinical and pathological details of this cohort have been described elsewhere 32.
HER4 immunohistochemistry was performed as described previously 17 using HER4 Ab-4 (Neomarkers, Clone HFR-1) directed against the carboxyl terminus of human HER4 at a concentration of 4.0 μg/ml. A tumor was considered positive for nuclear 4ICD if greater than 10% of tumor nuclei exhibited positive immunohistochemical staining. Associations between nuclear 4ICD and patient survival were determined using the Fisher’s exact test.
RESULTS
HER4 expression regulates tamoxifen induced apoptosis of ERα(+) breast tumor cells
Several recent clinical studies indicate that HER4 expression improves the prognosis of patients with ERα(+) breast tumors 13-15. Because the majority of these patients would have received endocrine therapy as part of their therapeutic regimen with tamoxifen being the most commonly prescribed endocrine therapy, we tested the possibility that HER4 regulates tamoxifen response of ERα(+) breast tumor cells. To this end, we examined the impact of tamoxifen cell-killing on the ERα(+) and HER4(+) MCF-7 and T47D breast cancer cell lines following RNA interference (RNAi) mediated knockdown of HER4 expression. Although tamoxifen has cytostatic effects, consistent with other reports we observed a dramatic increase in both MCF-7 and T47D cell-killing following treatment with physiological levels of tamoxifen (Fig. 1A). Interestingly, suppression of HER4 expression in each cell line by RNAi pre-treatment dramatically inhibited the ability of tamoxifen to induce apoptosis after 24 hrs and completely abolished tamoxifen cell-killing activity after 48 hrs (Fig. 1A). These results implicate HER4 as an important regulator of tamoxifen induced apoptosis of ERα(+) breast tumor cells and provide a molecular explanation for the improved prognosis of patients with ERα(+) tumors that also express HER4 13-15.
Figure 1.

HER4 expression regulates tamoxifen induced apoptosis of breast tumor cells. A, Suppression of HER4 expression by RNAi abolishes tamoxifen (5 μM) induced apoptosis of T47D and MCF-7 ERα(+) breast tumor cell lines. Cell lines were cultured in phenol red-free media containing 5% charcoal-stripped FBS 24 hrs prior to treatment with RNAi. Twelve hrs after RNAi treatment cells were left untreated or treated with 100 pM 17-β-estradiol (E2) alone or in combination with 5 μM 4-hydroxytamoxifen (TAM) for 24 and 48 hrs. Apoptosis was determined visually in a Nikon fluorescent microscope by staining the cells with DAPI and determining the percentage of cells with condensed nuclei (mean +/- SE of at least three experiments). Asterisks indicate significant differences in each data set as determined by student t-test. B, HER4 expression is suppressed in tamoxifen resistant MCF-7 cell variants. Western blot analysis of HER4 expression in cell lysates prepared from the parental MCF-7 cell line and three tamoxifen resistant variants TamR, LCC2, and HER2Δ16 each developed in an independent laboratory. Analysis of α-tubulin expression is included as a loading control. C, MCF-7 variants lacking HER4 expression are resistant to tamoxifen induced apoptosis. Each indicated cell line was cultured for 48 hrs in phenol red-free MEM containing 5% charcoal-stripped FBS and then treated for 24 hrs with 100 pM 17-β-estradiol (Estrogen) alone or in combination with 5 μM 4-hydroxytamoxifen (Tamoxifen). Apoptosis was determined visually in a Nikon fluorescent microscope by staining the cells with DAPI and determining the percentage of cells with condensed nuclei.
Our results coupled with clinical data suggest that loss of HER4 expression may contribute to tamoxifen refractory breast cancer. To test this hypothesis we examined HER4 expression in three established preclinical cellular models of tamoxifen resistance. The tamoxifen resistant LCC2 28 and TamR 27 cell lines were generated in independent laboratories by continuous exposure of MCF-7 cells to tamoxifen. The tamoxifen resistant HER2Δ16 cell line was derived in our laboratory by overexpressing an oncogenic isoform of HER2 in the MCF-7 cell line. When compared to parental MCF-7 cells HER4 expression was abolished in each tamoxifen resistant cell line examined (Fig. 1B) and as predicted each cell line was resistant to tamoxifen induced apoptosis (Fig. 1C). Taken together our results strongly implicate HER4 as an important mediator of tamoxifen induced cell-killing of breast tumor cells. By extension, loss of HER4 expression may represent a clinically important mechanism contributing to tamoxifen refractory breast tumors.
Tamoxifen disrupts the ERα/4ICD transcriptional coactivator complex
We next determined the molecular mechanism underlying the ability of HER4 to regulate tamoxifen response of breast tumor cells. HER4 is a unique multifunctional member of the EGFR-family that undergoes proteolytic processing at the cell surface to release the independently signaling 4ICD. We have shown that 4ICD is a potent ERα coactivator that directly interacts with ERα 19. Tamoxifen functions, in part, by altering ERα structure thereby occluding interactions between ERα and coactivators. We therefore determined if tamoxifen disrupts the interaction between ERα and the 4ICD coactivator. Consistent with our published results 19, 4ICD and ERα were coimmunoprecipitated from transfected HEK 293T cells and the amount of 4ICD/ERα coimmunoprecipitated complexes was augmented by the addition of estrogen (Fig. 2A, compare lanes 10 and 11). Tamoxifen treatment, however, disrupted immunoprecipitable 4ICD/ERα complexes (Fig. 2A, compare lanes 11 and 12). These results provide further evidence that 4ICD functions as a canonical ERα coactivator whose interaction with ERα is disrupted by tamoxifen.
Figure 2.
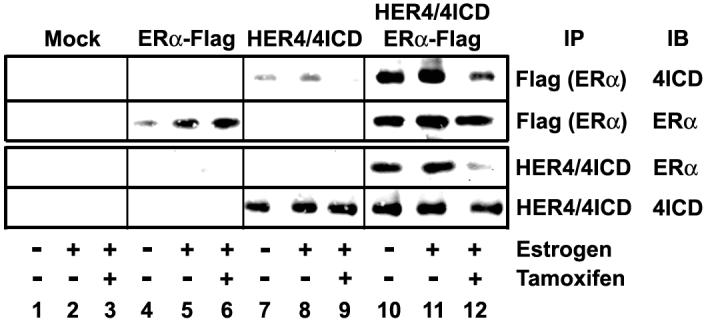
Tamoxifen disrupts the ERα/4ICD coactivator complex. HEK 293T cells were cultured in phenol red-free DMEM containing 5% charcoal-stripped FBS for 24 hrs then transfected with ERα-flag and/or HER4 expression plasmids. At 24 hrs post-transfection cells were left untreated or treated with 100 pM 17-β-estradiol (Estrogen) alone or in combination with 5 μM 4-hydroxytamoxifen (Tamoxifen) for 1 hr. Co-immunoprecipitation of ERα and 4ICD was analyzed by western blot. Tamoxifen disrupts both the basal and estrogen stimulated ERα interaction with 4ICD.
Tamoxifen promotes mitochondrial accumulation of the 4ICD BH3-only protein and activation of the intrinsic apoptotic pathway
In addition to the coactivator function of 4ICD, we have also demonstrated that 4ICD translocates to mitochondria where it functions as a pro-apoptotic BH3-only protein 17, 18. By disengaging 4ICD coactivator activity tamoxifen may induce cell-killing of breast tumor cells by serendipitous activation of the untethered 4ICD BH3-only protein. In response to a specific apoptotic signal BH3-only proteins initiate cell-killing by translocating to the mitochondria where they promote activation of the apoptotic gateway proteins BAX and BAK 33.
To determine if tamoxifen promotes 4ICD BH3 domain mediated cell-killing we first examined mitochondrial accumulation of 4ICD in response to tamoxifen. Western blot analysis of mitochondrial extracts prepared from T47D cells revealed low basal levels of mitochondrial 4ICD which was depleted following estrogen treatment (Fig. 3A). The addition of tamoxifen however resulted in a dramatic increase in 4ICD mitochondrial accumulation (Fig. 3A). Tamoxifen treatment of MCF-7 or T47D cells did not however alter HER4 expression levels or 4ICD processing when compared to estrogen treated cells (Supplementary Fig. 1B). Taken together, these observations suggest that tamoxifen disruption of the ERα/4ICD coactivator complex results in mitochondrial localization of the untethered 4ICD BH3-only protein.
Figure 3.

Tamoxifen activates the proapoptotic 4ICD BH3-only protein. A, Tamoxifen stimulates mitochondrial accumulation of the 4ICD BH3-only protein. T47D cells were cultured in phenol red-free RPMI containing 5% charcoal-stripped FBS for 48 hrs then were left untreated or treated with 100 pM 17-β-estradiol (Estrogen) alone or in combination with 5 μM 4-hydroxytamoxifen (Tamoxifen) for 1 hr. Mitochondria were isolated and analyzed by western blot for accumulation of 4ICD. Analysis of TOM40 was included as a loading control. B, Inhibitors of γ-secretase and BCL-2 expression suppress tamoxifen induced apoptosis of T47D cells. T47D cells were cultured in phenol red-free RPMI containing 5% charcoal-stripped FBS for 48 hrs and treated with 100 pM 17-β-estradiol (Estrogen) alone or in combination with 5 μM 4-hydroxytamoxifen (Tamoxifen) for 48 hrs. HER4 processing to generate 4ICD was prevented by the addition of 20 nM γ-secretase Inhibitor XXI to the 17-β-estradiol and tamoxifen treatments. Apoptosis was determined visually in a Nikon fluorescent microscope by staining the cells with DAPI and determining the percentage of cells with condensed nuclei (mean +/- SE of at least three experiments). C, Tamoxifen stimulates dimerized activation of mitochondrial BAK. T47D cells were cultured in phenol red-free RPMI containing 5% charcoal-stripped FBS for 48 hrs then were left untreated or treated with 50 ng/ml of heregulin β1 (HRG), or 100 pM 17-β-estradiol (Estrogen) alone or in combination with 5 μM 4-hydroxytamoxifen (Tamoxifen) for 1 hr. Mitochondrial extracts were treated with 1 mM of BMH crosslinker for 30 min. at room temperature and BAK monomers or cross-linked activated BAK dimers were detected by western blot analysis. The HER4 ligand HRG and tamoxifen independently activated dimerization of the mitochondrial dysfunction gateway protein BAK. D, Tamoxifen fails to activate BAX. T47D cells were cultured in phenol red-free RPMI containing 5% charcoal-stripped FBS for 48 hrs then left untreated or treated with 1 μM staurosporine (STS) for 12 hrs, 50 ng/ml of heregulin β1 (HRG), or 100 pM 17-β-estradiol (Estrogen) alone or in combination with 5 μM 4-hydroxytamoxifen (Tamoxifen) for 1 hr. Mitochondrial extracts were analyzed by western blot for BAX dimerized activation.
To confirm that tamoxifen induced cell-killing involved proteolytic processing of HER4 to generate 4ICD we blocked HER4 processing by incubating T47D and MCF-7 cells with γ-secretase inhibitor XXI (Calbiochem) in combination with estrogen and/or tamoxifen. In both cell lines tamoxifen induced apoptosis was reduced to 17% in the presence of the γ-secretase inhibitor (Fig. 3B and results not shown) suggesting that proteolytic processing of HER4 to liberate 4ICD is required for tamoxifen action. To confirm that the cell-killing activity of tamoxifen is regulated by a mechanism involving the mitochondrial dysfunction pathway we determined the ability of anti-apoptotic BCL-2 to suppress tamoxifen induced apoptosis in T47D cells. BCL-2 suppresses BH3-only protein mediated mitochondrial dysfunction by interacting with and sequestering proapoptotic BH3-only proteins 33. Similarly, we have previously demonstrated that BCL-2 interacts with and suppresses the apoptotic activity of 4ICD 17. BCL-2 overexpression completely abolished tamoxifen induced apoptosis of T47D cells implicating a role for a BH3-only protein in tamoxifen mediated breast tumor cell-killing (Fig. 3B). Similar results were observed with tamoxifen treated MCF-7 cells overexpressing BCL-2 (data not shown). BH3-only proteins initiate mitochondrial dysfunction and apoptosis through activation of both BAK and BAX mitochondria pore forming complexes. Interestingly, 4ICD is unique among BH3-only protein family members in that 4ICD mediates cell-killing exclusively through BAK activation while BAX remains unaffected. We therefore examined BAK and BAX dimerized activation in T47D cells following tamoxifen treatment. Consistent with our previous results, stimulation of T47D cells with the HER4 ligand heregulin, which promotes 4ICD apoptotic activity 17, resulted in dramatic conversion of monomeric BAK to the activated dimer form (Fig. 3C and Supplementary Fig. 2) but had minimal impact on BAX dimerization (Fig. 3D and Supplementary Fig. 2). Similarly, tamoxifen also induced significant levels of BAK dimerization but failed to activate BAX (Fig. 3C,D and Supplementary Fig. 2). Staurosporine which activates the intrinsic apoptotic pathway in a BH3-only protein dependent manner was included as a positive control for BAX dimerization (Fig. 3D). Taken together these results provide compelling evidence that tamoxifen induced cell-killing of breast tumor cells involves mitochondrial dysfunction mediated by a BAK activating BH3-only protein. Tamoxifen induced mitochondrial localization of 4ICD coupled with selective activation of BAK further implicates 4ICD as the critical BH3-only protein regulating tamoxifen induced apoptosis.
HER4 with an intact 4ICD BH3-domain reverts tamoxifen refractory breast tumor cells to a tamoxifen sensitive phenotype
We have shown that independently isolated MCF-7 cell line variants with acquired tamoxifen resistance also exhibit suppressed HER4 expression (Fig. 1B). To determine if 4ICD BH3 domain activity can restore tamoxifen sensitivity, wild-type HER4 or HER4 with BH3 domain inactivating mutations (muBH3) were stably reintroduced into the tamoxifen resistant MCF-7 variant TamR. Significantly, TamR cells with reintroduced HER4 reverted to a tamoxifen sensitive phenotype with levels of tamoxifen induced apoptosis equivalent to the parental MCF-7 cells line (Fig. 4A). In striking contrast, TamR cells expressing HER4 harboring mutations inactivating BH3 domain mediated 4ICD cell-killing, remained refractory to tamoxifen induced apoptosis (Fig. 4A). Taken together, these results demonstrate that reintroduced HER4 expression can restore tamoxifen sensitivity and strongly implicate the 4ICD BH3-only protein as an essential mediator of tamoxifen regulated apoptosis of breast tumor cells.
Figure 4.

An intact HER4/4ICD proapoptotic BH3 domain restores tamoxifen sensitivity in a refractory cell line. A, Reintroduction of HER4 with an intact BH3-domain restores tamoxifen sensitivity to the TamR cell line. The MCF-7 tamoxifen resistant variant TamR was stably transfected with vector, HER4, or HER4muBH3 lacking a functional BH3 domain. Each cell line was cultured for 48 hrs in phenol red-free MEM containing 5% charcoal-stripped FBS and then treated for 24 hrs with 100 pM 17-β-estradiol (Estrogen) alone or in combination with 5 μM 4-hydroxytamoxifen (Tamoxifen). Apoptosis was determined visually in a Nikon fluorescent microscope by staining the cells with DAPI and determining the percentage of cells with condensed nuclei (mean +/- SE of at least three experiments). Asterisks indicate significant differences in each data set as determined by student t-test. B, HER4 with an intact BH3-domain causes tumor regression in response to tamoxifen in a xenograph model. Nude mice implanted with estrogen pellets were injected with the indicated cell line and after 15 days mice with established tumors were treated with implanted tamoxifen pellets. After 21 days of tamoxifen treatment mice were sacrificed and photographed. TamR cells expressing HER4, but not HER4 with a mutated BH3 domain, fully regressed following tamoxifen treatment. C, Xenograph tumor growth kinetics demonstrates HER4 mediated tumor regression in response to tamoxifen. The volume of xenograph tumors was measured every 3 days before and after tamoxifen treatment at day 15. The mean +/- SE from at least five independent tumor sites was plotted. TamR cells expressing HER4, but not HER4 with a mutated BH3 domain, began to regress following 6 days of tamoxifen treatment. Growth of tamoxifen treated TamR/HER4 induced tumors was significantly lower than TamR and TamR/HER4muBH3 induced tumors (p = 0.043).
Tamoxifen induced tumor regression is mediated by the HER4/4ICD BH3-only protein
We extended our in vitro results to a xenograph tumor model and determined if HER4 also mediated tumor regression following tamoxifen treatment. The modified TamR cell lines were injected into the flanks of estrogen primed nude mice and tumors were allowed to develop to approximately 100 mm3. After 15 days established tumors were exposed to tamoxifen or placebo and tumor volumes were recorded during the 21 day experiment. As expected, the TamR tumors were resistant to tamoxifen treatment with a final tumor volume equivalent to the placebo treated TamR tumors (Fig. 4B,C). Although TamR tumors expressing HER4 continued to grow for six days following tamoxifen exposure, these tumors rapidly regressed by day nine of tamoxifen treatment (Fig. 4B). Significantly, TamR cells expressing HER4 with an inactivated BH3 domain continued to grow in the presence of tamoxifen (Fig. 4B,C) providing compelling evidence that the HER4/4ICD BH3-only protein is a critical regulator of tamoxifen activity in vivo. TamR cells expressing HER4 or HER4 with a mutated BH3 domain and treated with placebo both exhibited growth kinetics similar to tamoxifen treated TamR/muBH3 cells (data not shown).
To determine if tumor regression in HER4 expressing TamR cells was due to increased tumor apoptosis we examined xenograph tumors histologically for proliferation by BrdU incorporation and apoptosis by TUNEL. A significant difference in the levels of tamoxifen induced apoptosis was observed, with levels of apoptosis in TamR/HER4 tumors greater than 4% compared to less than 1% observed in TamR and TamR/muBH3 tumors (Fig. 5A,B). The TamR and TamR/muBH3 tumors were however highly proliferative with 14 and 18% of nuclei incorporating BrdU, respectively. In contrast, we rarely observed BrdU positive nuclei in the TamR/HER4 tumors (Fig. 5A,B). These results suggest that TamR/HER4 tumor regression in response to tamoxifen is due to a combination of increased apoptosis and impaired proliferation.
Figure 5.

HER4 induced tumor regression in response to tamoxifen is associated with an increase in tumor apoptosis and a reduction in proliferation. A, Prior to sacrifice mice bearing xenograph tumors were injected with BrdU Cell Labeling Reagent. Histological sections of xenograph tumors following tamoxifen treatment were examined for apoptosis by in situ TUNEL assay or proliferation by BrdU immunohistochemistry. Several TUNEL positive nuclei are indicated by arrowheads in the TamR/HER4 tumors. Rare TUNEL positive nuclei in the TamR and TamR/muBH3 tumors are indicated by arrowheads. An example of the extensive BrdU positive nuclei in TamR and TamR/muBH3 tumors is indicated by arrowheads along with a rare BrdU positive nuclei observed in TamR/HER4 tumors. B, Quantitation of BrdU and TUNEL positive nuclei following histochemical analysis. The percentage of BrdU or TUNEL positive nuclei in random microscope fields was determined. Data represents the mean +/- SE from at least three different tumors. The levels of TamR/HER4 tumor proliferation (BrdU) and apoptosis (TUNEL) were significantly different from both TamR and TamR/muBH3 cells as determined by student t test.
Nuclear 4ICD expression improves patient response to tamoxifen therapy
To determine the clinical impact of nuclear 4ICD expression on survival of breast cancer patients treated with tamoxifen as a single agent, we analyzed HER4 expression in a cohort of 791 patients. To eliminate complicating and unpredictable factors associated with a combined therapeutic regimen we focused our analysis on ER/PgR(+) patients that were treated with tamoxifen as a single agent. This is a relatively rare cohort and was limited to 42 patients with 17 patients expressing nuclear 4ICD (Fig. 6B) and 25 patients lacking nuclear 4ICD expression (Fig. 6A). Despite the small sample size the results were quite striking with no therapeutic failures observed in the 17 tamoxifen treated patients with tumor expression of nuclear 4ICD (Fig. 6C). In contrast, 20% (5/25) of tamoxifen treated patients lacking HER4/4ICD tumor expression succumbed to their disease within 10 years of diagnosis (Fig. 6C). These data approached significance with a Fisher’s exact test result of p = 0.06. These clinical observations support our preclinical data and further suggest that tamoxifen disruption of the nuclear 4ICD and ERα transcriptional complex results in tumor cell killing mediated by the untethered 4ICD proapoptotic protein. Our data is in concordance with another recent clinical study implicating HER4/4ICD expression as an independent tumor marker for patient response to tamoxifen 34.
Figure 6.

Nuclear 4ICD expression predicts patient response to tamoxifen. A, Example of primary breast tumor staining negative and B, positive for nuclear 4ICD by immunohistochemistry. C, Kaplan-Meier survival curves for tamoxifen treated ER/PgR(+) patients by presence or absence of nuclear 4ICD tumor expression. Statistical analysis requires at least one failure in each group. None of the patients with nuclear 4ICD (n = 17) failed tamoxifen therapy with 14 years of follow-up.
DISCUSSION
Despite widespread clinical use for over 30 years the molecular regulators of tamoxifen action remain poorly understood. On the one hand tamoxifen exerts cytostatic effects on some ER(+) breast tumors which can be explained in part by the ability of tamoxifen to disrupt growth-promoting ERα and coactivator complexes. On the other hand a molecular mechanism explaining how tamoxifen disruption of ERα and coactivators contributes to tamoxifen induced apoptosis of breast tumor cells is less clear. In the current study, we provide multiple lines of experimental evidence, including a preclinical xenograph model, to implicate the 4ICD BH3-only protein as a critical mediator of tamoxifen induced breast tumor cell-killing. For example, in the ER(+)/HER4(+) and tamoxifen sensitive MCF-7 and T47D breast tumor cell lines, de novo tamoxifen resistance was induced in each cell line when HER4 expression was suppressed using an RNAi strategy. Furthermore, we found that HER4 expression was suppressed in multiple MCF-7 models of acquired tamoxifen resistance developed in independent laboratories with different experimental manipulations including continuous tamoxifen exposure (TamR and LCC2) or HER2 overexpression (HER2Δ16). Indeed, one of the most common models of acquired tamoxifen resistance, the MCF-7/HER18 model developed by the Osborne laboratory also exhibits suppressed HER4 expression 35. Significantly, when we reintroduced HER4 but not HER4 with a mutated BH3 domain into the TamR MCF-7 variant, tamoxifen sensitivity was restored both in vitro and in a preclinical xenograph model. Taken together, our data establishes an important role for the 4ICD BH3 cell-killing domain in tamoxifen response.
As a cell-surface receptor HER4 must employ novel molecular mechanisms to regulate tamoxifen action. HER4 is a unique member of the EGFR-family and undergoes proteolytic processing at the cell surface to release the 4ICD transcriptional coactivator. Indeed, in multiple experimental systems the ability of HER4 to modulate gene expression requires presenilin-dependent γ-secretase cleavage to release the soluble 4ICD transcriptional regulator 18, 19, 21, 24. We have previously demonstrated that similar to other ERα coactivators 4ICD directly interacts with ERα and binds with ERα at selective estrogen regulated gene promoters in an estrogen dependent manner. Tamoxifen functions in part by disrupting ERα and coactivator complexes 4. Consistent with the role of 4ICD as an ERα coactivator, we show here that tamoxifen significantly impairs basal and estrogen stimulated 4ICD complex formation with ERα. It remains unclear however if tamoxifen disrupts a cytosolic or DNA bound nuclear ERα/4ICD complex.
Our current results suggest that disruption of the 4ICD/ERα transcriptional complex contributes to tumor regression in response to tamoxifen through activation of the proapoptotic 4ICD BH3 only protein. Indeed, we show that tamoxifen induced cell-killing can be abolished by overexpression of BCL-2. BCL-2 protects tumor cells from apoptosis by sequestering proapoptotic BH3-only protein members, thus implicating a role for a BH3-only protein in tamoxifen cell-killing. Importantly we have previously demonstrated that 4ICD harbors many of the functional characteristics of a proapoptotic BH3-only protein member of the BCL-2 family. For example, when localized to mitochondria 4ICD induces apoptosis in a BH3-domain dependent manner through activation of the apoptosis gateway protein BAK. In addition, the cell-killing activity of 4ICD is abolished in cells overexpressing BCL-2 17. Significantly, we show here that tamoxifen stimulates mitochondrial accumulation of 4ICD presumably through disruption of the 4ICD/ERα transcriptional complex. Subsequent activation of mitochondrial BAK but not BAX in response to tamoxifen further implicates the 4ICD BH3-only protein as the proapoptotic mediator of tamoxifen induced cell-killing. Indeed, selective activation of BAK is a property unique to the 4ICD BH3-only protein 17. Furthermore, an intact 4ICD BH3 domain was required for ectopic HER4 to reestablish tamoxifen sensitivity in a resistant cell line both in vitro and in a xenograph model. In fact, xenograph tumors of HER4 expressing TamR cells fully regressed with a significant increase in tumor apoptosis when compared to control or tumors expressing HER4 with a mutated 4ICD BH3 domain. Taken together these results provide important in vitro and preclinical in vivo evidence for 4ICD BH3-domain mediated cell-killing activity as an important regulator of tamoxifen response in breast cancer.
Based upon our hypothesis that disruption of the nuclear ERα/4ICD transcriptional complex mediates tamoxifen induced cell-killing through release of the 4ICD pro-apoptotic protein one would predict that patients with nuclear 4ICD expression will exhibit improved response to tamoxifen. Our clinical data provide compelling support for this hypothesis. After 14 years of follow-up we have not observed a single failure in our patients whose tumors express nuclear 4ICD and received tamoxifen as their sole therapeutic intervention. Activation of the 4ICD BH3-only protein through disruption of ERα/4ICD transcriptional complexes may emerge as a common mechanism of action for endocrine therapies that target ERα function. For example, HER4 expression was also suppressed in a panel of fulvestrant resistant breast tumor cell lines 36. Fulvestrant treatment, which leads to ERα degradation, may also disengage ERα/4ICD complexes and promote fortuitous activation of 4ICD cell-killing activity.
There exists a formal possibility that loss of HER4 expression contributes to tamoxifen resistance through disengaged 4ICD coactivation of estrogen regulated genes. However, mutation of the 4ICD BH3 domain failed to impact HER4 stimulation of estrogen induced gene expression (SupFig 1C), while this same functional alteration of HER4 abolished the ability of HER4 to restore tamoxifen sensitivity in a resistant cell line. These results further implicate a role for the 4ICD BH3-only protein in tamoxifen response independent of 4ICD coactivator function.
In summary, we demonstrate that suppression of HER4 expression in multiple breast tumor model systems results in tamoxifen resistance and we further show that the proapoptotic BH3 domain of 4ICD is an important mediator of tamoxifen induced cell-killing and tumor regression in a preclinical xenograph model. In direct corroboration with our preclinical data a recent analysis of primary breast tumors from patients treated with tamoxifen as a single agent revealed that suppressed HER4 expression was an independent tumor marker for tamoxifen resistance in a multivariate analysis 34. Our clinical results indicating a profound impact of nuclear 4ICD expression on breast cancer patient survival following tamoxifen treatment further implicates 4ICD as a critical mediator of tamoxifen activity and as an important tumor marker for predicting patient response to endocrine therapy. Emerging evidence suggests that the subcellular distribution of cell-surface receptors and their intracellular domains has a profound effect on tumor biology 13, 15, 17, 37, 38. Here we provide a proof-of-hypothesis to support further studies aimed at deciphering and clinically validating the molecular mechanisms regulating 4ICD subcellular localization and divergent 4ICD activities directly influencing breast cancer progression and therapeutic response.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank members of the Jones laboratory past and present for insightful discussions and experimental advice. This work was supported in part by the Tulane Cancer Center. This work is dedicated to June Allison, loving wife and mother currently undergoing tamoxifen therapy for recurrent breast cancer.
Financial Support: Supported by NCI/NIH grants RO1CA95783 (FEJ) and RO1CA96717 (FEJ) and US AMRMC grant DAMD170610418 (AN).
REFERENCES
- 1.Early Breast Cancer Trialists’ Collaborative Group Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351:1451–67. [PubMed] [Google Scholar]
- 2.Normanno N, Di Maio M, De Maio E, et al. Mechanisms of endocrine resistance and novel therapeutic strategies in breast cancer. Endocr Relat Cancer. 2005;12:721–47. doi: 10.1677/erc.1.00857. [DOI] [PubMed] [Google Scholar]
- 3.Osborne CK, Schiff R. Estrogen-receptor biology: continuing progress and therapeutic implications. J Clin Oncol. 2005;23:1616–22. doi: 10.1200/JCO.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 4.Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–37. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 5.Sutherland RL, Hall RE, Taylor IW. Cell proliferation kinetics of MCF-7 human mammary carcinoma cells in culture and effects of tamoxifen on exponentially growing and plateau-phase cells. Cancer Res. 1983;43:3998–4006. [PubMed] [Google Scholar]
- 6.Ellis PA, Saccani-Jotti G, Clarke R, et al. Induction of apoptosis by tamoxifen and ICI 182780 in primary breast cancer. Int J Cancer. 1997;72:608–13. doi: 10.1002/(sici)1097-0215(19970807)72:4<608::aid-ijc10>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001;6:469–77. doi: 10.1023/a:1012437607881. [DOI] [PubMed] [Google Scholar]
- 8.Kallio A, Zheng A, Dahllund J, Heiskanen KM, Harkonen P. Role of mitochondria in tamoxifen-induced rapid death of MCF-7 breast cancer cells. Apoptosis. 2005;10:1395–410. doi: 10.1007/s10495-005-2137-z. [DOI] [PubMed] [Google Scholar]
- 9.Obrero M, Yu DV, Shapiro DJ. Estrogen receptor-dependent and estrogen receptor-independent pathways for tamoxifen and 4-hydroxytamoxifen-induced programmed cell death. J Biol Chem. 2002;277:45695–703. doi: 10.1074/jbc.M208092200. [DOI] [PubMed] [Google Scholar]
- 10.Diel P, Smolnikar K, Michna H. The pure antiestrogen ICI 182780 is more effective in the induction of apoptosis and down regulation of BCL-2 than tamoxifen in MCF-7 cells. Breast Cancer Res Treat. 1999;58:87–97. doi: 10.1023/a:1006338123126. [DOI] [PubMed] [Google Scholar]
- 11.Thiantanawat A, Long BJ, Brodie AM. Signaling pathways of apoptosis activated by aromatase inhibitors and antiestrogens. Cancer Res. 2003;63:8037–50. [PubMed] [Google Scholar]
- 12.Zhang GJ, Kimijima I, Onda M, et al. Tamoxifen-induced apoptosis in breast cancer cells relates to down-regulation of bcl-2, but not bax and bcl-X(L), without alteration of p53 protein levels. Clin Cancer Res. 1999;5:2971–7. [PubMed] [Google Scholar]
- 13.Junttila TT, Sundvall M, Lundin M, et al. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005;65:1384–93. doi: 10.1158/0008-5472.CAN-04-3150. [DOI] [PubMed] [Google Scholar]
- 14.Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003;200:290–7. doi: 10.1002/path.1370. [DOI] [PubMed] [Google Scholar]
- 15.Tovey SM, Dunne B, Witton CJ, Cooke TG, Bartlett JM. HER4 in breast cancer: comparison of antibodies against intra- and extra-cellular domains of HER4. Breast Cancer Res. 2006;8:R19. doi: 10.1186/bcr1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carpenter G. ErbB-4: mechanism of action and biology. Exp Cell Res. 2003;284:66–77. doi: 10.1016/s0014-4827(02)00100-3. [DOI] [PubMed] [Google Scholar]
- 17.Naresh A, Long W, Vidal GA, et al. The ERBB4/HER4 intracellular domain 4ICD is a BH3-only protein promoting apoptosis of breast cancer cells. Cancer Res. 2006;66:6412–20. doi: 10.1158/0008-5472.CAN-05-2368. [DOI] [PubMed] [Google Scholar]
- 18.Vidal GA, Naresh A, Marrero L, Jones FE. Presenilin-dependent γ-secretase processing regulates multiple ERBB4/HER4 activities. J Biol Chem. 2005;280:19777–83. doi: 10.1074/jbc.M412457200. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Y, Sullivan LL, Nair SS, et al. Coregulation of estrogen receptor by estrogen-inducible ERBB4/HER4 establishes a growth promoting autocrine signal in breast cancer. Cancer Res. 2006;66:7991–8. doi: 10.1158/0008-5472.CAN-05-4397. [DOI] [PubMed] [Google Scholar]
- 20.Long W, Wagner K-U, Lloyd KCK, et al. Impaired differentiation and lactational failure in ErbB4-deficient mammary glands identify ERBB4 as an obligate mediator of Stat5. Development. 2003;130:5257–68. doi: 10.1242/dev.00715. [DOI] [PubMed] [Google Scholar]
- 21.Muraoka-Cook RS, Sandahl M, Husted C, et al. The intracellular domain of ErbB4 induces differentiation of mammary epithelial cells. Mol Biol Cell. 2006;17:4118–29. doi: 10.1091/mbc.E06-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sundvall M, Peri L, Maatta JA, et al. Differential nuclear localization and kinase activity of alternative ErbB4 intracellular domains. Oncogene. 2007;26:6905–14. doi: 10.1038/sj.onc.1210501. [DOI] [PubMed] [Google Scholar]
- 23.Tidcombe H, Jackson-Fisher A, Mathers K, Stern DF, Gassmann M, Golding JP. Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proc Natl Acad Sci U S A. 2003;100:8281–6. doi: 10.1073/pnas.1436402100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams CC, Allison JG, Vidal GA, et al. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol. 2004;167:469–78. doi: 10.1083/jcb.200403155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwong KY, Hung M-C. A novel splice variant of HER2 with increased transformation activity. Mol Carcinog. 1998;23:62–8. doi: 10.1002/(sici)1098-2744(199810)23:2<62::aid-mc2>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 26.Riese DJ, II, van Raaij TM, Plowman GD, Andrews GC, Stern DF. Cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol Cell Biol. 1995;15:5770–6. doi: 10.1128/mcb.15.10.5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rayala SK, Talukder AH, Balasenthil S, et al. P21-activated kinase 1 regulation of estrogen receptor-alpha activation involves serine 305 activation linked with serine 118 phosphorylation. Cancer Res. 2006;66:1694–701. doi: 10.1158/0008-5472.CAN-05-2922. [DOI] [PubMed] [Google Scholar]
- 28.Brunner N, Frandsen TL, Holst-Hansen C, et al. MCF7/LCC2: a 4-hydroxytamoxifen resistant human breast cancer variant that retains sensitivity to the steroidal antiestrogen ICI 182,780. Cancer Res. 1993;53:3229–32. [PubMed] [Google Scholar]
- 29.Jones FE, Welte T, Fu X-Y, Stern DF. ErbB4 signaling in the mammary gland is required for lobuloalveolar development and Stat5 activation during lactation. J Cell Biol. 1999;147:77–87. doi: 10.1083/jcb.147.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Amer Statistical Assoc. 1952;47:583–621. [Google Scholar]
- 31.Li L, Cleary S, Long W, Mandarano MA, Birchmeier C, Jones FE. The breast protooncogene, HRGα regulates epithelial proliferation and lobuloalveolar development in the mouse mammary gland. Oncogene. 2002;21:4900–7. doi: 10.1038/sj.onc.1205634. [DOI] [PubMed] [Google Scholar]
- 32.Liu S, Edgerton SM, Moore DH, 2nd, Thor AD. Measures of cell turnover (proliferation and apoptosis) and their association with survival in breast cancer. Clin Cancer Res. 2001;7:1716–23. [PubMed] [Google Scholar]
- 33.Adams JM. Ways of dying: multiple pathways to apoptosis. Genes and Dev. 2003;17:2481–95. doi: 10.1101/gad.1126903. [DOI] [PubMed] [Google Scholar]
- 34.Guler G, Iliopoulos D, Guler N, Himmetoglu C, Hayran M, Huebner K. Wwox and Ap2γ Expression Levels Predict Tamoxifen Response. Clin Cancer Res. 2007;13:6115–21. doi: 10.1158/1078-0432.CCR-07-1282. [DOI] [PubMed] [Google Scholar]
- 35.Kurokawa H, Lenferink AE, Simpson JF, et al. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res. 2000;60:5887–94. [PubMed] [Google Scholar]
- 36.Frogne T, Benjaminsen RV, Sonne-Hansen K, et al. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. 2008 doi: 10.1007/s10549-008-0011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC. Novel Prognostic Value of Nuclear Epidermal Growth Factor Receptor in Breast Cancer. Cancer Res. 2005;65:338–48. [PubMed] [Google Scholar]
- 38.Wang SC, Lien HC, Xia W, et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell. 2004;6:251–61. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.