

Abstract
In order to develop targeted pharmaceutical carriers additionally capable of responding certain local stimuli, such as decreased pH values in tumors or infarcts, targeted long-circulating PEGylated liposomes and PEG-phosphatidylethanolamine (PEG-PE)-based micelles have been prepared with several functions. First, they are capable of targeting a specific cell or organ by attaching the monoclonal antimyosin antibody 2G4 to their surface via pNP-PEG-PE moieties. Second, these liposomes and micelles were additionally modified with biotin or TAT peptide (TATp) moieties attached to the surface of the nanocarrier by using biotin-PE or TATp-PE or TATp-short PEG-PE derivatives. PEG-PE used for liposome surface modification or for micelle preparation was made degradable by inserting the pH-sensitive hydrazone bond between PEG and PE (PEG-Hz-PE). Under normal pH values, biotin and TATp functions on the surface of nanocarriers were “shielded” by long protecting PEG chains (pH-degradable PEG2000-PE or PEG5000-PE) or by even longer pNP-PEG-PE moieties used to attach antibodies to the nanocarrier (non-pH-degradable PEG3400-PE or PEG5000-PE). At pH 7.5–8.0, both liposomes and micelles demonstrated high specific binding with 2G4 antibody substrate, myosin, but very limited binding on an avidin column (biotin-containing nanocarriers) or internalization by NIH/3T3 or U-87 cells (TATp-containing nanocarriers). However, upon brief incubation (15-to-30 min) at lower pH values (pH 5.0–6.0) nanocarriers lost their protective PEG shell because of acidic hydrolysis of PEG-Hz-PE and acquired the ability to become strongly retained on avidin-column (biotin-containing nanocarriers) or effectively internalized by cells via TATp moieties (TATp-containing nanocarriers). We consider this result as the first step in the development of multifunctional stimuli-sensitive pharmaceutical nanocarriers.
INTRODUCTION
Ideally, a nanoparticular drug delivery system (DDS) should be able: (a) to specifically accumulate in the required organ or tissue, and then (b) penetrate inside target cells delivering its load (drug or DNA) intracellularly. Organ or tissue (tumor, infarct) accumulation could be achieved by the passive targeting via the enhanced permeability and retention (EPR) effect (1, 2); or by the antibody-mediated active targeting (3, 4), while the intracellular delivery could be mediated by certain internalizable ligands (folate, transferrin) (5, 6) or by cell-penetrating peptides (CPPs, such as TAT or polyArg) (7, 8). Such a DDS should simultaneously carry on its surface various active moieties, i.e. be multifunctional and possess the ability to switch on and switch off certain functions when necessary, under the action of local stimuli characteristic of the target pathological zone (first of all, increased temperature or lowered pH values characteristic of inflamed, ischemic, and neoplastic tissues). Another important requirement is that different properties of the multifunctional DDS are coordinated in an optimal fashion. Thus, for example, if the system is to be constructed that can provide the combination of the longevity allowing for the target accumulation via the EPR effect and specific cell surface binding allowing for its internalization by target cells, two requirements have to be met. First, the half-life of the carrier in the circulation should be long enough to fit EPR effect requirements, and second, the internalization of the DDS by the target cells should proceed fast enough not to allow for the carrier degradation and drug loss in the interstitial space. However, development of systems like this still represents a challenge.
Intracellular transport of different biologically active molecules is one of the key problems in drug delivery in general. Nanoparticular DDS, such as liposomes and micelles, are frequently used to increase the efficacy of drug and DNA delivery and targeting (9, 10). So far, multiple and not always successful attempts have been made to deliver various drug carriers directly into the cell cytoplasm, bypassing the endocytic pathway, to protect drugs and DNA from the lysosomal degradation, thus enhancing drug efficiency and DNA incorporation into the cell genome (11–14). Within the paradigm of multifunctional DDS, one can think about the development of a DDS built in such a way that during the first phase of delivery, a non-specific cell-penetrating function is shielded by the function providing organ/tissue-specific delivery (sterically-protecting polymer or antibody). Upon accumulating in the target, protecting polymer or antibody attached to the surface of the DDS via the stimuli-sensitive bond should detach under the action of local pathological conditions (abnormal pH or temperature) and expose the previously hidden second function allowing for the subsequent delivery of the carrier and its cargo inside cells. While such DDS should be stable in the blood for a long time (hours) to allow for an efficient target accumulation, it has to lose the protective coat inside the target almost instantly to allow for the fast internalization (minutes) to minimize the washing away of the released drug or DNA. The schematic pattern of such system is shown in Figure 1. Intracellular trafficking, distribution and fate of the carrier and its cargo can be additionally controlled by its charge and composition, which can drive it to the nuclear compartment or towards other cell organelles.
Figure 1.

Interaction of the multifunctional pH-responsive pharmaceutical nanocarrier with the target cell. Local stimuli-dependent removal of protecting PEG chains or mAb-PEG moieties allows for the direct interaction of the CPP moiety with the cell membrane.
Within last few years, it has been demonstrated that certain proteins and peptides (CPPs, such as TAT peptide, TATp) can enter cell cytoplasm directly and even target cell nuclei (15, 16). CPPs have also been successfully used for the intracellular delivery of small drug molecules, large molecules (enzymes, DNA) and nanoparticulates (quantum dots, iron oxide nanoparticles, liposomes) (13, 17–22). The mechanism of this phenomenon is still under investigation, although an important progress has been already made and it was shown that electrostatic interactions and hydrogen bonding lay behind the CPP-mediated direct transduction of small molecules (23, 24), while the energy-dependent macropinocytosis is responsible for the CPP-mediated intracellular delivery of large molecules and nanoparticulates with their subsequent enhanced release from endosomes into the cell cytoplasm (25, 26).
In this study, we have attempted to prepare and test in vitro stable targeted PEGylated DDS (liposomes and micelles) containing the second specific function (i.e. the intracellular internalization of the DDS), which, under normal conditions, is shielded by the protecting polymer or polymer-antibody conjugate, however can be “developed” upon brief incubation at lowered pH values characteristic of inflamed or neoplastic areas. As the first step on the way to multifunctional drug delivery systems, we have prepared liposomes and micelles sterically protected by the low pH-cleavable PEG chains and additionally containing a monoclonal antibody attached to the surface of the DDS via non-cleavable longer PEG spacer and an additional function (biotin or TATp) attached to the surface of the DDS via the non-cleavable anchor shorter than the cleavable PEG chain.
MATERIALS AND METHODS
Materials
Cell lines, mouse fibroblast NIH 3T3 and human astrocytoma U-87 MG, were purchased from the American Type Culture Collection (Manassas, VA). All cell culture media, DMEM, and RPMI 1640, heat-inactivated fetal bovine serum (FBS), and concentrated solutions of sodium pyruvate and penicillin/streptomycin stock solutions were purchased from Cellgro® (Herndon, VA). TAT-peptide (11-mer: TyrGlyArgLysLysArgArgGlnArgArgArg; molecular mass, 1,560 Da; three reactive amino groups) was prepared by Research Genetics (Huntsville, AL). Monoclonal antibody (mAb) 2G4 was produced and purified in our laboratory. Also, pNP-PEG3400-PE was synthesized and purified according to an established method in our laboratory (27). Egg phosphatidylcholine (PC), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-750] (ammonium salt) (mPEG750-PE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (ammonium salt) (mPEG2000-PE), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (sodium salt) (biotin-PE) 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (sodium salt) (PE-SH) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (Rh-PE) were obtained from Avanti Polar Lipids, Inc. (Alabaster, AL). mPEG2000-butyraldehyde (mPEG2000-CHO) and mPEG5000-butyraldehyde (mPEG5000-CHO) was obtained from Nektar™ (Huntsville, AL). Control bovine antibody IgG was obtained from Serologicals Proteins, Inc. (Kankakee, IL). 3-(2-Pyridyldithio) propionyl hydrazide (PDPH) and Immobilized NeutrAvidin™ protein was purchased from Pierce Biotechnology, Inc. (Rockford, IL). Bovine serum albumin and all other chemicals and buffer solution components were from Sigma (St. Louis, MO) and were of analytical grade.
Synthesis of pH-cleavable mPEG2000-hydrazone-phospatidyl ethanolamine (mPEG2000-Hz-PE)
Developing the method of coupling oxidized antibody to the PEG terminus through a hydrazone bond as suggested by Hansen et al (28), we devised our own scheme of conjugate reaction for synthesis of pH-cleavable PEG-PE. The reaction was performed in two steps: first, the activation of mPEG2000-CHO with PDPH, and second, the conjugation of 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (sodium salt) (PE-SH) to activated mPEG-CHO.
For the step I of the synthesis, 150 mg (64 μmole) of mPEG2000-CHO was dissolved in dry chloroform containing 3.5 molar excess of 3-(2-pyridyldithio) propionyl hydrazide (PDPH) to obtain 50 mg/ml solution of mPEG2000-CHO. The mixture was incubated for 48 h at room temperature with stirring under argon. Thin layer chromatography (TLC) (CHCl3 : CH3OH : H2O - 80:20:2) revealed that the reaction was complete. The starting material mPEG2000-CHO did not absorb UV and was positive to Dragendorff spray, while PDPH absorbed UV and was negative for Dragendorff spray. The product, mPEG2000-Hz-PDP, absorbed UV and was positive to Dragendorff spray. Organic solvents were then removed using a rotary evaporator. mPEG2000-Hz-PDP was then dissolved in deionized water (adjusted to pH 10–11 using 1 M NaOH) and purified from unreacted PDPH using Sepharose G25 column and deionized water (adjusted to pH 10.5 using 1 M NaOH). Pooled fractions containing mPEG2000-Hz-PDP (Dragendorff and UV positive) were freeze-dried.
For the step II of the reaction, 20 mg (26 μmole) of 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (sodium salt) (PE-SH) was dissolved in dry chloroform containing 1.5 molar excess of mPEG2000-Hz-PDP, to get a 10 mg/mL solution of PE-SH. The solution was supplemented with 15 μL (approx. 3-fold molar excess over PEG) of triethylamine (TEA). The sample was incubated overnight at room temperature with stirring under argon. TLC (CHCl3:CH3OH:H2O - 80:20:2) revealed that the reaction was complete. The starting material mPEG2000-Hz-PDP was positive to Dragendorff spray and negative for molybdenum blue, while PE-SH was positive to molybdenum blue and negative for Dragendorff spray. The product, mPEG2000-Hz-PE, was positive to both Dragendorff spray and molybdenum blue. The organic solvents were then removed using a rotary evaporator. The mPEG2000-Hz-PE micelles were formed in deionized water (adjusted to pH 10.5 using 1 M NaOH) by vortexing. The micelles were separated from the unbound PEG and released pyridine-2-thione on CL-4B column using deionized water (adjusted to pH 10.5 using 1 M NaOH) as an eluent. Pooled fractions containing mPEG2000-Hz-PE were freeze-dried and extracted with chloroform. mPEG2000-Hz-PE was stored as 10 mg/mL chloroform solution at −80° C under argon until further use. mPEG5000-Hz-PE used in some experiments was synthesized in the same way starting with mPEG5000-CHO.
Acidic pH cleavability of mPEG2000-Hz-PE
TLC analysis
TLC-verified degradation of the polymer conjugates after pH treatment and spots corresponding to plain PEG and plain PE were observed after incubation of the polymers at pH 5.0 for 15 minutes at 37° C.
HPLC analysis
Micelles of mPEG2000-Hz-PE were prepared containing 1 mol % of rhodamine-PE as fluorescent marker as follows. Lipid film was prepared by mixing chloroform solutions of both the lipids in a round bottom flask and then removing chloroform using rotary evaporator. To ensure complete removal of any traces of chloroform further drying was done using lyophilizer. Appropriate volume of pH 8.5 phosphate buffer (100 mM phosphate, 150 mM sodium chloride) was added and vortexed for 2 min to form 0.5 mM solution of mPEG2000-Hz-PE micelles. Sample was then divided into equal volumes and treated for different pH incubation. For pH 7.4 treatment, a 50 μL aliquot of the first half of the micelle formulation was applied, as is, to a Shodex KW-804 size exclusion column at regular intervals using pH 7.4 Phosphate buffer (100 mM phosphate, 150 mM sodium sulfate) was used as the eluent and run at 1.0 ml/min. Both UV (from 200 to 400 nm) and fluorescence (550/590) was used to monitor the micelles. To the second half of the micelle formulation an appropriate volume of 1N HCl was added to get a final pH of ~ 5.0; aliquots of which were then analyzed as above at different intervals. As a control, micelles of mPEG2000-PE (non-pH-sensitive micelles) were prepared and analyzed after treatment at both the pH values as above.
Kinetics of the pH-dependent degradation of mPEG2000-Hz-PE
The degradation of the micelles spontaneously formed by mPEG2000-Hz-PE under the action of the acidic pH was studied by following the presence or absence of micelles over the period of time in buffer solutions of different pHs (i.e. pH values 6.0, 7.0, 8.0 and 10). Rh-PE-labeled micelles of mPEG2000-Hz-PE conjugate were prepared in phosphate buffer (10mM phosphate, 150mM NaCl) solutions of different pH values. The pH of the solution was adjusted using appropriate amounts of either 1N HCl or NaOH. 50 μl aliquots were sampled out at different time intervals for size exclusion chromatographic analysis in 100mM phosphate buffer (pH 7.0) containing 150mM sodium sulphate using fluorescence detector (EX: 550 nm, EM: 590 nm). The area under micelle peak (mean retention time: 9.35 min) was determined for each chromatogram.
Preparation of pH-sensitive DDS
Micelles
For micelle preparations, a mixture of mPEG750-PE, pH-sensitive mPEG2000-Hz-PE, biotin-PE (or TATp-PE), and Rh-PE at molar ratio of 40:54:5:1 was prepared in chloroform. Chloroform was removed on a rotary evaporator followed by freeze-drying on a Freezone 4.5 (Labconco, Kansas City, MO). The film was hydrated with PBS, pH 8.0 (10 mM phosphate, 150 mM sodium chloride) at room temperature and vortexed for 5 min. Micelle size was controlled by using a Coulter N4 Plus submicron particle analyzer.
Liposomes
For liposome preparations, a mixture of phosphatidylcholine and cholesterol in 6:3 molar ratio and with the addition of various quantities (up to 18% mol) of PEG5000-Hz-PE or mPEG5000-PE was prepared in chloroform. When required, the composition for liposome preparation was supplemented with 0.5 to 1 % mol of TATp- PEG2000-PE (prepared as described earlier in (13)) and with 0.5 % mol of Rh-PE (for the fluorescent labeling). Chloroform was removed on a rotary evaporator followed by freeze-drying on a Freezone 4.5 (Labconco, Kansas City, MO). The film obtained was hydrated with HBS buffer (pH 8.0) at room temperature for 5 min. The lipid dispersion was extruded 20 times through polycarbonate filters (pore size 200 nm) by using a Micro extruder (Avanti). Vesicle size was controlled by using a Coulter N4 Plus submicron particle analyzer.
Preparation of immunocarriers
First, mAb 2G4 or nonspecific control bovine IgG was conjugated to pNP-PEG3400-PE as in (27) with some modifications. Briefly, pNP-PEG3400-PE and mPEG750-PE was dried in a rotary evaporator and freeze-dryer to form a thin film. The film was hydrated with 5mM citrate buffered saline, pH 5.0, and vortexed. Antibody solution was prepared in 50mM tris-buffered saline, pH 8.7 and incubated with a 10-fold molar excess of pNP-PEG3400-PE for 24 h at 4°C to allow the attachment of the antibody to the activated PEG terminus with the simultaneous hydrolysis of non-reacted pNP groups, thus forming the antibody-micelle solution. Then, the required aliquot of this solution was added to liposomes or micelles prepared as described above and incubated for about an hour to allow for the quantitative incorporation of the modified antibody into the appropriate DDS (29).
ELISA
An ELISA assay (indirect, using an enzyme-tagged secondary Ab) was performed to show the ability of the pH-sensitive immunocarriers to recognize the target antigen at different pH values (pH 8.0 and 5.0).
First, ELISA plates were coated with 50 μl of 10 μg/ml cardiac myosin and incubated overnight at 4°C. Then, each well was washed three times with 200 μl of TBST (TBS containing 0.05% w/v Tween-20), and incubated with 50 μl of serial dilutions of 2G4 antibody (or non-specific IgG) in TBST-casein (TBST with 2 mg/mL casein) for 1 h at RT. After incubation, the wells were washed as before and incubated with 50 μl/well of 1:5000 dilution of goat anti-mouse IgG peroxidase conjugate (ICN Biomedicals, Inc., Aurora, OH) in TBST-Casein for 1 h at RT. The wells were again washed as before, and each well was incubated with 100 μl of enhanced Kblue® TMB peroxidase substrate (Neogen Corporation, Lexington, KY) for 15 min. The microplate was read at a dual wavelength of 620nm with the reference filter at 492nm using a Labsystems Multiskan MCC/340 microplate reader installed with GENESIS-LITE windows based microplate software.
Biotin-avidin binding
To test the binding of biotin-bearing Rh-PE-labeled DDS before and after incubation at lowered pH values, the corresponding samples were kept for 15 min at pH 8.0 or pH 5.0 and then applied onto the Immobilized NeutrAvidin™ protein column. The degree of the retention of the corresponding preparation on the column was estimated following the decrease in the sample rhodamine fluorescence at 550/590 nm after passing through the NeutrAvidin™ column.
Interaction of TATp-containing pH-sensitive DDS with cells
For experiments with the micelles, NIH 3T3 cells (fibroblasts) have been chosen. After the initial passage in tissue culture flasks, NIH 3T3 cells were grown on coverslips in 6-well tissue culture plates (100,000 cells per well) in DMEM with 10% BSA. After 48 h the plates were washed twice with PBS, pH 7.4, and then treated with various Rh-PE-labeled micelle samples (without and with pre-incubation for 15 min at pH 5.0) in serum-free medium (2 ml/well, 30 mg total PEG-PE/ml). After a 1 h incubation period, the media were removed and the plates washed with serum-free medium three times. Individual coverslips were mounted cell-side down onto fresh glass slides with PBS. Cells were viewed with a Nikon Eclipse E400 microscope under bright light, or under epifluorescence with rhodamine/TRITC. For experiments with the liposomes, U-87 MG cells (astrocytoma) have been chosen.
After the initial passage in tissue culture flasks, U-87 MG cells were grown on coverslips in 6-well tissue culture plates (20,000 cells per well) in DMEM with 10% BSA. After 48 h the plates were washed twice with PBS, pH 7.4, and then treated with various Rh-PE-labeled liposome samples (with and without pre-incubation for 20 min at pH 5.0) in serum-free medium (2 ml/well, 30 mg total lipid/ml). After a 1 h incubation period, the media were removed and the plates washed with serum-free medium three times. Individual coverslips were mounted cell-side down onto fresh glass slides with PBS. Cells were viewed with a Nikon Eclipse E400 microscope under bright light, or under epifluorescence with rhodamine/TRITC.
RESULTS AND DISCUSSION
The particular design of the multifunctional DDS we have used in this study is presented in Figure 2. The suggested DDS (liposome or micelle) bears on its surface a “hidden” function (we have used biotin and TATp moieties for this purpose) inserted into the liposome membrane or micelle core via its modification with PE moiety; protecting PEG chains (PEG2000) attached to the surface via the pH-cleavable bond; and specific antibody attached to the surface via non-cleavable long PEG spacer (PEG3400). In some experiments with liposomes, cleavable PEG5000-Hz-PE and non-cleavable TATp-PEG2000-PE conjugates have been used. Such DDS are expected to demonstrate specific targeted properties (via antibody-mediated recognition) at both normal (7.5–8.0) and acidic (5.0–6.0) pH values, however, the incubation of the DDS at lowered pH should eliminate (detach) protecting PEG chains and de-shield the second function. i.e., after the exposure to the lowered pH, in addition to the immune recognition, such DDS should acquire the ability to bind with avidin column if the second, “hidden” function is biotin, or to demonstrate better internalization by cells if the second, “hidden” function is TATp. As an antibody, we have used the cardiac myosin-specific monoclonal 2G4 antibody (mAb 2G4) (30). The coupling of mAb 2G4 and biotin or TATp to the DDS surface was performed using the reactive derivative of poly(ethylene glycol)-phosphatidyl ethanolamine conjugate (PEG-PE) activated at free PEG terminus with p-nitrophenylcarbonyl (pNP) group (pNP-PEG-PE) according to the protocol developed by us earlier (27).
Figure 2.

Schematic for the design of the multifunctional DDS used in this study that includes pH-cleavable PEG-Hz-PE (a), temporarily “shielded” biotin or TATp (b), and monoclonal antibody (c) attached to the surface of DDS via pH-non-cleavable spacer.
Synthesis of pH-cleavable PEG-PE conjugate
The scheme of the synthesis of PEG-PE conjugated via the pH-cleavable hydrazone group (PEG-Hz-PE) is shown in the Figure 3. The synthesis was carried out in 2 steps. The first step involves the conjugation of 3-(2-pyridyldithio) propionyl hydrazide (PDPH) to mPEG2000-CHO. The hydrazide group in PDPH reacts with the aldehyde group of mPEG2000-CHO to form the acidic-pH-labile hydrazone bond. Since this bond is vulnerable to hydrolysis, efforts were taken to carry out the reaction in anhydrous conditions. Use of PDPH as the cross linker not only offers the advantage of forming the hydrazone bond but also introducing pyridyldisulfanyl groups for subsequent conjugation of PEG to thiol component of 1,2-dipalmitoyl-sn-glycero-3-phosphothioethanol (PE-SH) in the second step of the synthesis. The PE-SH in the conjugate serves as a hydrophobic anchor to assure its association with the lipid bilayer of liposome or the hydrophobic core of micelles. All steps of the reaction were followed on TLC to confirm the progress of the reaction. The structure of the final conjugate, mPEG2000-phospatidyl ethanolamine hydrazone (mPEG2000-Hz-PE) was confirmed by the proton NMR characterization: 1H NMR (500 MHz, CDCl3) δ (ppm) for mPEG2000-Hz-PE: 0.87 (t, CH3 of lipid, 6H), 1.27 (b, s, CH2, ≈ 56H), 2.29 (t, OCOCH2, 4H), 2.40 (t, COCH2CH2S, 2H), 2.45 (t, SCH2CH2O, 2H), 2.5 (t, COCH2CH2S, 2H), 2.59 (m, CH2CH=N, 2H), 3.1 (t, CH2CH, 2H), 3.39 (s, OCH3 of PEG, 3H) and 3.5 (bm, PEG, ≈ 184H).
Figure 3.

Schematic description of the conjugation reaction.
The stability of the conjugate and the kinetics of its degradation were analyzed by the size-exclusion HPLC following the area under the micelle peak on the chromatogram after conjugate incubation for different time intervals at different pH values (the conjugate spontaneously forms micelles in aqueous solutions similar to “normal” PEG-PE conjugate). Rhodamine-PE was incorporated into the micelles as a fluorescent tag, and the sample was monitored using fluorescence detector with excitation at 550 nm and emission at 590 nm. As a typical example, HPLC for mPEG2000-Hz-PE is shown in Figure 4; after appropriate pH treatment the peak at retention time 9.7 min (peak for micelle) observed in case of intact micelle (pH 8.0) disappears after the incubation at pH 5.0. This disappearance of the peak is indicative of the destruction of the micelle structure due to the loss of PEG corona, while non-pH-sensitive micelles produce the peak at both pH values. Table I shows the representative data for the degradation kinetics of PEG2000-Hz-DPPE micelles at different pH values. It is clearly seen from there that such micelles are quite stable at high pH value (8 and above), they disintegrate within few minutes at pH 5.0. This result was also confirmed by the TLC, when two different spots of PEG and PE were observed after few minutes of incubation at pH 5.0 with no spot of PEG-PE seen after the incubation (data not shown).
Figure 4.

HPLC analysis of the pH-sensitive mPEG2000-Hz-PE micelles after incubation in pH 8.0 (A) and after incubation in pH 5 (B) at room temperature.
Table I.
PEG2000-Hz-PE micelle stability at different pH values (as per cent of remaining micelles)
| pH value | 5.0 | 7.0 | 8.0 | 10.0 |
|---|---|---|---|---|
| Incubation Time | ||||
| 20 min | 3 | 56 | 94 | 99 |
| 40 min | 2.5 | 28 | 62 | 99 |
| 60 min | 2 | 10 | 53 | 99 |
Characterization of liposomes and micelles
According to the particle size analysis data, all prepared liposomes had the size within the 120-to-160 nm interval; micelle size was within the 7-to-15 nm interval. Critical micellation concentration (CMC) for both hydrolizable and non-hydrolizable PEG-PE conjugates determined by the pyrene method as in (31) was around 1.1 × 10−5 M. The average number of antibody molecule per single 150 nm liposome determined as in (29) was between 60 and 80; the average number of antibody molecules per single 10 nm micelle was around 5.
Immunoreactivity of multifunctional DDS
Fully assembled 2G4 antibody-bearing DDS demonstrated clear immunoreactivity towards the antigen, the monolayer of dog cardiac myosin in the standard ELISA test at both tested pH values, 8.0 and 5.0, as can be seen in Figure 5 (the results for PEG-PE-based 2G4-immunomicelles are shown). Although, one can observe some affinity decrease for the antibodies modified with the pNP-PEG-PE anchor and incorporated into the micelle structure (the same pattern is observed for immunoliposomes), this decrease is more apparent than real, since not all DDS-attached antibodies, even if they remain active, can interact with the substrate because of their steric orientation on the DDS surface. In a previous study it was shown that this reduction of activity in some of the antibodies is well compensated by the multipoint interaction of the DDS with the target (32,33). Thus, the systems prepared are immunologically active at both chosen pH values. Control preparations bearing a non-specific IgG exhibited no binding to myosin at any pH.
Figure 5.

Binding of antimyosin mAb 2G4-PEG2000-Hz-PE-immunomicelles to a monolayer of dog cardiac myosin in comparison to the native mAb 2G4 at corresponding pH values.
Avidin binding of multifunctional biotin-containing DDS
The avidin-biotin complexation was used initially as an easy-to-handle test system to follow the shielding and de-shielding of the second hidden function in pH-sensitive DDS. Liposomes and micelles containing 5 % mol of the biotin-PE in addition to 2G4-PEG3400-PE and pH-sensitive PEG2000-Hz-PE have been prepared and labeled with rhodamine-PE, and their ability to interact with avidin (NeutrAvidin affinity column) was investigated at pH 8.0 and after the brief (15 min) exposure at the lowered pH of 5.0. It was found that although biotin-containing 2G4-DDS have demonstrated identical immunoreactivity at both pH values, their ability to bind with avidin was dramatically different at pH 8.0 and after 15 min incubation at pH 5.0, which was expected to cleave away a substantial portion of the shielding PEG2000 micelle corona (or liposome coating). The data in Figure 6 (for micellar DDS) clearly show that while at pH 8.0 only about 15% of micelles were retained by the avidin column, after 15 min incubation at pH 5.0 about 75% of micelles were retained (the degree of the binding was estimated following the decrease in the sample rhodamine fluorescence at 550/590 nm after passing through the avidin column). This result clearly confirms that the elimination of the pH-cleavable PEG coat de-shields the hidden biotin function and allows for more biotin moieties to interact with avidin on the column.
Figure 6.
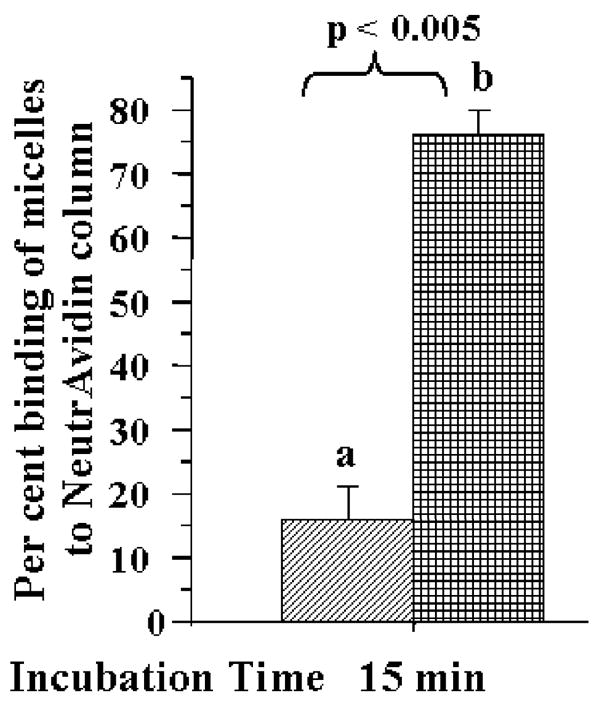
Binding of pH-sensitive biotin-micelles to NeutrAvidin columns after 15 min incubation at room temperature at pH 8.0 (a) and at pH 5.0 (b).
TATp-mediated interaction of multifunctional DDS with cells in vitro
For cell culture experiments, we have used rhodamine-labeled DDS similar to those described above, but containing TATp moieties attached to their surface instead of biotin groups. In this case, we have investigated DDS internalization by various cells (non-targets for the 2G4 antibody) at pH 8.0 and after the brief (20–30 min) exposure at the pH 5.0. It was found that TATp-containing DDS have also demonstrated a dramatically different ability to interact with cells at pH 8.0 and after the incubation at pH 5.0, which was expected to cleave away a substantial portion of the shielding PEG2000 micelle corona (or liposome coating). The data in Figure 7 clearly show that while cleavable PEG-PE-based TATp-containing micelles kept at pH 8.0 show only marginal association with NIH-3T3 murine fibroblasts, the same micelles pre-incubated for 30 min at pH 5.0 demonstrated dramatically enhanced association with the cells (higher fluorescence), i.e. better accessibility of TATp moieties for cell interaction. In case of TAT-bearing liposomes (Fig. 8), we have shown that the incorporation of 9 % mol of PEG-PE strongly diminished TATp-uptake of liposomes (Fig. 8B), and the incorporation of 18 % mol of PEG-PE completely eliminated it (Fig. 8C). However when pH-degradable PEG-PE was used, 20 min preincubation of both preparations at pH 5.0 significantly increased the association of both preparations with cells, bringing the cell binding of the liposomes with 9 % mol PEG (Fig. 8D) almost back to the level of PEG-free TATp-liposomes (Fig. 8A), and significantly improving the cell binding of the TATp-liposomes with 18 % mol of the initial PEG (Fig. 8E). These results clearly confirm that the elimination of the pH-cleavable PEG coat de-shield the hidden TATp function and allows for better association of DDS with cells.
Figure 7.
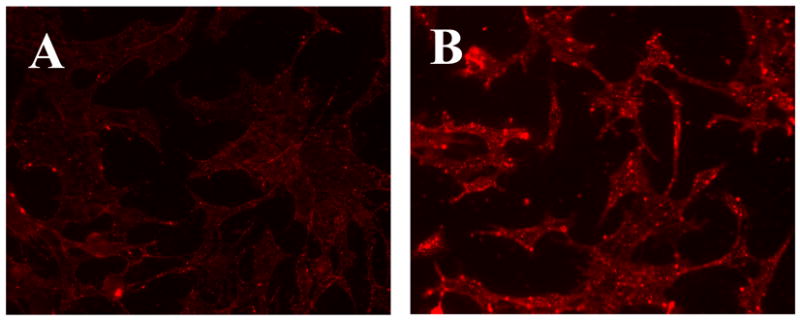
Fluorescence microscopy showing internalization of Rh-PE-labeled-TATp containing micelles by NIH 3T3 fibroblast cells after incubating micelles at pH 8.0 (A) and pH 5.0 (B) for 30 min.
Figure 8.
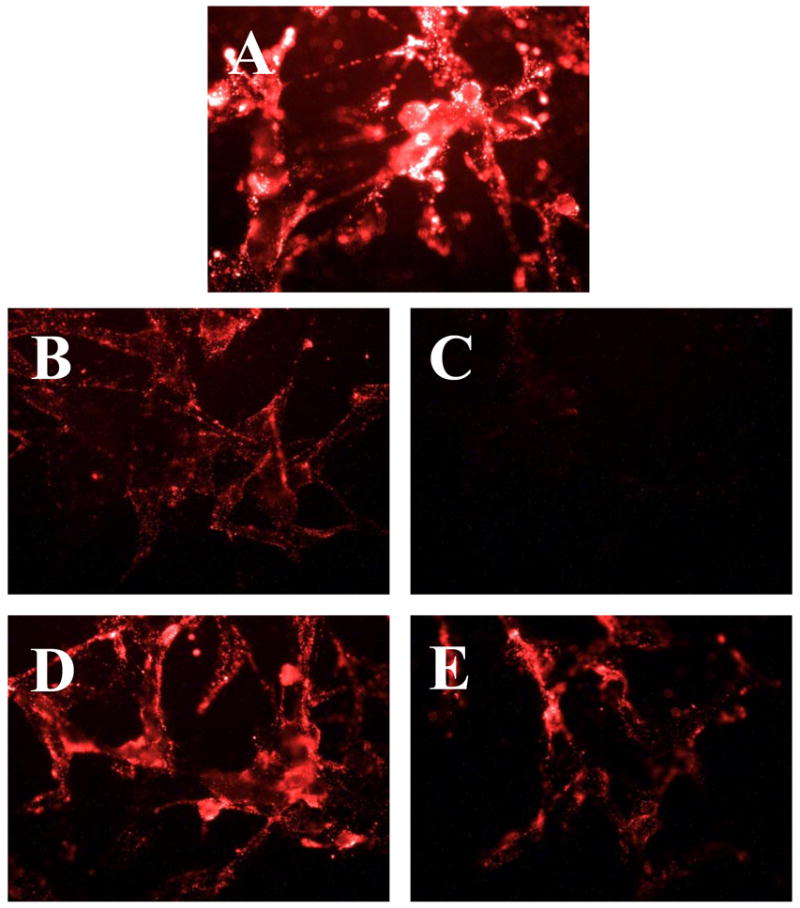
Fluorescence microscopy showing internalization of Rh-PE-labeled-TATp containing liposomes by U-87 MG astrocytoma cells using: PEG-free TATp-liposomes (A); 9 % mol pH-non-cleavable PEG-PE at pH 7.4 (B); 18 % mol pH-non-cleavable PEG-PE at pH 7.4 (C); 9 % mol pH-cleavable PEG-Hz-PE after incubation at pH 5.0 for 20 min (D)§, 18 % mol pH-cleavable PEG-Hz-PE after incubation at pH 5.0 for 20 min (E)§. §The pH of these formulations was raised back to pH 7.4 after their incubation at pH 5.0 and prior to incubation with cells.
The development of the double-targeted drug carriers, which, in addition to prolonged circulation (via the attached PEG) and target recognition (via the attached antibody), carry the temporarily hidden function that can develop under the action of certain local stimuli (such as lowered pH), represent a significant step on the way towards “smart” multifunctional pharmaceutical nanocarriers capable of both target accumulation and intracellular penetration in a controlled fashion.
Acknowledgments
This work was supported by the NIH grants R01 HL55519 and R01 EB001961 to Vladimir P. Torchilin.
References
- 1.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–84. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 2.Palmer TN, Caride VJ, Caldecourt MA, Twickler J, Abdullah V. The mechanism of liposome accumulation in infarction. Biochim Biophys Acta. 1984;797:363–8. doi: 10.1016/0304-4165(84)90258-7. [DOI] [PubMed] [Google Scholar]
- 3.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13:5043–54. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 4.Torchilin VP. Targeted polymeric micelles for delivery of poorly soluble drugs. Cell Mol Life Sci. 2004;61:2549–59. doi: 10.1007/s00018-004-4153-5. [DOI] [PubMed] [Google Scholar]
- 5.Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev. 2004;56:1177–92. doi: 10.1016/j.addr.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 6.Widera A, Norouziyan F, Shen WC. Mechanisms of TfR-mediated transcytosis and sorting in epithelial cells and applications toward drug delivery. Adv Drug Deliv Rev. 2003;55:1439–66. doi: 10.1016/j.addr.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57:637–51. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Lochmann D, Jauk E, Zimmer A. Drug delivery of oligonucleotides by peptides. Eur J Pharm Biopharm. 2004;58:237–51. doi: 10.1016/j.ejpb.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 9.Torchilin VP. Lipid-core micelles for targeted drug delivery. Curr Drug Deliv. 2005;2:319–27. doi: 10.2174/156720105774370221. [DOI] [PubMed] [Google Scholar]
- 10.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–60. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 11.Maheshwari A, Mahato RI, McGregor J, Han S, Samlowski WE, Park JS, Kim SW. Soluble biodegradable polymer-based cytokine gene delivery for cancer treatment. Mol Ther. 2000;2:121–30. doi: 10.1006/mthe.2000.0105. [DOI] [PubMed] [Google Scholar]
- 12.Tachibana R, Harashima H, Shono M, Azumano M, Niwa M, Futaki S, Kiwada H. Intracellular Regulation of Macromolecules Using pH-Sensitive Liposomes and Nuclear Localization Signal: Qualitative and Quantitative Evaluation of Intracellular Trafficking. Biochemical and Biophysical Research Communications. 1998;251:538–544. doi: 10.1006/bbrc.1998.9460. [DOI] [PubMed] [Google Scholar]
- 13.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci U S A. 2001;98:8786–91. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wattiaux R, Laurent N, Wattiaux-De Coninck S, Jadot M. Endosomes, lysosomes: their implication in gene transfer. Advanced Drug Delivery Reviews. 2000;41:201–208. doi: 10.1016/s0169-409x(99)00066-6. [DOI] [PubMed] [Google Scholar]
- 15.Caron NJ, Torrente Y, Camirand G, Bujold M, Chapdelaine P, Leriche K, Bresolin N, Tremblay JP. Intracellular delivery of a Tat-eGFP fusion protein into muscle cells. Mol Ther. 2001;3:310–8. doi: 10.1006/mthe.2001.0279. [DOI] [PubMed] [Google Scholar]
- 16.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–7. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 17.Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A. 1994;91:664–8. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudolph C, Plank C, Lausier J, Schillinger U, Muller RH, Rosenecker J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J Biol Chem. 2003;278:11411–8. doi: 10.1074/jbc.M211891200. [DOI] [PubMed] [Google Scholar]
- 19.Santra S, Yang H, Holloway PH, Stanley JT, Mericle RA. Synthesis of water-dispersible fluorescent, radio-opaque, and paramagnetic CdS:Mn/ZnS quantum dots: a multifunctional probe for bioimaging. J Am Chem Soc. 2005;127:1656–7. doi: 10.1021/ja0464140. [DOI] [PubMed] [Google Scholar]
- 20.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–72. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 21.Torchilin VP, Levchenko TS, Rammohan R, Volodina N, Papahadjopoulos-Sternberg B, D’Souza GG. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc Natl Acad Sci U S A. 2003;100:1972–7. doi: 10.1073/pnas.0435906100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao M, Kircher MF, Josephson L, Weissleder R. Differential conjugation of tat peptide to superparamagnetic nanoparticles and its effect on cellular uptake. Bioconjug Chem. 2002;13:840–4. doi: 10.1021/bc0255236. [DOI] [PubMed] [Google Scholar]
- 23.Mai JC, Shen H, Watkins SC, Cheng T, Robbins PD. Efficiency of protein transduction is cell type-dependent and is enhanced by dextran sulfate. J Biol Chem. 2002;277:30208–18. doi: 10.1074/jbc.M204202200. [DOI] [PubMed] [Google Scholar]
- 24.Vives E, Richard JP, Rispal C, Lebleu B. TAT peptide internalization: seeking the mechanism of entry. Curr Protein Pept Sci. 2003;4:125–32. doi: 10.2174/1389203033487306. [DOI] [PubMed] [Google Scholar]
- 25.Snyder EL, Dowdy SF. Cell penetrating peptides in drug delivery. Pharm Res. 2004;21:389–93. doi: 10.1023/B:PHAM.0000019289.61978.f5. [DOI] [PubMed] [Google Scholar]
- 26.Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10:310–5. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 27.Torchilin VP, Levchenko TS, Lukyanov AN, Khaw BA, Klibanov AL, Rammohan R, Samokhin GP, Whiteman KR. p-Nitrophenylcarbonyl-PEG-PE-liposomes: fast and simple attachment of specific ligands, including monoclonal antibodies, to distal ends of PEG chains via p-nitrophenylcarbonyl groups. Biochim Biophys Acta. 2001;1511:397–411. doi: 10.1016/s0005-2728(01)00165-7. [DOI] [PubMed] [Google Scholar]
- 28.Hansen CB, Kao GY, Moase EH, Zalipsky S, Allen TM. Attachment of antibodies to sterically stabilized liposomes: evaluation, comparison and optimization of coupling procedures. Biochim Biophys Acta. 1995;1239:133–44. doi: 10.1016/0005-2736(95)00138-s. [DOI] [PubMed] [Google Scholar]
- 29.Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos-Sternberg B. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc Natl Acad Sci U S A. 2003;100:6039–44. doi: 10.1073/pnas.0931428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liang W, Levchenko T, Khaw BA, Torchilin V. ATP-containing immunoliposomes specific for cardiac myosin. Curr Drug Deliv. 2004;1:1–7. doi: 10.2174/1567201043480063. [DOI] [PubMed] [Google Scholar]
- 31.Lukyanov AN, Gao Z, Mazzola L, Torchilin VP. Polyethylene glycol-diacyllipid micelles demonstrate increased accumulation in subcutaneous tumors in mice. Pharm Res. 2002;19:1424–9. doi: 10.1023/a:1020488012264. [DOI] [PubMed] [Google Scholar]
- 32.Klibanov AL, Muzykantov VR, Ivanov NN, Torchilin VP. Evaluation of quantitative parameters of the interaction of antibody-bearing liposomes with target antigens. Anal Biochem. 1985;150:251–7. doi: 10.1016/0003-2697(85)90507-x. [DOI] [PubMed] [Google Scholar]
- 33.Lukyanov AN, Elbayoumi TA, Chakilam AR, Torchilin VP. Tumor-targeted liposomes: doxorubicin-loaded long-circulating liposomes modified with anti-cancer antibody. J Control Release. 2004;100:135–44. doi: 10.1016/j.jconrel.2004.08.007. [DOI] [PubMed] [Google Scholar]