

Abstract
Vaults are 13 million Dalton ribonucleoprotein particles with a highly conserved structure. Expression and assembly by multimerization of an estimated 96 copies of a single protein, termed the major vault protein (MVP), is sufficient to form the minimal structure and entire exterior shell of the barrel-shaped vault particle. Multiple copies of two additional proteins, VPARP and TEP1, and a small untranslated vault RNA are also associated with vaults. We used the Sf9 insect cell expression system to form MVP-only recombinant vaults and performed a series of protein-mixing experiments to test whether this particle shell is able to exclude exogenous proteins from interacting with the vault interior. Surprisingly, we found that VPARP and TEP1 are able to incorporate into vaults even after the formation of the MVP vault particle shell is complete. Electrospray molecular mobility analysis and spectroscopic studies of vault-interacting proteins were used to confirm this result. Our results demonstrate that the protein shell of the recombinant vault particle is a dynamic structure and suggest a possible mechanism for in vivo assembly of vault-interacting proteins into preformed vaults. Finally, this study suggests that the vault interior may be functionally interactive with the cellular milieu.
Vaults are structurally conserved ribonucleoprotein (RNP)1 particles that have been implicated in multidrug resistance, nucleocytoplasmic transport, and as scaffolds for both epidermal growth factor signaling and interferon-gamma activated JAK/STAT signaling pathways; however, their precise function remains unclear (1-9). The particles have a capped-barrel morphology with dimensions of approximately 41 × 41 × 72.5 nm (10), and at 13 million Daltons, are the largest known RNP. Vaults have 8-fold symmetry around their longitudinal axis and each half-vault appears identical (8-2-2 symmetry). When plated onto poly-L-lysine coated electron microscopy grids and visualized by freeze-etch platinum shadowing, vaults tend to open and flatten into two half-vaults resembling flower-like structures with eight petals symmetrically arranged around a central ring (11). Vaults are formed from three proteins; the 96 kDa major vault protein (MVP) which accounts for greater than 70% of the particle mass, the 290 kDa telomerase-associated protein 1 (TEP1), and the 193 kDa vault poly(ADP-ribose) polymerase (VPARP). The RNA component of vaults is an untranslated small RNA, termed vault RNA (VR) (1, 12-14). Expression of MVP alone in insect cells using an MVP-encoding baculovirus leads to the assembly of recombinant MVP-only vaults (hence forth referred to as MVP recombinant vaults), demonstrating that multimerization of this single protein is sufficient to form the exterior shell of vaults (15). In cells, VPARP and TEP1 are also found in non-vault cytoplasmic and nuclear fractions, and although both proteins have been shown to associate with telomerase activity, neither is required (14, 16-18). TEP1 is an RNA binding protein that is required for the stable association of VR with vaults, and binds to VR and telomerase RNA in an indirect yeast three hybrid assay and directly in electrophoretic mobility shift assays (17, 19, 20). VPARP is a functional PARP family member that ADP-ribosylates itself and MVP, and is the first vault component demonstrated to have an enzymatic activity (13). The precise stoichiometry of VPARP, TEP1, and VR with respect to a single vault particle is not yet known, nor is it clear whether all vaults contain equal numbers of these vault associated components, as other cellular factors may influence their ability to assemble into vaults. Furthermore, some species such as human and bullfrog, express multiple related VRs that can interact with vaults (6, 12, 21).
The three dimensional image reconstruction of rat vaults at 22 Å resolution using cryoelectron microscopy (cryoEM) revealed that they consist of a protein shell, 2-5 nm thick, surrounding a large interior space (22, 23). Individual vaults contain interior mass as seen in cryoEM images or TEM of negative-stained samples; however these contents are either variable for a given vault, or irregular in structure or position and thus are subtracted out during reconstruction (23). Image reconstruction combined with difference mapping is a powerful tool for determining subunit locations. Difference mapping of untreated and RNase-treated vaults purified from rat liver localized VR to the interior of the vault caps (22). Recombinant vaults formed in insect cells have the same structure as native vaults. Cysteine peptide-tagged MVP (cpMVP) recombinant vaults were resolved to 16 Å, the best resolution vault structure obtained to date (10). Difference mapping of vaults comprised of N-terminal epitope-tagged MVP with those made from untagged MVP revealed that the N-terminus of each MVP monomer lies at the waist of the vault particle (10). Co-infection of insect cells with baculoviruses coding for MVP and either TEP1, VPARP, or both leads to the formation of recombinant vaults containing these proteins (10). In addition, when an MVP-interaction domain (INT - the C-terminus of VPARP, aa 1471-1724) is fused to heterologous proteins, either luciferase or a green fluorescent protein variant - green lantern (GL), the fusion proteins have been shown to assemble into MVP recombinant vaults (13, 24). CryoEM reconstructions indicate that all of the vault-interacting components are bound to the inner surface of the vault particle (10, 24).
The large interior volume of the vault (spacious enough to encompass an intact ribosome) supports the hypothesis that the function of vaults is related to scaffolding, sequestration or transport. However, nothing is known about how MVP monomers might assemble to form the exterior shell of vaults or how the other vault components assemble into the vault interior. Although none of the vault components are covalently associated, virtually all of the MVP in cells biochemically fractionates with the vault complex (1, 6). Following purification, vault particles are highly stable, able to resist dissociation after treatment with 2 M urea, 1.0% Triton X-100, or pH extremes ranging from 4-10 (11). Thus it seems unlikely that the MVP shell of vaults disassembles into subunits and then reassembles to allow entry of other proteins/contents. It is possible that vault contents are packaged simultaneously with the assembly of the vault MVP shell. Another non-mutually exclusive hypothesis is that intact vaults could open and close in response to cellular signals or that the vault shell in vivo is elastic, allowing relatively free exchange with the cytoplasmic environment. This latter possibility is reminiscent of findings that some viral capsids in solution are dynamic structures in which polypeptides and nucleic acids, mapped by x-ray crystallography as internal, are significantly more susceptible to external agents than would be expected (25-27).
In the current study we show that the vault particle is a dynamic structure allowing the vault-associated proteins VPARP and TEP1 to complex with recombinant vaults (expressed using the Sf9 insect cell system) even after the MVP shell of vaults has formed. These experiments were carried out by mixing vault-interacting proteins with pre-formed MVP recombinant vaults. In addition, we show that the vault-interacting proteins localize to the interior of the intact vault particle. Our results suggest that recombinant vault particles exhibit substantial flexibility that imparts access to the interior volume. The dynamic nature displayed by the recombinant vaults used in this study likely represents a key mechanism for the interaction of endogenous vaults with their in vivo environment.
EXPERIMENTAL PROCEDURES
Micrococcal nuclease digestion of purified vaults and subcellular extracts
Rat liver vaults (4 μg) were purified as previously described (1). Purified vaults or phenol extracted RNA from purified vaults were treated with 2000 u/ml of micrococcal nuclease for 0, 15, 30, or 60 min at 37° C. Crude vault extracts (100,000× g pellet, P100) were prepared from normal rat kidney epithelial cells (LR117) as previously described (6). The P100 pellet was resuspended by dounce homogenization in buffer [50 mM Tris-Cl (pH 7.4), 1.5 mM MgCl2, 75 mM NaCl] containing 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitors. Deproteinized RNA was isolated by phenol:chloroform extraction and ethanol precipitation. Equal volume amounts of P100 (RNP) or deproteinized P100 (RNA) extracts were treated with 2000 u/ml of micrococcal nuclease for 20 min at 25°C. All micrococcal nuclease digestions were terminated by the addition of EGTA to 8 mM final concentration. Reactions were phenol:chloroform extracted, ethanol precipitated, fractionated on a 10% acrylamide, 8M urea gel, electroblotted to Zeta GT+ membrane (Biorad) and probed for VR as previously described (6).
Plasmids and M-INT Cloning and Expression
Construction of the VPARP, TEP1, GL-INT and CP-MVP (cysteine rich N-terminal epitope tag-MVP)-expressing baculoviruses are described elsewhere (10, 24). The C-terminal MVP-interaction domain of VPARP (aa 1563-1724, M-INT) was inserted into the E.coli expression vector, pET28a (Novagen). M-INT was expressed in the E.coli strain BL21 CodonPlus (Stratagene) and purified on a His-bind column (Novagen). The eluted protein was dialyzed into buffer [20 mM Tris, pH 7.9, 50 mM KCl, 0.1 mM EDTA, 10% glycerol] containing 1 mM DTT and 1 mM PMSF. Purified protein was aliquoted and stored at −80°C.
Sf9 Insect Cells and Vault Purifications
Cells were maintained in Sf-900 II SFM media and grown at 27° C. Cultures were infected with MVP, TEP1, VPARP, or GL-INT viruses at a multiplicity of infection (MOI) of 0.01 for approximately 65 hours and then pelleted and lysed on ice in Buffer A [50mM Tris-HCl, pH 7.4; 75mM NaCl; 0.5mM MgCl2] + 1% Triton X-100, 1mM dithiothriotol, 0.5 mM PMSF, and protease inhibitor cocktail (Sigma). Vault purification from lysates was performed as previously described (15). In experiments involving the mixing of lysates from two separate infections, each cell pellet was lysed separately and then the two were mixed and incubated together on ice for 30 minutes before proceeding with the standard vault purification. Pellets from the 20-60% sucrose fractions were each resuspended in 20 mM MES buffer, pH 6.5 for further analysis.
Gas-Phase Electrophoretic Mobility Molecular Analyzer (GEMMA)
The details of the GEMMA have been described elsewhere (28-30). Briefly, the GEMMA instrument (TSI Inc., St. Paul, MN) consists of an electrospray ionization (ESI) unit with a neutralizing chamber, a differential mobility analyzer (DMA), and a condensation particle counter (CPC). The DMA voltage was scanned and data recorded by Aerosol Instrument Manager Software. The software corrects for the efficiency of the condensation particle counter as well as for a small fraction of multiply-charged particles assuming the bipolar charge distribution of Fuchs (31) and calculated by Wiedensohler’s formula (32). The electrophoretic mobility diameter maxima for peaks of interest were calculated from this corrected data. Igor Pro 4.08 software was used for further processing including smoothing of spectra.
From the GEMMA measurements by our laboratory (unpublished data) and from those reported by Bacher et al. (28) for over 40 protein complexes ranging in size from small protein dimers to complexes as large as the 700 kDa 20S proteasome and MDa-range viral particles, an average density of approximately 0.6 g/cc can be used to calculate the molecular mass of small and large protein complexes. Data presented for the molecular mass of vault particles are mean ± SE.
Prior to analysis, samples were desalted using Millipore Microcon Centrifugal Filter Devices (YM-100) in 20 mM ammonium acetate pH 7.4. Concentrations of analyses ranged from 400 ng/μl to 900 ng/μl. Protein solutions were introduced via the ESI source at a flowrate of approximately 70 nl/min. The electrophoretic mobilities for the samples used to estimate molecular weights were determined from three independent experiments of MVP vaults and MVP-VPARP vaults from lysate mixing, and from two independent experiments of MVP-VPARP from co-infection, in which the total protein concentration de-salted and analyzed ranged from 40 ng/μl to 600 ng/μl.
Negative Staining and Electron Microscopy
Vault containing samples were prepared for electron microscopy by absorption of samples onto fresh 1.8 nm thick carbon film mounted on carbon-coated holey-film grids for 5 min at 4°C. Following sample adsorption, grids were floated for 5 min on 1 ml of 1% uranyl acetate at 4°C and dried on filter paper prior to viewing in a JEM 1200EX microscope. The carbon coated holey-film grids and carbon film were prepared by Dr. Sergey Ryazantsev, Department of Biological Chemistry, UCLA School of Medicine. Micrographs were captured with a BioScan 600W digital camera (Gatan Inc. Pleasanton, CA) using Gatan’s DigitalMicrograph software version 3.7.1. Images were further processed using Adobe Photoshop 6.0 software.
Green Fluorescence Quenching
Quenching of purified GL-INT vaults and measurement of fluorescence intensity were carried out as previously described (24).
In Vitro Binding and Thrombin Cleavage
Purified vaults were incubated with 20, 40, or 100 ng M-INT for 1 hour at 25° in Buffer A + 1% Triton X-100 and 1 mM DTT in a 20 μl volume containing 1mg/ml BSA. Volumes were then adjusted to 70 μl in the same buffer and samples were pelleted at 60,000 rpm in a Beckman TLA 100.1 benchtop ultracentrifuge. Pellets were resuspended in 1X SDS sample buffer and all samples were loaded onto 12% SDS-PAGE gels and transferred to Hybond-C (Amersham Biosciences) for Western blotting using polyclonal antibodies to MVP and VPARP. For cleavage reactions, 20 ng M-INT was incubated with or without 1μg of purified CP-MVP vaults for 1 h at 25° C as above. The volume was then adjusted to 55μl and a 10μl aliquot was taken for the ‘0’ time point. Five μl of thrombin (0.0004 u/μl) or thrombin-agarose beads (0.01 u/μl) was added and the reaction was incubated at 25° C. Ten μl aliqouts were taken out at 1h and 2h and analyzed by Western blot. Thrombin-agarose beads were prepared using biotinylated thrombin and streptavidin-agarose beads as per the manufacturer’s instructions (Novagen). The beads were resuspended in buffer prior to use for the above experiment.
RESULTS
Vault Particles Differ in Susceptibility to Nuclease Treatment Depending Upon Their Level of Purity
It has been previously demonstrated that the VR component of purified rat liver vaults can be removed by digestion with a cocktail of RNase A and RNase T1, despite difference maps of 3-D cryoEM reconstructions of RNase-treated and untreated vaults that localize VR to the presumably protected interior of the vault caps (22). Complete VR removal typically requires high concentrations of RNases and extended incubations for efficient digestion, supporting the notion that either the MVP vault shell is partially protective, or that the vault-associated proteins (VPARP and/or TEP1) impede the ability of RNases to access the interior of the vault caps. To study the accessibility of VR in the vault particle complex we used micrococcal nuclease digestion. Micrococcal nuclease is a calcium-dependent nuclease that nonspecifically cleaves both RNA and DNA, and unlike RNAses, time course measurements of digestion can be carried out, by chelating the calcium with EGTA to stop the reaction. When purified rat liver vaults were treated with 2000 u/ml of micrococcal nuclease for up to 1 hour at 37° C, VR was substantially protected from digestion (Figure 1A, RNP). However, deproteinized VR extracted from rat liver vaults was susceptible to digestion under similar conditions (Figure 1A, RNA). Vault-containing fractions were then isolated from rat kidney epithelial cells by centrifugation of cell lysates at 100,000x g, which is known to pellet vault particles. A portion of the total cellular VR can be found in this high speed pellet (P100), and further fractionation of the pellet over a sucrose-equilibrium gradient results in co-fractionation of VR and MVP, indicating that the vast majority of P100 VR is vault-associated (6). VR is also found in the non-vault associated 100,000× g supernatant (S100), partly in association with the La RNA binding protein, and possibly bound to non-vault associated TEP1 (33). To determine the accessibility of VR in unpurified vaults we again used micrococcal nuclease digestion. In contrast to purified vaults, VR in both P100 (RNP) and deproteinized P100 (RNA) fractions was readily digested with micrococcal nuclease (Figure 1B). This suggests that the conformation of the vaults within the cell may be more open or dynamic than that of purified rat liver vaults, which are resistant to micrococcal nuclease treatment. This result, combined with the observation that VR is eventually digested by a combination of RNases even in purified vaults, prompted us to further examine the structural properties of the MVP vault shell.
FIGURE 1.
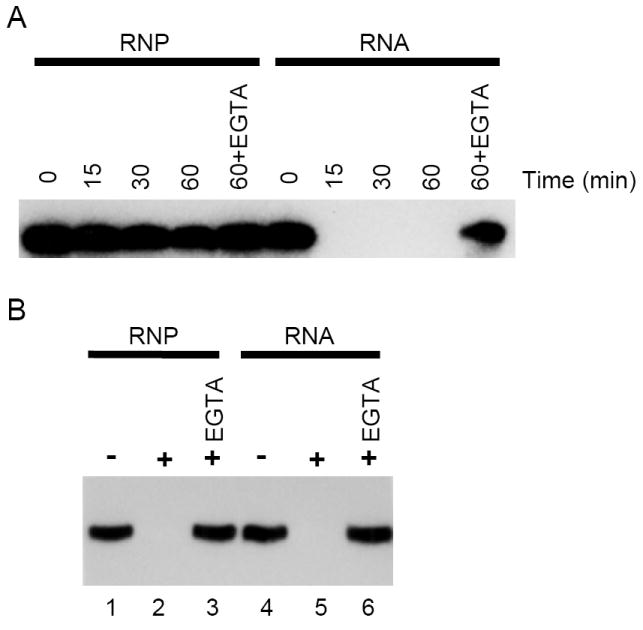
Micrococcal nuclease digestion of vault RNA. (A) Purified rat liver vaults (RNP) or RNA extracted from vaults were treated with 2000 u/ml of micrococcal nuclease for 0 (no enzyme), 15, 30, or 60 min. The reaction was terminated by the addition of EGTA. Control digests were incubated with micrococcal nuclease for 60 minutes in the presence of EGTA. (B) P100 (RNP) or RNA extracted from P100 were treated with 2000 u/ml for 20 min (lanes 2, 3, 5, and 6) or without enzyme (lanes 1 and 4) and digestion was terminated by the addition of EGTA. Control digests included both micrococcal nuclease and EGTA (lanes 3 and 6). Reactions were phenol:chloroform extracted, ethanol precipitated, fractionated on a 10% acrylamide-8 M urea gel, electroblotted to Zeta GT+ membrane, and probed for VR. Hybridized bands were visualized by autoradiography.
VPARP and TEP1 Associate with Pre-Formed MVP Recombinant Vaults
Vaults are predominantly cytoplasmic; however, it is unclear in which cellular compartment MVP, VPARP, TEP1 and VR assemble to form the complete vault particle or if the MVP vault shell is formed prior to its association with the other vault components. As the vault-associated components (VPARP, TEP1, and VR) are also found in non-vault associated fractions, it seems reasonable to propose that these components may shuttle into and out of the vault shell (6, 13, 14, 21). To determine whether this hypothesis is feasible given the closed capsule-like structure of the vault, we turned to the Sf9 insect cell expression system where we have shown that MVP alone is sufficient to form recombinant vaults (15). The MVP recombinant vaults assembled via baculovirus expression appear as structurally regular as vaults purified from rat liver. Using cryoEM it has been previously shown that co-infection of a single insect cell culture with baculoviruses expressing MVP and either VPARP, TEP1, or both proteins leads to the formation of vaults containing the expected protein components in the vault interior (10). The recombinant MVP-only vaults have also been shown to sequester co-expressed luciferase-INT or GL-INT described above (24). Furthermore, the activity of these two proteins could be shielded from the external environment by the thin protein shell of vaults.
We next determined whether the assembly of VPARP or TEP1 into a vault could occur after the MVP shell has been completely formed. To accomplish this, individual extracts containing either VPARP or TEP1 were mixed with MVP-only extracts containing pre-assembled recombinant vaults. Following incubation on ice to allow the assembly reaction to occur, vaults were purified from the mixed lysates (see Methods). Analysis of insect cells expressing MVP only indicates that all of the MVP is assembled into vaults; thus these crude extracts contain fully assembled MVP vault shells (15). When VPARP alone was expressed in Sf9 cells, the protein was found in both the S100 and P100 fractions (Figure 2A, left panel). The VPARP protein in the P100 fraction likely represents protein aggregates as it did not further purify by fractionation over a sucrose gradient. When VPARP and MVP lysates were mixed, VPARP in the S100 fraction shifted to the P100 (Figure 2A, right upper panel) and co-purified with MVP over a sucrose gradient (Figure 2A, right upper and lower panels), indicating association of VPARP with the vault particle. Identical results were obtained for TEP1-MVP lysate mixing experiments (Figure 2B). Importantly, lysate containing a TEP1 truncation that does not interact with MVP was not found in the sucrose gradient after mixing with MVP lysate (20), demonstrating that non-specific trapping of proteins in the vault interior does not occur. Thus, the fully formed MVP shell of recombinant vaults allows for the subsequent incorporation of VPARP or TEP1.
FIGURE 2.
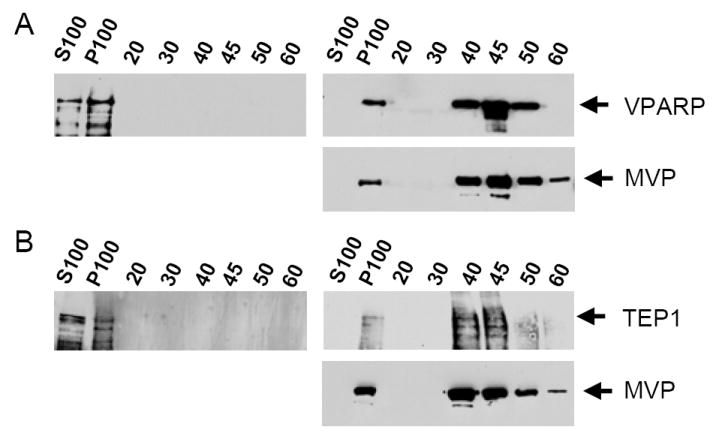
VPARP and TEP1 assemble into pre-formed recombinant MVP-only vaults in Sf9 insect cell extracts. (A) Sf9 cell lysates containing VPARP were subjected to the vault purification protocol with (right panels) or without (left panel) mixing with extracts containing MVP recombinant vaults. A 20,000× g supernatant was subjected to a 100,000× g centrifugation yielding a high speed supernatant (S100) and a high speed pellet (P100). The latter was retained for further purification. As a final purification step, samples were layered over a discontinuous sucrose gradient and an aliquot of the subsequently pelleted gradient fractions were analyzed by Western blot. (B) A similar analysis of TEP1 fractionation in the presence (right panel) or absence (left panel) of Sf9 cell lysates containing MVP recombinant vaults.
VPARP Assembles into the Interior of the Entire Population of Vaults
Although the vast majority of the recombinant vaults purified after mixing MVP lysates with VPARP or TEP1 lysates appeared morphologically normal by electron microscopy (see Figure 3B), it was possible that all of the VPARP and TEP1 could have assembled into a small subset of vaults that were still forming from precursors in cell lysates or from particles that were damaged during purification or incorrectly formed to some degree. To rule out these possibilities we first looked at the amounts of VPARP that could be assembled into vaults by Coomassie staining of vaults purified from mixed lysates. The maximum number of VPARP molecules per vault in endogenous mammalian vaults is not known; nor, as previously mentioned, is it clear whether all mammalian vaults contain an equal complement of VPARP molecules. However, recombinant vaults purified after lysate-mixing contain substantial amounts of VPARP (Figure 3A, right lane) that appear equal to those obtained from either co-infection of insect cell cultures, or mammalian vault purifications from tissues (1, 10). Mixing of purified MVP recombinant vaults with extracts containing VPARP followed by re-purification of these vaults yielded identical results (data not shown). The high molecular weight bands seen by Coomassie staining of purified MVP-only recombinant vaults (Figure 3A, left lane) are not VPARP since they are not detected by a VPARP antibody on Western blots (data not shown). These bands are, in fact, multimers of MVP, and are recognized by anti-MVP antibodies (data not shown). Purified vaults formed with or without VPARP were visualized by negative staining followed by electron microscopy in order to determine any differences in the interior morphology of the two species. Analysis of recombinant vaults derived from MVP-VPARP lysate-mixing, MVP-VPARP co-infection, or MVP infection alone revealed that despite the inherent variability of the negative staining technique, most vaults containing VPARP significantly excluded the uranyl acetate stain from the particle interior, indicating the presence of internal mass, and were thus easily differentiated from MVP-only vaults, which show prominent interior staining which is likely due to collapse of the hollow MVP-only particles onto the carbon grids (Figure 3B). No differences in staining were visible when comparing VPARP-containing vaults derived from lysate-mixing with those derived from co-infection (data not shown), indicating that VPARP localization is likely the same in the two samples.
FIGURE 3.
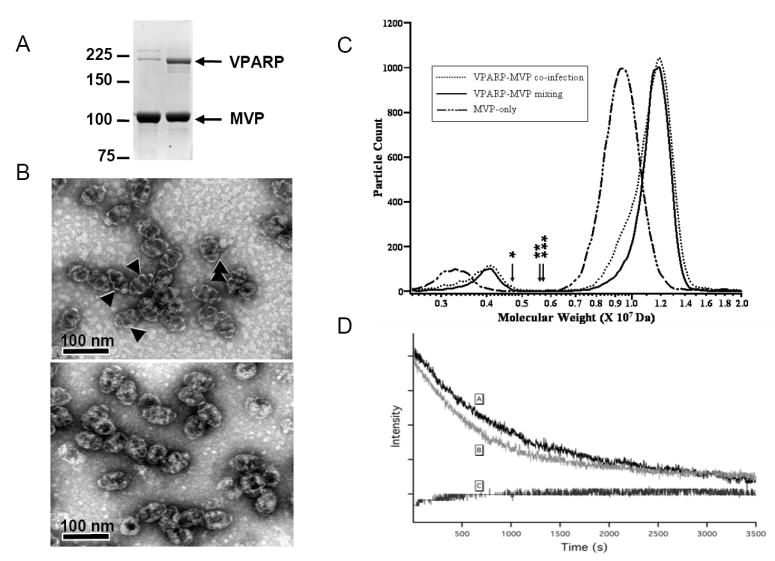
Pre-formed MVP-only recombinant vaults are uniformly able to take up exogenous VPARP. (A) Coomassie-staining of vaults purified from MVP-only lysates (left lane) and MVP-VPARP mixed lysates (right lane). To saturate the VPARP-binding sites of pre-formed MVP recombinant vaults, an MVP-containing lysate was mixed with a VPARP-containing lysate at a 1:5 ratio. The bands running just above and below 225 kDa marker in the left lane are multimers of MVP, as demonstrated by immunoreactivity with anti-MVP antibodies (not shown). (B) Electron micrograph of uranyl acetate-stained MVP-vaults (upper panel) and MVP-VPARP vaults purified from mixed lysates (lower panel). The arrows identify regions of the vaults that consistently show dark staining, due to the inclusion of stain, in MVP-only vaults but not in MVP-VPARP vaults. The lack of staining in the MVP-VPARP vaults suggests the presence of internal mass. (C) GEMMA analysis of vaults purified from MVP-only lystates, MVP-VPARP mixed lysates, and lysates from a culture co-infected with both VPARP and MVP baculoviruses. The GEMMA spectra of vaults show the distribution of predicted molecular weights calculated using an effective density of 0.57 g/cc. In this representative experiment, the calculated mass of MVP-only vaults is 9.3 MDa, MVP-VPARP vaults derived from co-infection is 11.2 MDa, and MVP-VPARP vaults derived from lysate-mixing is 11.4 MDa, which corresponds to an average VPARP content per vault of 9.9 and 11.0, respectively. Both the major and minor peaks (2.5 MDa to 5 MDa) in the spectra represent vaults with the same mass. The minor peaks represent doubly charged vaults, while the major peaks represent singly charged vaults. Half-vaults were not detected. The arrows above the x-axis correspond to the theoretical molecular weight of half-vaults (MVP-only (*), MVP-VPARP vaults derived from co-infection (**), and MVP-VPARP vaults derived from lysate-mixing (***). Samples were de-salted in 20 mM ammonium acetate, pH 7.4 and analyzed at a total protein concentration of 200 ng/μl. (D) Quenching of GL-INT fluorescence by KCl. The fluorescence of soluble GL-INT was quenched immediately after addition of KCl to a final concentration of 0.5M at t=0 [C], whereas that of GL-INT sequestered in vaults exhibits a much longer quenching time [A and B]. Differences in quenching between vaults purified from mixed Sf9 cell lysates [A] verses vaults purified from a co-infected Sf9 cell lysate [B] are not significant.
To provide additional evidence that the majority of preformed vaults were able to incorporate VPARP we turned to the use of electrospray mobility analysis of the vault complexes. GEMMA separates gas phase, predominately singly charged particles based on their differential electrophoretic mobility in air. Electrospray ionization with mass spectrometry has demonstrated capabilities to measure noncovalently-bound protein complexes (34, 35). With GEMMA, ESI generates gas phase ions representing the intact vault complexes from solution, and the ions are separated based on their electrophoretic mobilities and counted by the CPC (see Methods). Based on their measured mobilities, the electrophoretic mobility diameter and a molecular weight is calculated (28, 29). Although the electrophoretic mobility of a particle is governed by the size and shape of the particle, this method has also been used to characterize proteins and non-covalent protein complexes, showing a correlation between protein mass and the experimentally derived electrophoretic mobility diameter (28-30). This is based on a simple model relating molecular weight to the diameter of a sphere with an effective density (29). We reasoned that if exogenous VPARP was incorporated into only a subset of vaults, two peaks of apparent mass would be measured; one representing MVP-only vaults and one representing MVP-VPARP vaults. Analysis of vaults purified from MVP extracts with and without VPARP extract added (Figure 3C) indicate that most if not all of the vaults were able to incorporate VPARP, although the difference in apparent mass is too small to completely resolve the two peaks. The mass peak maxima were calculated from the distribution of electrophoretic mobilities corrected for the expected charge distribution of vault particles. The estimated number of VPARP copies incorporated into the vaults, were calculated from replicate measurements. Figure 3C shows overlaid spectra from one set of experiments. Based on the estimation that there are 96 copies of CP-MVP (MW 97 kDa) per vault and from the GEMMA measurement, the molecular weight of MVP-only vaults is approximately 9.3±0.1 MDa (n = 3). Using the same density applied to the MVP-only vault, the molecular weight of MVP-VPARP vaults from lysate mixing and MVP-VPARP vaults from co-infection is approximately 11.2± 0.3 MDa (n = 3) and an average of 11.0 MDa respectively. This corresponds to a calculated average of copies of 9.5 VPARP incorporated per MVP-VPARP vault from lysate mixing and 8.3 copies of VPARP per MVP-VPARP vault from co-infection. The molecular mass distribution of both the MVP-VPARP vaults from lysate mixing and those derived from co-infection show signs of species of lower molecular mass. This indicates that there is a small fraction of these MVP-VPARP vaults that contained less than saturating levels of VPARP, particularly in MVP-VPARP vaults derived from co-infection. These results suggest that the entire population of vaults is competent to incorporate VPARP after vault shell formation.
In order to confirm that vaults resulting from the assembly of vault-interacting proteins into MVP recombinant vaults by mixing (in vitro) are similar to those obtained by co-infection (in vivo), we carried out fluorescence quenching experiments using GL-INT as the vault interacting protein. GL-INT containing recombinant vaults were purified from mixed Sf9 cell lysates containing MVP only and GL-INT only, and Sf9 cells co-infected with MVP and GL-INT encoding baculoviruses (see Methods). Fluorescence quenching of GL upon addition of 0.5M KCl (final concentration) has been previously shown to be substantially delayed in GL-INT vaults derived from co-infection (24). We observed similar quenching curves for GL-INT vaults derived from mixed lysates (Figure 3D [A]) and those derived from co-infected cultures (Figure 3D [B]). In contrast, GL-INT alone in lysates was quenched immediately (Figure 3D [C]). The delay in quenching suggests that the vault shell acts as a barrier to protect the proteins assembled into the particle interior. This data indicates that there is no difference between vaults purified from co-infected verses mixed lysates and that the incorporated proteins are inside of the vault particles.
Cell Extracts Are Not Required for M-INT to Interact with MVP recombinant Vaults
We next examined whether other factors or components found in Sf9 cell extracts are required for the incorporation of exogenous proteins into the vault shell interior. For these experiments we decided to use the C-terminal region of VPARP (amino acids 1563-1724), that is the smallest domain identified to date, that interacts with MVP, termed M-INT (for minimal size interaction domain), as it could be easily expressed and purified from E. coli (Figure 4A) (13, 36). Addition of a hexahistidine tag to the M-INT construct allowed for purification of the expressed 22 kDa protein using Ni-NTA chromatography. Two micrograms of the highly purified protein was added to Sf9 cell lysate from either uninfected cells or cells infected with MVP-baculovirus. MINT fractionated in both the S100 and P100 fractions when added to extracts from uninfected cells, (Figure 4B left panel, S100 and P100 lanes) but did not enter the sucrose gradient (Figure 4B left panel, 20-60% sucrose fractions). In contrast, when M-INT was added to vault-containing extracts it fractionated entirely in the P100 fraction (Figure 4B, right upper panel, S100 and P100 lanes) and co-purified with MVP over a sucrose gradient (Figure 4B, right upper and lower panel, 20-60% sucrose fractions). We then determined whether extracts were strictly required for M-INT to assemble into vaults by incubating purified M-INT with purified MVP recombinant vaults followed by pelleting at 100,000× g. In the presence of MVP recombinant vaults, most of the M-INT shifted into the P100 fraction (Figure 4C, lanes 2, 6, and 10), while in the absence of vaults M-INT remained in the supernatant (Figure 4C, lanes 3, 7, and 11). Adding increasing amounts of M-INT to 1 μg of purified MVP recombinant vaults yielded an increasing amount of protein in the pellet (Figure 4C, lanes 1, 2, 5, 6, 9, and 10). The amount of M-INT in the supernatant also increased, and this is most likely due to saturation of the VPARP (M-INT) binding sites in the MVP recombinant vaults. In summary it appears that the interior surface of the vault barrel is accessible after a 1 hour incubation, even in the absence of cell extracts, for incorporation of this 22 kDa protein.
FIGURE 4.
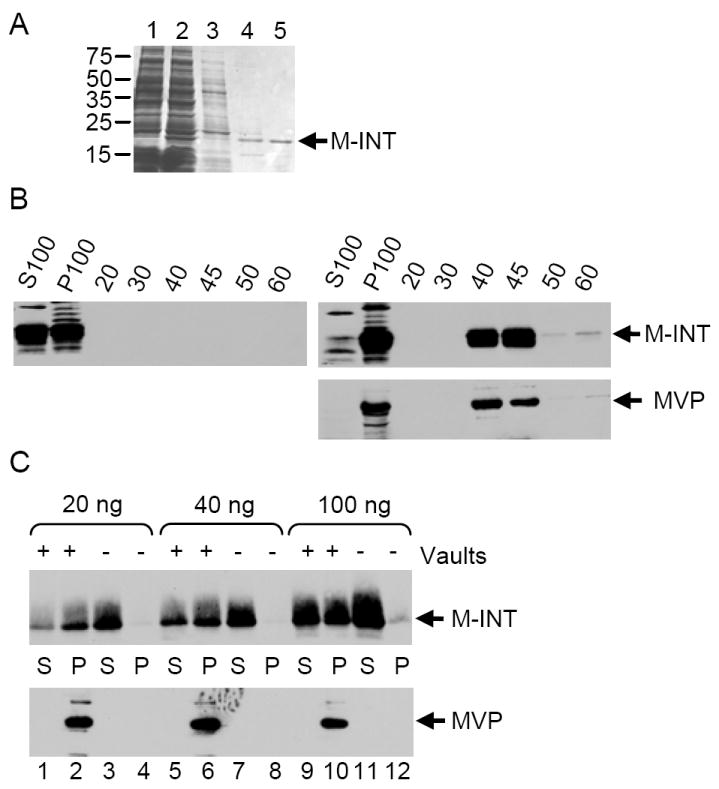
The minimal MVP interaction domain of VPARP (M-INT) does not require SF9 cell extracts to bind to vaults. (A) Coomassie stained gel of E. coli lysates: uninduced and induced (lanes 1 and 2), column flow through (lane 3), column wash (lane 4) and final elution of His-T7 epitope tagged M-INT protein off the Ni-NTA resin (lane 5), which was then dialyzed into storage buffer. (B) Western blot analysis of S100, P100 and sucrose gradient fractions (described in Figure 2) after mixing of purified M-INT into Sf9 cell extracts (left panel) or extracts containing MVP recombinant vaults (right panels). (C) Mixing of 20 ng, 40 ng, or 100 ng purified M-INT without (-) and with (+) 1μg of vaults followed by high-speed centrifugation (S=high speed supernatant, P=high speed pellet,). Pellets were resuspended in SDS sample buffer, and fractionated by SDS-PAGE followed by Western blot analysis.
Thrombin Cleavage of Vault-Associated M-INT Supports Particle Conformational Dynamics
Our data shows that M-INT was readily packaged into vault particle shells. However, the possibility still existed that the protein was bound to the outside of the particle. The restricted access of the small contact quenching reagent, KCl, to the particle interior (Figure 3D), led us to conclude that GL-INT was inside the vault shell. This together with the fact that luciferase-INT was localized to the interior of the vault barrel by cryoEM reconstruction (24) strongly suggests that M-INT is also on the inside of the vault particle. As M-INT contains a thrombin cleavage site derived from the pET28 bacterial expression vector, we were able to examine the accessibility of vault-associated M-INT to thrombin, a 36 kDa protein. This approach allowed us to probe the ability of larger substances lacking a vault targeting sequence to gain access to the vault interior. Thrombin treatment of M-INT removes the pET28 vector-derived hexahistidine tag of the protein (17 amino acid residues). The cleaved and uncleaved proteins can be resolved on a 12% SDS-polyacrylamide gel. Purified M-INT was pre-incubated with or without purified MVP-vaults for 1h, followed by a one or two hour incubation with thrombin. As expected, thrombin was able to efficiently cleave M-INT when incubated alone (Figure 5A, compare lanes 3 and 5 to lane 1). When thrombin was added to M-INT pre-incubated with vaults (and thus “packaged”), cleavage proceeded at a slightly but consistently reduced efficiency (Figure 5A, lanes 2,4,6,compare lane 3 with 4 and lane 5 with 6). This could be due to either a delayed access of thrombin to the vault interior or a change in conformation of M-INT upon binding to vaults. To confirm that the retardation of thrombin degradation of “packaged” M-INT was due to sequestration in the vault interior, the above experiment was repeated using thrombin-agarose beads. Under these conditions, there was a striking difference between the digestion of M-INT alone and M-INT associated with vaults (Figure 5B). After a 2h incubation with thrombin-agarose beads, more than 50% of the free M-INT was cleaved, whereas very little cleavage was seen for M-INT bound to vaults (Figure 5B, compare lane 3 with 4 and lane 5 with 6). The large thrombin-agarose particle appears to be almost entirely excluded from interacting with the particle interior. This data indicates that the M-INT domain can be assembled into the vault interior by simply mixing the purified proteins together and that the vault shell provides a protective environment.
FIGURE 5.
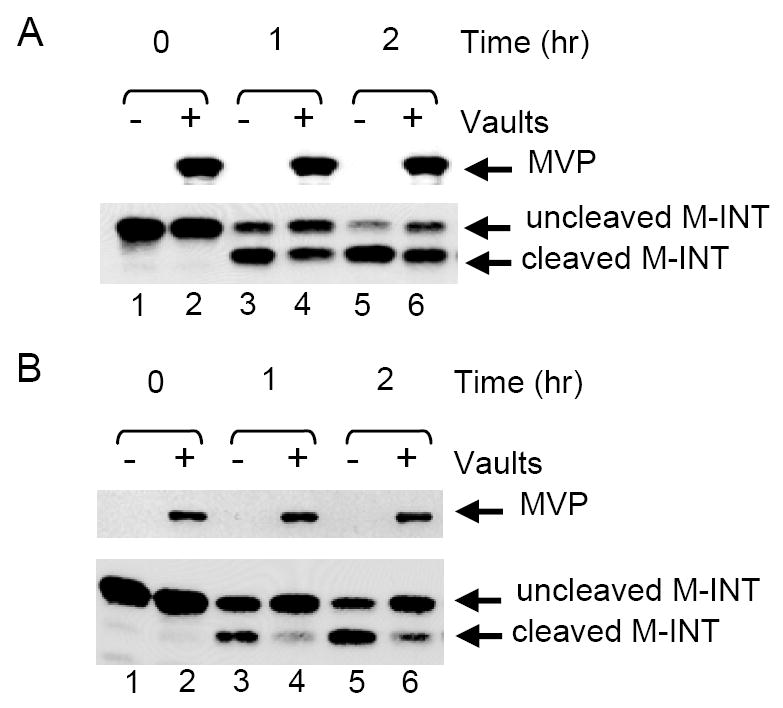
Vault-bound M-INT is less accessible to proteolytic cleavage. (A) Western blot analysis of thrombin-treated M-INT either pre-incubated with (+) or without (-) MVP recombinant vaults. Aliquots were collected at 0 (no enzyme), 1h, or 2h after treatment with thrombin, separated by 12% SDS-PAGE, transferred onto nitrocellulose and immunoblotted for MVP (top panel) and the M-INT domain of VPARP (bottom panel). Immunoblotting for M-INT shows both the intact and the cleaved protein. (B) Vaults treated with thrombin-agarose and analyzed as described above.
DISCUSSION
The present study demonstrates that the completely formed MVP shell of recombinant vaults allows the incorporation of the very large VPARP and TEP1 proteins into the vault interior. This result was somewhat unexpected given the nature of the three dimensional reconstructions of MVP recombinant vaults using cryoEM, which show a particle with a smooth outer surface that displays no large gaps. Thus far, the highest resolution reconstruction of vaults (16 Å) is from the CP-MVP construct used to form vaults in this study (10). The reconstruction of the CP-MVP recombinant vault appeared virtually identical on the exterior to reconstructions of vaults isolated from rat tissue (10, 23), and spectroscopic results from this and previous studies using luciferase assays and fluorescence quenching indicate that the vault interior is only accessible over time (24). Therefore, some mechanism must allow for a significant modification of the vault structure that is not readily apparent in the static 3-D image that enables entry of large proteins into its interior. Endogenous vaults have been shown to open at the particle’s waist into two half-vault flower-like structures when plated on poly-lysine coated EM grids, and a small number of open vault halves are also seen in normal negative-stain vault preparations from tissue and baculovirus-infected Sf9 cells (11, 15). In addition, when purified vaults are visualized by cryoEM after rapid freezing, some partially open vaults and half-vaults are seen (23). This suggests that although the vault structure overall is able to withstand a remarkable range of conditions in vitro (11), the interactions keeping the eight petals of one half-vault bound to the eight petals of the other half-vault may be less stable than the forces keeping a half-vault together. Thus a half-vault/vault equilibrium could allow entry of large molecules into the particle interior. An alternate explanation for the results obtained here is that single pairs of opposing petals (made up of multiple MVPs) on each half-vault could temporarily separate and then close, much as a door would swing open and shut, without disrupting the integrity of the particle (see Figure 6 for a schematic describing these two models). However, some other mechanism for the transient opening of holes in the vault shell may be occurring, perhaps between adjacent vault petals of the same half-vault. Insights into the precise mechanism of vault dynamics will have to await an atomic resolution structure.
FIGURE 6.
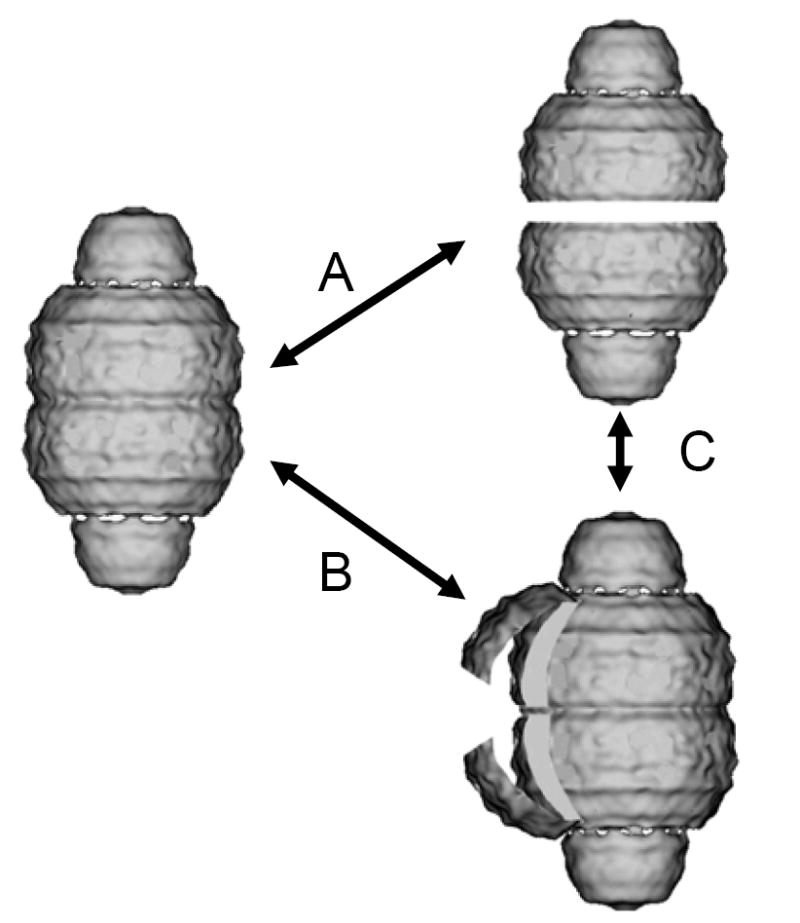
Model of proposed vault dynamics. Vault particle opening could occur by vault petals reversibly opening at the particle waist (B), or the particle could transiently separate into 2 half-vaults (A), which in some instances, may lead to transitions between these two forms (C). Vault structure adapted from 3-D image reconstructions of recombinant MVP-only vaults (P. Stewart, Vanderbilt University Medical Center).
The slow digestion of VR from within the caps of purified rat liver vaults using micrococcal nuclease, or more rapid digestion using high concentrations of RNase A/RNase T1 mixtures, suggests that the shell of the vault particle is partially, but not fully, protective. It is also possible that in purified vaults, more so than in vaults in extracts, the vault RNA is protected from RNases that enter the MVP vault shell by close association with the TEP1 RNA binding protein, or that the presence of TEP1, VPARP, and other vault-associating proteins physically impede the entrance of RNases into the vault interior. We have made numerous attempts to increase the accessibility of the interior and to “open” the relatively protected environment of purified rat liver vaults by treatment with 10 mM DTT, 1 mM spermidine, 10 mM ATP, 10 mM GTP, 0.5 mg/ml heparin, or 0.5% NP-40. All attempts have thus far been unsuccessful (data not shown). As MVP has been shown to bind calcium, we have also treated vaults with various concentrations of EGTA or calcium chloride to no avail (36). We considered the possibility that since endogenous vaults in extracts are much less protective of the associated VR than are purified vaults, a soluble factor that facilitates the opening of vaults could be present in extracts and then lost during purification. To test this hypothesis, we performed mixing experiments with pure rat vaults and increasing amounts of whole cell extract. These mixing experiments were also unsuccessful at “opening” the purified vaults.
Study of viral capsid structure lends plausibility to the hypothesis that vaults are dynamic particles, transiently opening and closing. In one example, the dynamics of flock house viral capsids was studied using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) after a proteolysis time course (26). The results of this study determined that regions near the N- and C-terminus of the capsid polypeptides were among the earliest sites cleaved, despite crystallographic data indicating that these regions are internal to the viral capsid. Proteolysis and related studies using chemical modification or neutralizing monoclonal antibodies to internal epitopes of poliovirus indicate that transient externalization of these internal epitopes occurs, with functional implications for cell attachment and RNA release (25). Interestingly, in flock house virus it has been shown that the association of viral RNA alters the dynamics of the capsid structure and appears to enhance the structural integrity of the particle, although this is not evident by crystallography (37). Although the function of vaults is not known, the structural similarities to the viral capsid as “containers” made up of multimers of one or few proteins are striking, and the dynamics of internal versus external domains likely has functional significance. Of interest, one study that conducted pulse-chase experiments combined with immunoprecipitations of MVP and VPARP suggests that incorporation of new VPARP molecules into vaults occurs earlier than does incorporation of new MVP molecules, indicating that newly synthesized VPARP may associate with pre-existing vaults (38). This result appears to be consistent with our own findings. Recently an NMR solution structure of two-repeat units found in MVP (domains 3 and 4) has been solved and M-INT was shown to bind to domain 4. Their model of the MVP repeats stacking suggests a distinct inner and outer surface in the vault particle with the putative VPARP binding surface on the inside of the vault (39). Additionally, the La RNA binding protein has been shown to bind to VR apart from the vault particle and to loosely associate with vaults during biochemical fractionation, leading us to hypothesize that there must be some form of VR cycling that deposits VR into the interior of the vault cap, which would seem to necessitate a dynamic vault shell (33).
Our results demonstrate that the vault is not a rigid, impermeable structure, but rather a dynamic entity. Vaults could have a dual function; first to allow proteins to enter under certain conditions, and second to restrict access during other conditions so that the internal contents are protected or sequestered from other cellular factors with which they could otherwise interact. Although relatively small proteins apparently do not require cell extracts to enter vaults, it remains to be seen whether assembly factors may be required for larger proteins like full length VPARP and TEP1 to enter the particle interior. It will be interesting to study the factors that control opening/closing of vaults or “breathing” of the vault structure, and to determine whether breathing is a important factor, not just in vault assembly, but in vault function. Vaults have been suggested to be carriers or transporters in the cell due to both their highly conserved capsule shape and their localization at such diverse regions including the nuclear pore complex, the leading edges of lamellapodia, adhesion plaques, the tips of neurites, and along microtubules (7, 40, 41). They have also been proposed to function as assembly scaffolds for signaling complexes (8, 9, 42). The present study demonstrates that recombinant vaults are dynamic particles and that the vault protein shell formed by MVP can transiently open to allow incorporation of other vault components and, potentially, other vault-interacting proteins. This data suggests a mechanism whereby endogenous vault can interact with the cellular milieu.
Acknowledgments
We would like to thank Stanley L. Kaufman (TSI, Inc.) for advice on the interpretation of the GEMMA data and Phoebe Stewart (Vanderbilt University) for helpful comments and critical reading of the manuscript.
Footnotes
This research was supported by grants from the National Science Foundation (MCB-0210690 and CHE-0507929) and the G. Harold and Leila Y. Mathers Charitable Foundation (01124244). The UCLA Functional Proteomics Center was established and equipped by a grant from the W. M. Keck Foundation. JAL acknowledges support from the Jonsson Comprehensive Cancer Center at UCLA, the UCLA-DOE Institute for Genomics and Proteomics, and the National Institutes of Health (RR 20004).
ABBREVIATIONS: RNP, ribonucleoprotein; MVP, major vault protein; TEP1, telomerase associated protein 1; VPARP, vault poly(ADP-ribose) polymerase; VR, vault RNA; cryoEM, cryoelectron microscopy; TEM, transmission electron microscopy; M-INT, minimal MVP interaction domain; GEMMA, gas-phase electrophoretic mobility molecular analyzer; ESI, electrospray ionization; DMA, differential mobility analyzer; CPC, condensation particle counter; GL-INT, green fluorescent protein variant fused to the MVP interaction domain.
References
- 1.Kedersha NL, Rome LH. Isolation and characterization of a novel ribonucleoprotein particle: large structures contain a single species of small RNA. J Cell Biol. 1986;103:699–709. doi: 10.1083/jcb.103.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kedersha NL, Miquel MC, Bittner D, Rome LH. Vaults. II. Ribonucleoprotein structures are highly conserved among higher and lower eukaryotes. J Cell Biol. 1990;110:895–901. doi: 10.1083/jcb.110.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mossink MH, van Zon A, Franzel-Luiten E, Schoester M, Kickhoefer VA, Scheffer GL, Scheper RJ, Sonneveld P, Wiemer EA. Disruption of the murine major vault protein (MVP/LRP) gene does not induce hypersensitivity to cytostatics. Cancer Res. 2002;62:7298–304. [PubMed] [Google Scholar]
- 4.Hamill DR, Suprenant KA. Characterization of the sea urchin major vault protein: a possible role for vault ribonucleoprotein particles in nucleocytoplasmic transport. Dev Biol. 1997;190:117–28. doi: 10.1006/dbio.1997.8676. [DOI] [PubMed] [Google Scholar]
- 5.Scheffer GL, Wijngaard PL, Flens MJ, Izquierdo MA, Slovak ML, Pinedo HM, Meijer CJ, Clevers HC, Scheper RJ. The drug resistance-related protein LRP is the human major vault protein. Nat Med. 1995;1:578–82. doi: 10.1038/nm0695-578. [DOI] [PubMed] [Google Scholar]
- 6.Kickhoefer VA, Rajavel KS, Scheffer GL, Dalton WS, Scheper RJ, Rome LH. Vaults are up-regulated in multidrug-resistant cancer cell lines. J Biol Chem. 1998;273:8971–4. doi: 10.1074/jbc.273.15.8971. [DOI] [PubMed] [Google Scholar]
- 7.Chugani DC, Rome LH, Kedersha NL. Evidence that vault ribonucleoprotein particles localize to the nuclear pore complex. J Cell Sci. 1993;106(Pt 1):23–9. doi: 10.1242/jcs.106.1.23. [DOI] [PubMed] [Google Scholar]
- 8.Steiner E, Holzmann K, Pirker C, Elbling L, Micksche M, Sutterluty H, Berger W. The major vault protein is responsive to and interferes with interferon-{gamma}-mediated STAT1 signals. J Cell Sci. 2006;119:459–69. doi: 10.1242/jcs.02773. [DOI] [PubMed] [Google Scholar]
- 9.Kolli S, Zito CI, Mossink MH, Wiemer EA, Bennett AM. The Major Vault Protein Is a Novel Substrate for the Tyrosine Phosphatase SHP-2 and Scaffold Protein in Epidermal Growth Factor Signaling. J Biol Chem. 2004;279:29374–85. doi: 10.1074/jbc.M313955200. [DOI] [PubMed] [Google Scholar]
- 10.Mikyas Y, Makabi M, Raval-Fernandes S, Harrington L, Kickhoefer VA, Rome LH, Stewart PL. Cryoelectron microscopy imaging of recombinant and tissue derived vaults: localization of the MVP N termini and VPARP. J Mol Biol. 2004;344:91–105. doi: 10.1016/j.jmb.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 11.Kedersha NL, Heuser JE, Chugani DC, Rome LH. Vaults. III. Vault ribonucleoprotein particles open into flower-like structures with octagonal symmetry. J Cell Biol. 1991;112:225–35. doi: 10.1083/jcb.112.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kickhoefer VA, Searles RP, Kedersha NL, Garber ME, Johnson DL, Rome LH. Vault ribonucleoprotein particles from rat and bullfrog contain a related small RNA that is transcribed by RNA polymerase III. J Biol Chem. 1993;268:7868–73. [PubMed] [Google Scholar]
- 13.Kickhoefer VA, Siva AC, Kedersha NL, Inman EM, Ruland C, Streuli M, Rome LH. The 193-kD vault protein, VPARP, is a novel poly(ADP-ribose) polymerase. J Cell Biol. 1999;146:917–28. doi: 10.1083/jcb.146.5.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kickhoefer VA, Stephen AG, Harrington L, Robinson MO, Rome LH. Vaults and telomerase share a common subunit, TEP1. J Biol Chem. 1999;274:32712–7. doi: 10.1074/jbc.274.46.32712. [DOI] [PubMed] [Google Scholar]
- 15.Stephen AG, Raval-Fernandes S, Huynh T, Torres M, Kickhoefer VA, Rome LH. Assembly of vault-like particles in insect cells expressing only the major vault protein. J Biol Chem. 2001;276:23217–20. doi: 10.1074/jbc.C100226200. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Snow BE, Hande MP, Baerlocher G, Kickhoefer VA, Yeung D, Wakeham A, Itie A, Siderovski DP, Lansdorp PM, Robinson MO, Harrington L. Telomerase-associated protein TEP1 is not essential for telomerase activity or telomere length maintenance in vivo. Mol Cell Biol. 2000;20:8178–84. doi: 10.1128/mcb.20.21.8178-8184.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington L, McPhail T, Mar V, Zhou W, Oulton R, Bass MB, Arruda I, Robinson MO. A mammalian telomerase-associated protein. Science. 1997;275:973–7. doi: 10.1126/science.275.5302.973. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Snow BE, Kickhoefer VA, Erdmann N, Zhou W, Wakeham A, Gomez M, Rome LH, Harrington L. Vault poly(ADP-ribose) polymerase is associated with mammalian telomerase and is dispensable for telomerase function and vault structure in vivo. Mol Cell Biol. 2004;24:5314–23. doi: 10.1128/MCB.24.12.5314-5323.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kickhoefer VA, Liu Y, Kong LB, Snow BE, Stewart PL, Harrington L, Rome LH. The Telomerase/vault-associated protein TEP1 is required for vault RNA stability and its association with the vault particle. J Cell Biol. 2001;152:157–64. doi: 10.1083/jcb.152.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poderycki MJ, Rome LH, Harrington L, Kickhoefer VA. The p80 homology region of TEP1 is sufficient for its association with the telomerase and vault RNAs, and the vault particle. Nucleic Acids Res. 2005;33:893–902. doi: 10.1093/nar/gki234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Zon A, Mossink MH, Schoester M, Scheffer GL, Scheper RJ, Sonneveld P, Wiemer EA. Multiple human vault RNAs. Expression and association with the vault complex. J Biol Chem. 2001;276:37715–21. doi: 10.1074/jbc.M106055200. [DOI] [PubMed] [Google Scholar]
- 22.Kong LB, Siva AC, Kickhoefer VA, Rome LH, Stewart PL. RNA location and modeling of a WD40 repeat domain within the vault. RNA. 2000;6:890–900. doi: 10.1017/s1355838200000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong LB, Siva AC, Rome LH, Stewart PL. Structure of the vault, a ubiquitous cellular component. Structure Fold Des. 1999;7:371–9. doi: 10.1016/s0969-2126(99)80050-1. [DOI] [PubMed] [Google Scholar]
- 24.Kickhoefer VA, Garcia Y, Mikyas Y, Johansson E, Zhou JC, Raval-Fernandes S, Minoofar P, Zink JI, Dunn B, Stewart PL, Rome LH. Engineering of vault nanocapsules with enzymatic and fluorescent properties. Proc Natl Acad Sci U S A. 2005;102:4348–52. doi: 10.1073/pnas.0500929102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broo K, Wei J, Marshall D, Brown F, Smith TJ, Johnson JE, Schneemann A, Siuzdak G. Viral capsid mobility: a dynamic conduit for inactivation. Proc Natl Acad Sci U S A. 2001;98:2274–7. doi: 10.1073/pnas.051598298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bothner B, Dong XF, Bibbs L, Johnson JE, Siuzdak G. Evidence of viral capsid dynamics using limited proteolysis and mass spectrometry. J Biol Chem. 1998;273:673–6. doi: 10.1074/jbc.273.2.673. [DOI] [PubMed] [Google Scholar]
- 27.Li Q, Yafal AG, Lee YM, Hogle J, Chow M. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J Virol. 1994;68:3965–70. doi: 10.1128/jvi.68.6.3965-3970.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bacher G, Szymanski WW, Kaufman SL, Zollner P, Blaas D, Allmaier G. Charge-reduced nano electrospray ionization combined with differential mobility analysis of peptides, proteins, glycoproteins, noncovalent protein complexes and viruses. J Mass Spectrom. 2001;36:1038–52. doi: 10.1002/jms.208. [DOI] [PubMed] [Google Scholar]
- 29.Kaufman SL, Kuchumov AR, Kazakevich M, Vinogradov SN. Analysis of a 3.6-MDa hexagonal bilayer hemoglobin from Lumbricus terrestris using a gas-phase electrophoretic mobility molecular analyzer. Anal Biochem. 1998;259:195–202. doi: 10.1006/abio.1998.2644. [DOI] [PubMed] [Google Scholar]
- 30.Kaufman SL, Skogan JW, Dorman FD, Zarrin F, Lewis KC. Macromolecule Analysis Based on Electrophoretic Mobility in Air: Globular Proteins. Analytical Chemistry. 1996;68:1895–1904. doi: 10.1021/ac951128f. [DOI] [PubMed] [Google Scholar]
- 31.Fuchs NA. On the stationary charge distribution on aerosol particles in a bipolar ionic atmosphere. Geofis Pura Appl. 1963;56:185–193. [Google Scholar]
- 32.Wiedensohler A. An approximation of the bipolar charge distribution for particles in the submicron size range. J Aerosol Sci. 1988;19:387–389. [Google Scholar]
- 33.Kickhoefer VA, Poderycki MJ, Chan EK, Rome LH. The La RNA-binding protein interacts with the vault RNA and is a vault-associated protein. J Biol Chem. 2002;277:41282–6. doi: 10.1074/jbc.M206980200. [DOI] [PubMed] [Google Scholar]
- 34.Loo JA. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom Rev. 1997;16:1–23. doi: 10.1002/(SICI)1098-2787(1997)16:1<1::AID-MAS1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 35.Rostom AA, Fucini P, Benjamin DR, Juenemann R, Nierhaus KH, Hartl FU, Dobson CM, Robinson CV. Detection and selective dissociation of intact ribosomes in a mass spectrometer. Proc Natl Acad Sci U S A. 2000;97:5185–90. doi: 10.1073/pnas.97.10.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Zon A, Mossink MH, Schoester M, Scheffer GL, Scheper RJ, Sonneveld P, Wiemer EA. Structural domains of vault proteins: a role for the coiled coil domain in vault assembly. Biochem Biophys Res Commun. 2002;291:535–41. doi: 10.1006/bbrc.2002.6472. [DOI] [PubMed] [Google Scholar]
- 37.Bothner B, Schneemann A, Marshall D, Reddy V, Johnson JE, Siuzdak G. Crystallographically identical virus capsids display different properties in solution. Nat Struct Biol. 1999;6:114–6. doi: 10.1038/5799. [DOI] [PubMed] [Google Scholar]
- 38.Zheng CL, Sumizawa T, Che XF, Tsuyama S, Furukawa T, Haraguchi M, Gao H, Gotanda T, Jueng HC, Murata F, Akiyama S. Characterization of MVP and VPARP assembly into vault ribonucleoprotein complexes. Biochem Biophys Res Commun. 2005;326:100–7. doi: 10.1016/j.bbrc.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Kozlov G, Vavelyuk O, Minailiuc O, Banville D, Gehring K, Ekiel I. Solution structure of a two-repeat fragment of major vault protein. J Mol Biol. 2006;356:444–52. doi: 10.1016/j.jmb.2005.11.064. [DOI] [PubMed] [Google Scholar]
- 40.Herrmann C, Golkaramnay E, Inman E, Rome L, Volknandt W. Recombinant major vault protein is targeted to neuritic tips of PC12 cells. J Cell Biol. 1999;144:1163–72. doi: 10.1083/jcb.144.6.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eichenmuller B, Kedersha N, Solovyeva E, Everley P, Lang J, Himes RH, Suprenant KA. Vaults bind directly to microtubules via their caps and not their barrels. Cell Motil Cytoskeleton. 2003;56:225–36. doi: 10.1002/cm.10147. [DOI] [PubMed] [Google Scholar]
- 42.Kim E, Lee S, Mian MF, Yun SU, Song M, Yi KS, Ryu SH, Suh PG. Crosstalk between Src and major vault protein in epidermal growth factor-dependent cell signalling. Febs J. 2006;273:793–804. doi: 10.1111/j.1742-4658.2006.05112.x. [DOI] [PubMed] [Google Scholar]