

Abstract
Electronic perturbation of quinone methides (QM) greatly influences their stability and in turn alters the kinetics and product profile of QM reaction with deoxynucleosides. Consistent with the electron deficient nature of this reactive intermediate, electron-donating substituents are stabilizing and electron-withdrawing substituents are destabilizing. For example, a dC N3-QM adduct is made stable over the course of observation (7 days) by the presence of an electron-withdrawing ester group that inhibits QM regeneration. Conversely, a related adduct with an electron donating methyl group is very labile and regenerates its QM with a half-life of approximately 5 hr. The generality of these effects is demonstrated with a series of alternative quinone methide precursors (QMP) containing a variety of substituents attached at different positions with respect to the exocyclic methylene. The rates of nucleophilic addition to substituted QMs measured by laser flash photolysis similarly span five orders of magnitude with electron rich species reacting most slowly and electron deficient species reacting most quickly. The reversibility of QM reaction can now be predictably adjusted for any desired application.
Introduction
Developing reagents for selective reaction with cellular components is often dismissed due to an unnecessary assumption that covalent processes are limited to irreversible chemistry. Reversible covalent reaction has the potential to overcome kinetic traps that limit both potency and selectivity. Reactive species may be regenerated repeatedly in a reversible process to escape trapping with non-targeted materials and even overcome cellular repair processes. The ability of the natural product Ecteinascidin 743 (Et-743) to bind covalently to duplex DNA and yet still migrate between various nucleotide sequences, for example, also allows Et-743 to reassociate with DNA after its initial adduct has been removed by excision repair (Scheme 1).1 In contrast, reagents such as nitrogen mustards react irreversibly with strong nucleophiles in DNA, most notably the N7 of guanine (Scheme 2) and other cellular compounds.2–4 The most significant effect of the mustards, DNA cross-linking, only proceeds if a second nucleophile is proximal to the initial site of attachment. However, this second nucleophile is not typically present and reaction is quenched before the mustard can migrate to a site in DNA that would support cross-linking.
Scheme 1.

Repair and Reformation of a DNA Adduct
Scheme 2.

Nucleobase Structure
Reversible alkylating agents may also react with a range of cellular components, but after multiple regenerations they may ultimately associate with their thermodynamically favorable targets (e.g., Et-743 with duplex DNA, Scheme 1). Reversibility then extends the effective lifetime of electrophiles and consequently enhances their potential for selective biological activity. Such activity has been investigated with carcinogens such as malondialdehyde5–8 and a variety of anticancer drugs candidates based on cyclopropylpyrroloindole.9,10 For two closely related drug candidates, duocarmycin SA (duoSA) is twenty-fold more cytotoxic than duocarmycin A (duoA). This result is likely a function of the slower reaction of duoSA vs. duoA with nucleophiles and its faster release from adducts formed by nucleophilic coupling.11,12 A direct correlation between reversibility of DNA adducts and cytotoxicity was also extended to a number of additional cyclopropylpyrroloindole derivatives as well.13
Our laboratories and others have applied the reversibility of quinone methide (QM) generation for efficient cross-linking and target-promoted alkylation of DNA.14–18 Related QMs had previously been implicated in the biological activation 2,6-ditert-butyl-4-methylphenol,19 tamoxifen20 and a variety of natural products including mitomycin21 and certain diterpenone catechols.22 Initial model studies suggested a selectivity of QM for weak nucleophiles and an ability of some adducts to form reversibly.19,23–26 Further investigation revealed that this unusual selectivity was a function of QM adduct stability rather than initial product formation.27–29 In contrast to earlier assumptions, the strongest nucleophiles of DNA reacted most quickly with a simple ortho-QM model to form kinetic products. However, these products are not stable and regenerate the ortho-QM for ultimate transfer to weaker nucleophiles that react irreversibly and form thermodynamic products (Scheme 3).27,30
Scheme 3.
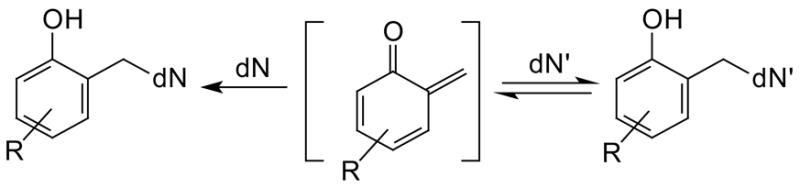
Reversible and Irreversible Formation of QM Adducts
Computational studies on this same ortho-QM model anticipated the experimental results with remarkable accuracy.28,29 A low value for ΔG‡ was calculated for both adduct formation and QM regeneration with strong nucleophiles such as dC N3, dA N1 and dG N7 (Scheme 2) whereas ΔG‡ values for addition of the exo-amino groups (dG N2 and dA N6) to ortho-QM were considerably higher and essentially irreversible. The free energy of activation should also be sensitive to variations in the electronic properties of the intermediate QMs. A relatively electron-rich derivative may be more stable and hence more readily generated than its electron-poor counterpart. Hence, electron withdrawing and donating groups attached directly on the QM should influence the rate of QM formation and adduct stability. A series of such derivatives has now been prepared to test this possibility and determine the sensitivity of the QM to a variety of substituents. A method for predictably manipulating QM stability will be indispensable for optimizing the kinetics and selectivity of target promoted alkylation of DNA15 and will additionally facilitate use of QM for mechanism-based inactivation of enzymes31 and drug release by self-immolative, or cascade-release, dendrimers.32–34 Quinone methides are also used as intermediates in organic synthesis35 and can be stabilized by metal coordination.36–39
Results and Discussion
A limited number of studies previously began to explore substituent effects on QM reactivity,40–43 but no data was available to estimate structural effects influencing the kinetics or product distribution of deoxynucleoside reaction. Electron donating and withdrawing groups have now been placed para and meta to the QM methylene to avoid complications from steric effects. The products of reversible reaction were expected to be most susceptible to changes in the QM stability since their presence depends on the rates of both nucleophilic addition to the QM as well as subsequent elimination to regenerate the QM. In contrast, the products formed irreversibly should depend on only the relative strength of their nucleophilic components to compete for initial addition to the QM.
Formation and Decomposition of dC Adducts Generated from QMs of Varying Reactivity
Our initial investigation to determine the extent to which QM reactivity can be controlled by aromatic substituents was performed with dC since this deoxynucleoside forms a single adduct upon incubation with QM1.44 The quinone methide precursor (QMP1) originally used to generate the unsubstituted QM1 was also modified alternatively with a methyl (QMP2) and methyl ester (QMP3) group to examine the effect of electron donating and withdrawing groups, respectively (Scheme 4). As described previously, fluoride was used to initiate deprotection of the QMPs.44 Subsequent elimination of acetate generated the QMs for dC alkylation. Both QM2 and QM3 also generated only a single adduct with dC under the same conditions used previously to form the dC N3 adduct of QM1.44 1H NMR data for these new derivatives are consistent in each case with linkage through the N3 of dC. The chemical shifts of the benzylic methylene protons are quite characteristic of QM adduct structure25,26 and benzylic signals for the dC N3-QM adducts of QM1, QM2 and QM3 differ by no more than approximately 0.1 ppm (Table 1).
Scheme 4.

QM generation from Silyl-protected Precursors
Table 1.
Benzylic proton shifts of substituted QM adducts.
| Adducts formed by | dC N3 | dA N1 | dA N6 | dG N1 | dG N2 | guanine N7 |
|---|---|---|---|---|---|---|
| QM1 | 4.96 | 5.26 | 4.76 | 5.28 | 4.58 | 5.49 |
| QM2 | 5.03 | ND | 4.72 | 5.24 | 4.33 | 5.30 |
| QM3 | 5.11 | 5.16 | 4.69 | 5.09 | 4.43 | 5.40 |
Kinetic measurement of dC addition to the QMs was performed under conditions identical to those first used to follow reaction of QM1.30 The maximum yield of adduct formed in each case was similar, but their rates of formation and stability varied greatly (Figure 1). The dC N3 adduct of the electron-rich QM2 formed most rapidly, and a maximum yield was achieved in less than 1 hr. Decomposition of this adduct was also very rapid with a half-life of approximately 5 hr and resulted in release of dC and presumably quenching of QM2 with water. In contrast, the unsubstituted QMP1 examined previously generated a maximum yield of dC N3-QM1 adduct after 4 – 10 hr. This product exhibited moderate stability under the reaction conditions and decomposed with a half-life of approximately 24 hr. Finally, the electron-poor QM3 formed very slowly over 40 hr, but the dC N3 adduct that resulted from its reaction persisted without change over a minimum of 72 hrs. No decomposition of the dC N3-QM3 adduct was even detected after an extended incubation of 7 days.
Figure 1.
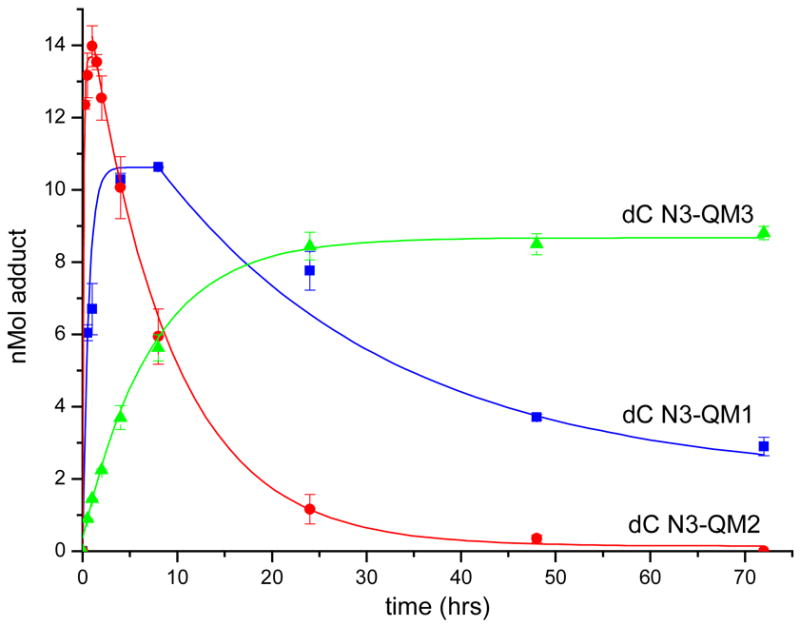
Formation and decomposition of quinone methide adducts of dC N3. Reaction conditions and product analysis are described in the Experimental Procedures. Each point represents the average of at least three independent determinations and was fit to exponential processes for highlighting the net trends of the data. The indicated error derives from the standard deviations.
The rate of adduct formation primarily reflects the rate of initial QM formation by elimination of the benzylic acetate after rapid desilylation. Electron donation of a single methyl substituent stabilizes QM2 sufficiently to promote its formation by at least 4-fold relative to formation of QM1 (Figure 1 and Supporting Information). Similarly, QM1 forms perhaps 10-fold more rapidly than QM3 that contains an electron withdrawing group. This same trend is apparent for QM regeneration from each of the dC N3 adducts. Thus, small changes in the electronics of the QMs affect their reactivity and adduct stability dramatically and now also predictably.
Deoxynucleoside Competition with an Electron-rich QM
The consequences of QM stability on the kinetics of forming all deoxynucleoside adducts was next examined with a competitive equimolar mixture of dA, dG, dC and T in analogy to earlier studies with the unsubstituted QM1.30 Product standards were first isolated after reaction with individual deoxynucleosides, and each product was characterized by 1H NMR, high-resolution mass spectrometry (HRMS) and UV absorbance. Once again, the chemical shift of the benzylic protons was diagnostic of the linkage between the QMs and deoxynucleosides (Table 1). Characteristic changes in the UV absorbance maxima further confirmed the assignments (Supporting Information). Reaction of QM2 with dA yielded two adducts linked through its N1 and N6 positions. Only the N6 adduct was stable enough for isolation and characterization. However, the UV maximum of the second product was recorded during HPLC analysis and is consistent with an N1 adduct (Supporting Information). Three products were generated by QM2 and dG and identified as adducts of its N1, N2 and N7 positions. Both the N1 and N2 adducts were characterized as their dG derivatives. The N7 adduct of dG was evident during chromatographic separation but spontaneously deglycosylated to form its guanine derivative. This secondary product was stable and subject to standard analysis. Similar results had previously been observed with reaction of dG and QM1.26,30 No adducts of T were observed under the range of conditions examined in this work.
Monitoring the competitive reaction of dA, dC, dG and T with QMP2 over time resulted in the same trends observed previously with QMP1 except the timeframe of the evolving products was significantly compressed (Figure 2). The initial two hrs were dominated by reaction of the strong nucleophiles, dA N1 and dC N3. However, the resulting adducts decomposed with half-lives of less than 4 hrs. The weaker nucleophiles in turn formed adducts more slowly over 8 hr and most likely as a result of QM2 regeneration from the adducts of dA N1 and dC N3. QM2 generation from its initial QMP2 precursor is nearly complete within the first 2 hrs (Supporting Information). This again illustrates the ability of reversible adducts to extend the effective lifetime of an otherwise highly transient intermediate. The leaving group abilities of dA N6, dG N1 and dG N2 are not sufficient to regenerate the QM, and thus their adducts remained stable over the course of observation.
Figure 2.

Time-dependent profile of adducts formed by the electron-rich QM2 and deoxynucleosides. Reaction conditions and product analysis are identical to those used for Figure 1. Again, each point represents the average of at least three independent determinations and was fit to exponential processes for highlighting the net trends of the data. The indicated error derives from the standard deviations.
Initial reaction at dG N7 is less than might be expected from its strong nucleophilicity. However, its adduct has the unique ability to deglycosylate as well as eliminate QM2. At the deoxynucleoside level, partitioning between deglycosylation and QM regeneration had previously been observed to occur at equivalent rates.30 The deglycosylation product from reaction with QM2 accumulated over approximately 15 hrs and remained stable since guanine N7 does not share the strong leaving group ability of dG N7. Although the kinetics of alkylation with QM2 were significantly faster than with QM1, all of the irreversible adducts were stable for at least six days beyond the observations of Figure 2. The nucleophilic sites used to create these adducts therefore still lacked the leaving group ability to eliminate from a QM with the added electron density provided by a pendent methyl group. This group was, however, sufficient to accelerate QM regeneration from adducts that had previously exhibited lability with QM1.30 Finally, the overall yields of stable adducts were similar for QM1 and QM2, and only minor differences in their relative formation was detected.
Deoxynucleoside Competition with an Electron-poor QM
Individual deoxynucleosides were also treated with the electron-poor QMP3 in order to generate synthetic standards for structural and chromatographic markers. Incubation of dA with QMP3 and fluoride resulted in adducts formed by its N1 and N6 positions. This is consistent with reactions of both QM127 and QM2. Additionally, the 1H NMR signals for the benzylic positions were again diagnostic of their structures (Table 1). Comparable reaction of QMP3 with dG yielded only two products. These were isolated and identified as adducts of guanine N7 and dG N2. The dG N7 adduct did not accumulate since its deglycosylation is likely faster than its formation due to the slow generation of QM3. The product of dG N1 alkylation was also not initially apparent but could later be formed when the concentration of dG was raised from the standard 0.25 mM to 5.0 mM. Computational studies for QM1 indicated that reaction at dG N1 has an activation energy approximately 3 kcal/mol higher than that of dG N2 as modeled with 9-methylguanine in water.29 This can be used to explain the greater yield of the dG N2 adduct over the yield of the dG N1 adduct with QM1 and QM3. Surprisingly, QM2 exhibited the opposite selectivity. The origin of this difference is currently subject to additional computational analysis. Once again, no reaction of T was detected.
Reaction of QMP3 with dNs was significantly slower than reaction with QMP1 due to electronic destabilization of QM3 from the attached methyl ester (Figure 3). Maximum accumulation of the dC N3 adduct required more than 24 hr and reflected a slow generation of QM3 from its desilylated derivative of QMP3 (Supporting Information). Additionally, this adduct was stable well beyond the 120 hr period of observation (see above). The leaving group ability of dC N3 was thus insufficient to overcome the barrier to regenerate QM3 in contrast to its behavior with the more electron-rich QM1 and QM2 derivatives. The change in electronics between QM2 and QM3 did not greatly effect the maximum yield of the dC N3 adduct despite its conversion from a reversible to irreversible adduct, respectively.
Figure 3.

Time-dependent profile of adducts formed by the electron-poor QM3 and deoxynucleosides. Reaction conditions and product analysis are identical to those used for Figure 1. Again, each point represents the average of at least three independent determinations and was fit to exponential processes for highlighting the net trends of the data. The indicated error derives from the standard deviations.
Formation of the dA N1 adduct similarly reflected the slow generation of QM3 (Figure 3). A maximum yield for this adduct required approximately 10 hrs with QMP3 while less than 0.5 hr was necessary with the electron rich QMP2. Reversibility of the dA N1 adduct persisted despite the electron deficiency of QM3, although regeneration of QM3 (half-life > 40 hr, Figure 3) was greatly suppressed relative to comparable regeneration of QM2 (half-life < 2 hr, Figure 2). Behavior of the dC N3 and dA N1 adducts is consistent with the relative strength of dC N3 and dA N1 as leaving groups.30 The slow formation of the dA N1 adduct combined with its remaining lability prevented its accumulation to the same level as the dC N3 adduct or related adducts with QM2. The guanine N7 adduct resulting from QM alkylation and deglycosylation also formed slowly with QMP3, but its yield was similar with both QM2 and QM3.
Destabilization of the QM affects both the kinetics of its formation and the resulting product profile as evident by the stability of the dC N3-QM3 adduct and the complete absence of a dG N1 adduct. Adducts of dG N2 and dA N6 were still generated by QM3 in similar yields and approach the levels of accumulation measured with QM2 (Figure 2). However, these QM3 adducts continue to increase for more than 120 hrs (rather than 10 hr for QM2). The original source of QM3 is mostly consumed by 30 hrs, and thus continued generation of the dG N2 and dA N6 adducts likely derives from the regeneration of QM3 from its dA N1 adduct.
Effects of Aromatic Substituents on QM Generation
A methoxy group was originally attached to the model QM1 in order to examine a broad range of substituent effects on deoxynucleoside adduct stability. Preliminary study of the adduct formed between this electron-rich intermediate and dC N3 indicated that it was too labile for convenient analysis. Thus, the weaker electron-donating methyl group was used in place of the MeO group as described above. As a complement to the range of derivatives available for deoxynucleoside study, a series of N-morpholino derivatives were prepared to extend correlations between QM stability and its rate of (re)generation. The morpholino-QM adducts were chosen to replace deoxynucleoside-QM adducts based on the pKa of morpholine’s conjugate acid. This property reflects the nucleophile’s leaving group ability that, along with QM stability (see above), determines the rate of adduct collapse and QM regeneration (Scheme 3).30,45 For nucleophiles with conjugate acids exhibiting pKa’s between approximately 4 and 8, QM regeneration has been detected over hours under physiological conditions.30 Morpholine has a conjugate acid pKa of 8.4 which is at the high end of this acidity range and should consequently provide some stability to even highly electron-rich QM precursors. Accordingly, morpholino adducts provided a basis for determining electronic effects of QM reversibility over a greater range of electronic perturbations than available with the deoxynucleosides.
The desired morpholino adducts of substituted QMs were synthesized by a Mannich reaction of 4-methylenemorpholinium halide and the appropriate hydroxylated aromatic prescursor. Substituents influencing QM stability ranged from the strongly electron-donating -OMe group to the electron-withdrawing -COOMe group (Table 2). These groups were placed at the 5-position like QMP2 and QMP3 as well as at the 4-position to compare positional differences. The overall trend of adduct stability (QMP4-QMP8) was monitored by conversion of the morpholino adducts to their water adducts (benzylic alcohols, QMA). The range of reactivities observed for this series was even more dramatic than that observed for the deoxynucleoside adducts above. An aqueous solution of the electron-poor adduct QMP4 was fully stable after incubation at 100 °C for 3 hrs (Table 2). No consumption of QMP4 or formation of QMA3 was detected by HPLC after this harsh treatment. This suggests that QM3 was not generated under these conditions nor was QMP4 subject to direct substitution through a SN2 mechanism. This alternative process should not be significantly influenced by remote aromatic substitution, and thus QMA formation from the other adducts is indicative of QM generation as detected in a number of related systems14,15,25,45–47 rather than direct substitution. Similarly, an alternative SN1 mechanism is not consistent with the lack of reactivity of O-silyl QMPs and their O-methyl derivatives compared to that of their parent phenols14,48 since these substituent should all promote such a process almost equally.49
Table 2.
quinone methide precursor quinone methide benzylic alcohol
 | ||||||
|---|---|---|---|---|---|---|
| QMP | X | Y | QM | QMA | T/°C | t1/2 of QMP (min) in water |
| QMP4 | COOMe | H | QM3 | QMA3 | 100 | Stablea |
| QMP5 | H | COOMe | QM5 | QMA5 | 100 | 149 |
| QMP6 | H | H | QM1 | QMA1 | 50 | 115 |
| QMP7 | H | OMe | QM7 | QMA7 | 22 | 11 |
| QMP8 | OMe | H | QM8 | QMA8 | 22 | 4 |
No benzylic alcohol QMA3 was detected by HPLC.
An electron-withdrawing –COOMe group placed at the 4-position was not as effective at destabilizing its QMP (QMP5) as the same group placed at the 5-position (QMP4, Table 2). QMP5 was slowly converted to its benzylic alcohol QMA5 albeit heating to 100 °C was still necessary. The unsubstituted parent QMP6 was somewhat more labile. Formation of QMA1 from trapping of the transient QM1 was observed after heating to only 50 °C. In sharp contrast, the electron rich derivatives containing methoxy substituents (QMP7, QMP8) were very labile with half-lives of only minutes at 22 °C. The sensitivity of QM generation to electronic effects that was first observed for deoxynucleoside adducts is consequently quite general and remarkable for other QM-producing adducts as well. Furthermore, the site of substitution within the QM adduct also plays an important role in its stability. The ortho-QMs characterized in this study were more strongly influenced by substitution at the 5- than 4-position (QMP4 vs. QMP5 and QMP8 vs. QMP7). Inductive and resonance effects from both positions apparently modulate QM stability, but only the 5-position can participate directly in resonance with the nascent exocyclic methylene.
Substituent Effects on Nucleophilic Addition to Quinone Methides
Substituents used to tune the stability of QM adducts should concurrently effect their electrophilicity and lifetime. The magnitude of these perturbations has now been quantified by laser flash photolysis (LFP). Photochemical activation of hydroxybenzyl alcohols and phenolic Mannich bases (Scheme 5, L = OH and NMe2, respectively) has been well described as a mild source of QMs by Wan,50–52 Kresge53–55 and Saito.56 Recently, one of our laboratories has developed an alternative photochemical precursor to QMs, benzyl ammonium salts of phenolic Mannich bases (Scheme 5, L = +NMe3I−).45 In contrast to Mannich bases, these salts are not nucleophilic and do not react with the transient QM. Also, these salts display higher photochemical quantum yields than the corresponding alcohols for QM generation.45 The quaternary ammonium salts are therefore ideal for generating a series of o-QMs and monitoring their consumption over time in the presence of various nucleophiles.
Scheme 5.

Photogeneration of QM
While Wan,50–52 Kresge,53–55,57 Richard58–60 and our laboratories30,45 have investigated the kinetic behavior of the parent o-QM (QM1), its para isomer (p-QM) and a few of their derivatives under aqueous conditions, no systematic analysis of substituent effects on QM reactivity has yet been published to our knowledge. Consequently, QMP9-QMP14 were synthesized to examine a diverse range of electronic perturbations, and QM reaction was tested with three prototypical nucleophiles.
Each QM intermediate was generated from its corresponding QMP (0.2 mM) by flash photolysis at 266 nm. QM consumption was then monitored at its λmax under aqueous conditions (pH 7) in the absence and presence of 0.2 – 50 mM morpholine (buffered at pH 9 with Na2CO3) and 2-mercaptoethanol (buffered at pH 7 with sodium phosphate), alternatively (Table 3). Second order decay rate constants (k2) were calculated from the concentration dependence of pseudo-first order kinetic measurements. The rate constants determined under each of the three conditions exhibit parallel trends that are consistent with the behavior of the deoxynucleoside and morpholino adducts. Electron-donating groups significantly stabilize QM. Such groups facilitate QM regeneration from their adducts (Figures 1–3, Table 2) and are now shown to slow nucleophilic addition to the nascent QM as well (Table 3). In a complementary manner, electron-withdrawing groups suppress QM regeneration and strongly promote nucleophilic addition to the nascent QMs.
Table 3.
 | ||||||
|---|---|---|---|---|---|---|
| Quaternary ammonium salts | Y | Quinone Methide | λmax/nm | k2(H2O)/M−1s−1 | k2(morpholine)a/105M−1s−1 | k2(RSH)/105M−1s−1 |
| QMP9 | OMe | QM7 | 410 | 1.9 | 5.5 | 0.40 |
| QMP10 | H | QM1 | 400 | 7.8 | 23 | 1.9 |
| QMP11 | Cl | QM11 | 400 | 22.8 | 36 | 4.4 |
| QMP12 | COOMe | QM5 | 420 | 363 | 160 | 59 |
| QMP13 | CN | QM13 | 415 | 1999 | 505 | 501 |
| QMP14 | NO2 | QM14 | 435 | 25400 | -b | - b |
Keeping pH 9.0 with Na2CO3.
The transient absorbance of QM bearing the strongest NO2 electron withdrawing substituent in the presence of morpholine and thiol were completely quenched. This could be due to an electron transfer mechanism from the nucleophile to the QM (work in progress).
The presence of an increasingly strong electron-withdrawing group (5-Cl, 5-COOMe, 5-CN and 5-NO2) progressively decreased QM lifetime as evident by the increasing values of k2. The 5-NO2 (QM14) in particular resulted in a k2 for the hydration reaction that was several orders of magnitude greater than that of the parent QM1. In contrast, the electron-donating ability of the 5-MeO group decreased the rate of its QM (QM7) consumption vs. the parent QM1 by 4- to 5-fold. Although the rates of QM decay were much faster in the added presence of strong nucleophiles represented here by morpholine and 2-mercaptoethanol, their general sensitivity to electron-withdrawing and donating substituents remains nearly equivalent. Still, the relatively selectivity for the individual nucleophiles decreases with increasing reactivity of the QM. Such data is consistent with the much maligned reactivity-selectivity principle despite the lack of reaction controlled by diffusion.61
The sensitivity of QM reaction to substituents was also examined by its free energy relationship [ln(ky/kH)] for addition of morpholine (Figures 4a,a′), water (Figure 4b,b′) and 2-mercaptoethanol (Figure 4c,c′). Linear correlations were slightly better with Hammett σp− constants (Figures 4a,b,c) than with Hammett σp constants (Figures 4a′,b′,c′). The σp− constants were originally set by changes in the pKa of para substituted phenols and are now used for systems with strong resonance interaction between substituent and reaction center. Such a case is proposed for reactive QMs that appear to develop a degree of aromaticity in the transition-state (TS) with negative charge efficiently stabilized by delocalization over the former oxygen carbonyl group and Y substituent (Scheme 6). Such resonance contributions are likely even greater for substituents at the 5-position since they may influence the exocyclic methylene of the QM more directly. Preliminary support for this prediction is evident by the greater influence of substituents at the 5- vs. 4-position as summarized in Table 2.
Figure 4.

Free energy relationships ln(ky/kH) vs Hammet σp− (a,b,c) and Hammet σp (a′,b′,c′) for the addition reactions of morpholine (a, a′), water (b, b′) and thiol (c, c′).
Scheme 6.

Aromaticity Develops in the Transition-state of QM Addition
Hansch had previously noted that σp− constants often yield better correlations than σp when resonance interaction develops between the substituent and the reaction center, and the resulting magnitude of ρ has provided insight into reaction mechanisms.49 For QM addition, the linear free energy correlations are positive and large. The values of ρ equal 3.0, 5.9 and 5.4 for morpholine, water and 2-mercaptoethanol, respectively (Figure 4a,b,c). In comparison, ρ values for the equilibrium acidity of phenol in water and DMSO are 2.1 and 5.3, respectively.62,63 The equilibrium acidity of p-substituted benzyltriphenylphosphonium cations to form the corresponding ylide in DMSO exhibited a ρ of 4.78.64 Much larger values of ρ (12 and 10) were determined for substituent effects on the acidity of a benzylic hydrogen in toluene and 10-substituted-9-methylanthracene derivatives in DMSO, respectively.65,66 The intermediate values of ρ for QM addition in water is then suggestive of significant anionic character in the TS. The lower value for the phosphonium salt vs. toluene was considered by Bordwell to indicate a diminished charge delocalization on the salt due to coulombic and polarization contributions by the triphenylphosphonium group.64 The smaller ρ value of morpholine vs. those of water and 2- mercaptoethanol may also be explained by the electrostatics of the developing positive charge on the nitrogen nucleophile.
Conclusions
Formation of QMs had previously demonstrated a dependence on the strength of the leaving group attached to the benzylic position of its precursor when studying a model o-QM in the presence of biological nucleophiles (Scheme 3).30,45 Now, both the formation and subsequent reaction of QMs have also been shown to be highly responsive to the presence of electron-withdrawing and donating groups. All trends in reactivity are consistent with the electron deficient nature of the QM intermediate. Electron donating groups greatly facilitate initial QM generation and its subsequent regeneration from adducts formed by the nucleophiles of deoxynucleosides that also function as good leaving groups. Conversely, electron withdrawing groups greatly suppress initial formation of QM and suppress its regeneration from the reversible deoxynucleoside adducts. These properties similarly extend to nucleophiles and leaving groups beyond those of DNA and provide a foundation for predicting the general behavior of QM stability and selectivity. In a complementary manner, electron-rich QMs react much more slowly but more selectively with nucleophiles than do the electron-poor QMs. These fundamental characteristics provide the basis for explaining the product profiles observed after DNA has been exposed to various QMs. In the future, these same characteristics may be used to fine-tune the stability and reactivity of QM conjugates for target-promoted and gene-specific alkylation.15 An equivalent approach may also be used to optimize the kinetics and sensitivity of polymers based on QMs to disassemble for drug release.32–34
Experimental Procedures for Quinone Methide Reaction
Kinetic Studies with Individual Deoxynucleosides
To a aqueous solution (70 μL) of 4 mM phenol, 0.5 mM deoxynucleoside, 10 mM potassium phosphate pH 7, 500 mM KF was added the quinone methide precursor (QMP1, QMP2 and QMP3 alternatively) in DMF (30 μL) yielding a final concentration of 25 mM. The reactions were incubated at 37 °C and, at the indicated times, analyzed directly by reverse phase HPLC (C-18, Varian, Microsorb-MV 300, 5 μm particle size, 250 mm × 4.6 mm) using a gradient of 3 % CH3CN, 9.7 mM TEAA, to 25 % CH3CN, 7.5 mM TEAA at 1 mL/min over 66 min. For QMP1 reaction, TEAA was adjusted to pH 4, and for QMP2 and QMP3, the TEAA was adjusted to pH 6. Product formation was monitored by integrating the elution profile at 260 nm generated by a diode array detector and normalizing its signal by the relative ε260 of each product and absorbance of an internal standard (phenol).
Competition Studies in the Presence of all Deoxynucleosides
To a aqueous solution (70 μL) of 4 mM phenol, 0.25 mM of dA, dC, dG, and T (1 mM total dN), 10 mM potassium phosphate pH 7, 500 mM KF, was added the quinone methide precursor (QMP1, QMP2 and QMP3 alternatively) in DMF (30 μL) yielding a final concentration of 25 mM. Reactions were incubated at 37°C and analyzed as described above.
Kinetics of Quinone Methide Regeneration and Benzylic Alcohol Formation from the Morpholino-quinone Methide Adducts
A stock solution of QMP6 in acetonitrile was diluted with water:acetonitrile (1:1) to yield final concentration of 1.0 mM, and anisole (0.6 mM) was added as internal standard. The resulting solution was incubated for 240 min at 50 °C. Every 5 min, an aliquot (200 μL) was removed and cooled at −20 °C in a ice-salt bath prior to analysis by reverse phase HPLC (C-18, Intersil ODS-2, 5 μm, column dimension: φ = 4.6 mm, length = 250 mm, φ = 10.0 mm, length = 250 mm) with 50% aq CH3CN (1 mL/min). The benzylic alcohol (QMA1) was detected with a variable-wavelength detected the alcohol and compared to an authentic sample. Reaction of the adducts (QMP4, QMP5, QMP7 and QMP8) was monitored similarly although reaction time and temperature as indicated in Table 1 to detect the benzylic alcohols (QMA3, QMA5, QMA7 and QMA8).
Laser Flash Photolysis (LFP) to Determine the Second Order Rate Constants for Addition of Water, Morpholine and 2-Mercaptoethanol to a Range of Quinone Methides
Kinetic studies of nucleophilic addition to QMs were carried out at 25 °C, in pure water for the hydration reaction and in the added presence of 2-mercaptoethanol (pH 7) and morpholine (adjusted to pH 9 by addition of Na2CO3) alternatively. The pH of each solution was measured using an Orion SA520 pH meter with a 8102 Ross electrode. QM1, QM5, QM8, QM11, QM13 and QM14 were generated by flashing a dilute solutions of QMP9-QMP14 (2–5 × 10−4 M). Disappearance of the QMs was followed under pseudo first-order conditions by monitoring the absorbance decrease at the maximum wavelength reported in Table 3. Pseudo-first-order rate constants (kobsd) were obtained from the fitting the absorbance data to a single exponential function and were reproducible to +/− 5%. The second-order rate constants k2 (M−1 s−1) for addition of nucleophiles to the QMs were determined as the least-squares linear slopes of kobsd vs total concentration of the nucleophile varied between 5×10−2 to 2×10−4 M.
Laser Flash Photolysis Apparatus
The laser pulse photolysis apparatus consisted of a Nd:YAG laser used at the fourth harmonic of its fundamental wavelength. It delivers a maximum power of 10 mJ at 266 nm with 10 ns pulse duration. The monitoring system was arranged in a cross-beam configuration and consisted of a 275 W Xe arc lamp, an F/3.4 monochromator, and a five-stage photomultiplier supplied by Applied Photophysics. Signals were captured by a Hewlett-Packard 54510A digitizing oscilloscope, and the data was processed on a 486-based computer system using software developed in-house. Solutions for analysis were placed in a fluorescence cuvette (d=10 mm) and adjusted to an absorbance of 1.5.
Supplementary Material
All synthetic protocols and product characterization; extinction coefficients and λmax values for deoxynucleoside-QM adducts; time-dependent consumption of the deprotected derivatives of QMP1, QMP2 and QMP3 to form QM1, QM2 and QM3, respectively; sample HPLC chromatograms used to monitor deoxynucleoside adducts formed by QM2 and QM3. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported in part by the National Institutes of Health (CA81571, SER), the Howard Hughes Medical Institute through the Undergraduate Biological Sciences Education Program (KNF, CHM) and Pavia University (FAR 2004, MF). We thank Dr. Yui-fai Yam for help with NMR characterization of the deoxynucleoside adducts.
References
- 1.Zewail-Foote M, Hurley LH. J Am Chem Soc. 2001;123:6485–6495. doi: 10.1021/ja004023p. [DOI] [PubMed] [Google Scholar]
- 2.Mattes WB, Hartley JA, Kohn KW. Nucleic Acids Res. 1986;14:2971–2987. doi: 10.1093/nar/14.7.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohn KW, Hartley JA, Mattes WB. Nucleic Acids Res. 1987;15:10531–10549. doi: 10.1093/nar/15.24.10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colvin OM, Friedman HS, Gamcsik MP, Fenselau C, Hilton J. Adv Enz Regula. 1993;33:19–26. doi: 10.1016/0065-2571(93)90006-y. [DOI] [PubMed] [Google Scholar]
- 5.Dedon PC, Plastaras JP, Rouzer CA, Marnett LJ. Proc Natl Acad Sci USA. 1998;95:11113–11116. doi: 10.1073/pnas.95.19.11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mao H, Schnetz-Boutaud NC, Weisenseel JP, Marnett LJ, Stone MP. Proc Natl Acad Sci USA. 1999;96:6615–6620. doi: 10.1073/pnas.96.12.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Plastaras JP, Riggins JN, Otteneder M, Marnett LJ. Chem Res Toxicol. 2000;13:1235–1242. doi: 10.1021/tx0001631. [DOI] [PubMed] [Google Scholar]
- 8.Riggins JN, Daniels S, Rouzer CA, Marnett LJ. J Am Chem Soc. 2004;126:8237–8243. doi: 10.1021/ja040009r. [DOI] [PubMed] [Google Scholar]
- 9.Boger DL, Johnson DS. Angew Chem, Int Ed Engl. 1996;35:1438–1474. [Google Scholar]
- 10.Lee SJ, Seaman FC, Sun D, Xiong H, Kelly RC, Hurley LH. J Am Chem Soc. 1997;119:3434–3442. [Google Scholar]
- 11.Boger DL, Yun W. J Am Chem Soc. 1993;115:9872–9873. [Google Scholar]
- 12.Boger DL, Garbaccio RM. Acc Chem Res. 1999;32:1043–1052. [Google Scholar]
- 13.Asai A, Nagamura S, Saito H, Takahashi I, Nakano H. Nucleic Acids Res. 1994;22:88–93. doi: 10.1093/nar/22.1.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veldhuyzen WF, Pande P, Rokita SE. J Am Chem Soc. 2003;125:14005–14013. doi: 10.1021/ja036943o. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Q, Rokita SE. Proc Natl Acad Sci USA. 2003;100:15452–15457. doi: 10.1073/pnas.2533112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P, Liu R, Wu X, Ma H, Cao X, Zhou P, Zhang J, Weng X, Zhang XL, Zhou X, Weng L. J Am Chem Soc. 2003;125:1116–1117. doi: 10.1021/ja029040o. [DOI] [PubMed] [Google Scholar]
- 17.Richter SN, Maggi S, Mels SC, Palumbo M, Freccero M. J Am Chem Soc. 2004;126:13973–13979. doi: 10.1021/ja047655a. [DOI] [PubMed] [Google Scholar]
- 18.Wang P, Song Y, Zhang L, He H, Zhou X. Curr Med Chem. 2005;12:2893–2913. doi: 10.2174/092986705774454724. [DOI] [PubMed] [Google Scholar]
- 19.Lewis MA, Yoerg DG, Bolton JL, Thompson JA. Chem Res Toxicol. 1996;9:1368–1374. doi: 10.1021/tx960115+. [DOI] [PubMed] [Google Scholar]
- 20.Fan PW, Zhang F, Bolton JL. Chem Res Toxicol. 2000;13:45–52. doi: 10.1021/tx990144v. [DOI] [PubMed] [Google Scholar]
- 21.Tomasz M. Chem Biol. 1995;2:575–579. doi: 10.1016/1074-5521(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Q, Zuniga MA. Chem Res Toxicol. 2005;18:382–388. doi: 10.1021/tx049703a. [DOI] [PubMed] [Google Scholar]
- 23.Angle SR, Yang W. J Org Chem. 1992;57:1092–1097. [Google Scholar]
- 24.Angle SR, Rainer JD, Woytowicz C. J Org Chem. 1997;62:5884–5892. [Google Scholar]
- 25.Pande P, Shearer J, Yang J, Greenberg WA, Rokita SE. J Am Chem Soc. 1999;121:6773–6779. [Google Scholar]
- 26.Veldhuyzen W, Lam Y-f, Rokita SE. Chem Res Toxicol. 2001;14:1345–1351. doi: 10.1021/tx0101043. [DOI] [PubMed] [Google Scholar]
- 27.Veldhuyzen WF, Shallop AJ, Jones RA, Rokita SE. J Am Chem Soc. 2001;123:11126–11132. doi: 10.1021/ja011686d. [DOI] [PubMed] [Google Scholar]
- 28.Freccero M, Di Valentin C, Sarzi-Amadè M. J Am Chem Soc. 2003;125:3544–3553. doi: 10.1021/ja028732+. [DOI] [PubMed] [Google Scholar]
- 29.Freccero M, Gandolfi R, Sarzi-Amadè M. J Org Chem. 2003;68:6411–6423. doi: 10.1021/jo0346252. [DOI] [PubMed] [Google Scholar]
- 30.Weinert EE, Frankenfield KN, Rokita SE. Chem Res Toxicol. 2005;18:1364–1370. doi: 10.1021/tx0501583. [DOI] [PubMed] [Google Scholar]
- 31.Wakselman M. New J Chem. 1983;7:439–447. [Google Scholar]
- 32.de Groot FMH, Albrecht C, Koekkoek R, Beusker PH, Scheeren HW. Angew Chem Int Ed. 2003;42:4490–4494. doi: 10.1002/anie.200351942. [DOI] [PubMed] [Google Scholar]
- 33.Li SJ, Szalai ML, Kevwitch RM, McGrath DV. J Am Chem Soc. 2003;125:10516–10517. doi: 10.1021/ja0349960. [DOI] [PubMed] [Google Scholar]
- 34.Shamis M, Lode HN, Shabat D. J Am Chem Soc. 2004;126:1726–1731. doi: 10.1021/ja039052p. [DOI] [PubMed] [Google Scholar]
- 35.Van De Water RW, Pettus TRR. Tetrahedron. 2002;58:5367–5405. [Google Scholar]
- 36.Amouri H, Besace Y, Le Bras J. J Am Chem Soc. 1998;120:6171–6172. [Google Scholar]
- 37.Amouri H, le Bras J. Acc Chem Res. 2002;35:501–510. doi: 10.1021/ar010105m. [DOI] [PubMed] [Google Scholar]
- 38.Rabin O, Vigalok A, Milstein D. Chem Eur J. 2000;6:454–457. doi: 10.1002/(sici)1521-3765(20000204)6:3<454::aid-chem454>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 39.Vigalok A, Milstein D. Acc Chem Res. 2001;34:798–807. doi: 10.1021/ar970316k. [DOI] [PubMed] [Google Scholar]
- 40.Filar LJ, Winstein S. Tet Lett. 1960:9–16. [Google Scholar]
- 41.Loubinoux B, Miazimbakana J, Gerardin P. Tetrahedron Let. 1989;30:1939–1942. [Google Scholar]
- 42.Bolton JL, Valerio LG, Thompson JA. Chem Res Toxicol. 1992;5:816–822. doi: 10.1021/tx00030a014. [DOI] [PubMed] [Google Scholar]
- 43.Bolton JL, Comeau E, Vukomanovic V. Chem Biol Interact. 1995;95:279–290. doi: 10.1016/0009-2797(94)03566-q. [DOI] [PubMed] [Google Scholar]
- 44.Rokita SE, Yang J, Pande P, Greenberg WA. J Org Chem. 1997;62:3010–3012. doi: 10.1021/jo9700336. [DOI] [PubMed] [Google Scholar]
- 45.Modica E, Zanaletti R, Freccero M, Mella M. J Org Chem. 2001;66:41–52. doi: 10.1021/jo0006627. [DOI] [PubMed] [Google Scholar]
- 46.Zhou Q, Pande P, Johnson AE, Rokita SE. Bioorg Med Chem. 2001;9:2347–2354. doi: 10.1016/s0968-0896(01)00151-1. [DOI] [PubMed] [Google Scholar]
- 47.Kumar D, Veldhuyzen WF, Zhou Q, Rokita SE. Bioconj Chem. 2004;15:915–922. doi: 10.1021/bc049941h. [DOI] [PubMed] [Google Scholar]
- 48.Zeng Q, Rokita SE. J Org Chem. 1996;61:9080–9081. [Google Scholar]
- 49.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. [Google Scholar]
- 50.Wan P, Barker B, Diao L, Fischer M, Shi Y, Yang C. Can J Chem. 1996;74:465–475. [Google Scholar]
- 51.Diao L, Yang C, Wan P. J Am Chem Soc. 1995;117:5396–5370. [Google Scholar]
- 52.Brousmiche DW, Xu M, Lukerman M, Wan P. J Am Chem Soc. 2003;125:12961–12970. doi: 10.1021/ja036808b. [DOI] [PubMed] [Google Scholar]
- 53.Chiang Y, Kresge AJ, Zhu Y. J Am Chem Soc. 2001;123:8089–8094. doi: 10.1021/ja010826g. [DOI] [PubMed] [Google Scholar]
- 54.Chiang Y, Kresge AJ, Zhu Y. J Am Chem Soc. 2002;124:717–722. doi: 10.1021/ja0120375. [DOI] [PubMed] [Google Scholar]
- 55.Chiang Y, Kresge AJ, Zhu Y. J Am Chem Soc. 2002;124:6349–6356. doi: 10.1021/ja020020w. [DOI] [PubMed] [Google Scholar]
- 56.Nakatani K, Higashida N, Saito I. Tet Lett. 1997;38:5005–5008. [Google Scholar]
- 57.Chiang Y, Kresge AJ. Org Biomol Chem. 2004;2:1090–1092. doi: 10.1039/b400098f. [DOI] [PubMed] [Google Scholar]
- 58.Toteva MM, Moran M, Amyes TL, Richard JP. J Am Chem Soc. 2003;125:8814–8819. doi: 10.1021/ja029588v. [DOI] [PubMed] [Google Scholar]
- 59.Richard JP, Toteva MM, Crugeiras J. J Am Chem Soc. 2000;122:1664–1674. [Google Scholar]
- 60.Toteva MM, Richard JP. J Am Chem Soc. 2000;122:11073–11083. [Google Scholar]
- 61.Mayr H, Ofial AR. Angew Chem Int Ed. 2006;45:1844–1854. doi: 10.1002/anie.200503273. [DOI] [PubMed] [Google Scholar]
- 62.Hammett LP. Physical Organic Chemistry. McGraw-Hill; New York: 1940. Ch 7 [Google Scholar]
- 63.Bordwell FG, McCallum RJ, Olmstead WN. J Org Chem. 1984;49:1424–1427. [Google Scholar]
- 64.Zhang XM, Fry AJ, Bordwell FG. J Org Chem. 1996;61:4101–4106. doi: 10.1021/jo951862z. [DOI] [PubMed] [Google Scholar]
- 65.Zhang XM, Bordwell FG, Bares JE, Cheng JP, Petrie B. J Org Chem. 1993;58:3051–3059. [Google Scholar]
- 66.Bordwell FG, Zhang XM, Cheng JP. J Org Chem. 1993;58:6410–6416. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All synthetic protocols and product characterization; extinction coefficients and λmax values for deoxynucleoside-QM adducts; time-dependent consumption of the deprotected derivatives of QMP1, QMP2 and QMP3 to form QM1, QM2 and QM3, respectively; sample HPLC chromatograms used to monitor deoxynucleoside adducts formed by QM2 and QM3. This material is available free of charge via the Internet at http://pubs.acs.org.